
Study on the Alkylation Reactions of N(7)-Unsubstituted 1,3-Diazaoxindoles
by
 ,
,
Eszter Kókai
1,2,3, 
Judit Halász
1,
András Dancsó
1,
József Nagy
2,
Gyula Simig
1 and
Balázs Volk
1,*
1
Directorate of Drug Substance Development, Egis Pharmaceuticals Plc., P.O. Box 100, 1475 Budapest, Hungary
2
Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, P.O. Box 91, 1521 Budapest, Hungary
3
Department of Materials Technology, GAMF Faculty of Engineering and Computer Science, Pallasz Athéné University, P.O. Box 700, 6001 Kecskemét, Hungary
*
Author to whom correspondence should be addressed.
Molecules 2017, 22(5), 846; https://doi.org/10.3390/molecules22050846
Submission received: 9 April 2017
/
Revised: 13 May 2017
/
Accepted: 16 May 2017
/
Published: 19 May 2017
(This article belongs to the Collection Heterocyclic Compounds)
Abstract
:The chemistry of the 5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (1,3-diazaoxindole) compound family, possessing a drug-like scaffold, is unexplored. In this study, the alkylation reactions of N(7)-unsubstituted 5-isopropyl-1,3-diazaoxindoles bearing various substituents at the C(2) position have been investigated. The starting compounds were synthesized from the C(5)-unsubstituted parent compounds by condensation with acetone and subsequent catalytic reduction of the 5-isopropylidene moiety. Alkylation of the thus obtained 5-isopropyl derivatives with methyl iodide or benzyl bromide in the presence of a large excess of sodium hydroxide led to 5,7-disubstituted derivatives. Use of butyllithium as the base rendered alkylation in the C(5) position possible with reasonable selectivity, without affecting the N(7) atom. During the study on the alkylation reactions, some interesting by-products were also isolated and characterized.
1. Introduction
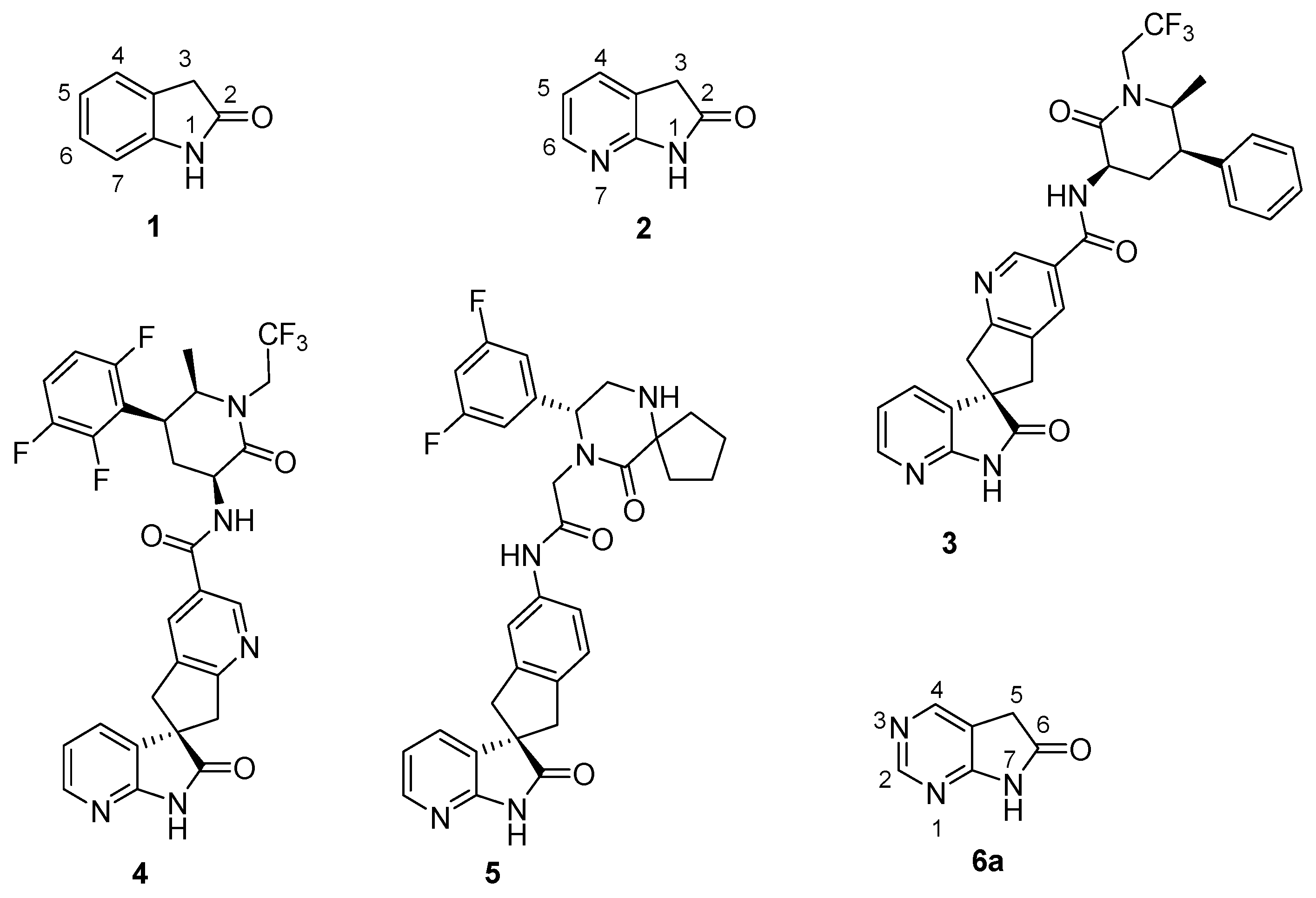
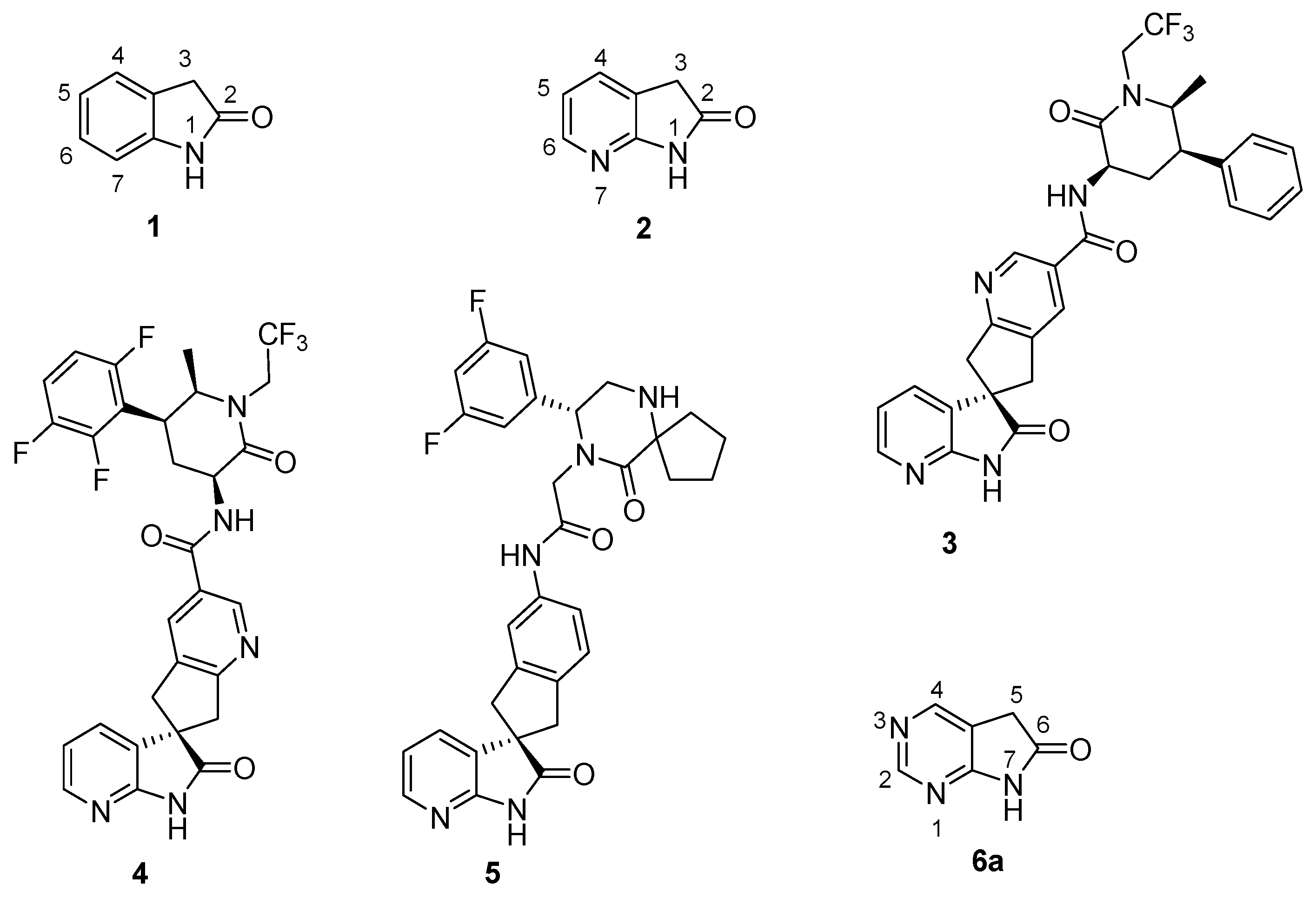
The biological activity of 1,3-dihydro-2H-indol-2-one (oxindole, 1, Figure 1) derivatives is best demonstrated by launched drugs possessing this skeleton: the dopamine agonist ropinirole [1,2] for the treatment of Parkinson's disease and restless leg syndrome; the atypical antipsychotic ziprasidone [3]; and two oncology drugs from the tyrosine kinase inhibitor family, sunitinib [4] and the recently launched nintedanib [5]. The 7-aza analogue of oxindole, 1,3-dihydro-2H-pyrrolo[2,3-b]pyridin-2-one (7-azaoxindole, 2) is also a skeleton of biological importance. Oral calcitonin gene-related peptide (CGRP) antagonists ubrogepant (MK-1602, 3) [6] and MK-8031 (4) [6] are in phase III clinical development, while MK-3207 (5) [7,8,9] had been in phase II, all three for the acute treatment of migraine attacks (Figure 1).
Bioisosteric replacement of the aromatic carbon atom C(5) of 7-azaoxindole (2) with a nitrogen atom, i.e., that of the pyridine ring by a pyrimidine nucleus [10,11,12] formally leads to 5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (6a, 1,3-diazaoxindole, Figure 1).
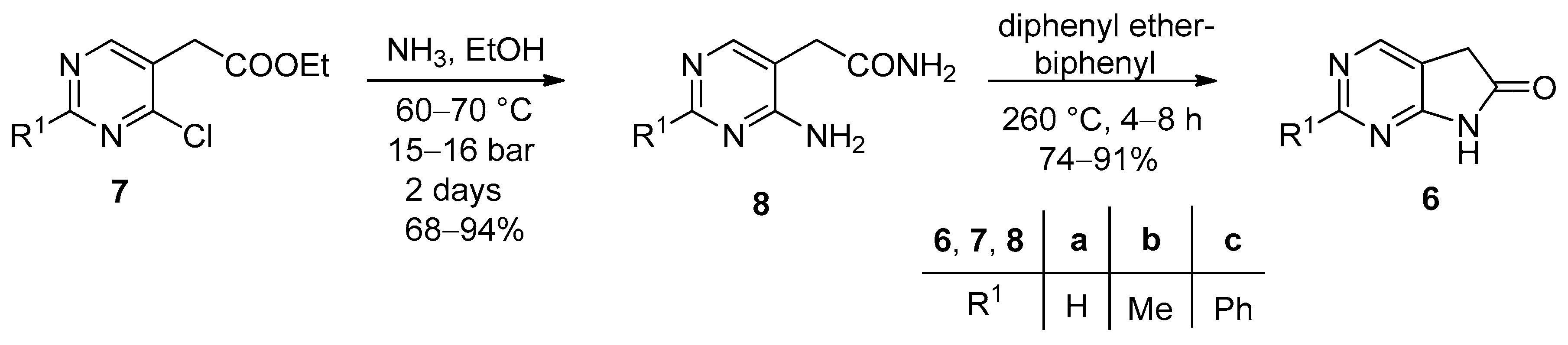
Recently we have reported a new synthetic route to 1,3-diazaoxindoles 6 (Scheme 1). Treatment of ethyl 2-(4-chloropyrimidin-5-yl)acetate derivatives 7 with ethanolic ammonia at 60–70 °C afforded the corresponding 2-(4-aminopyrimidin-5-yl)acetamides 8, which were finally cyclized to the target compounds 6 in a mixture of diphenyl ether and biphenyl at 260 °C [13].
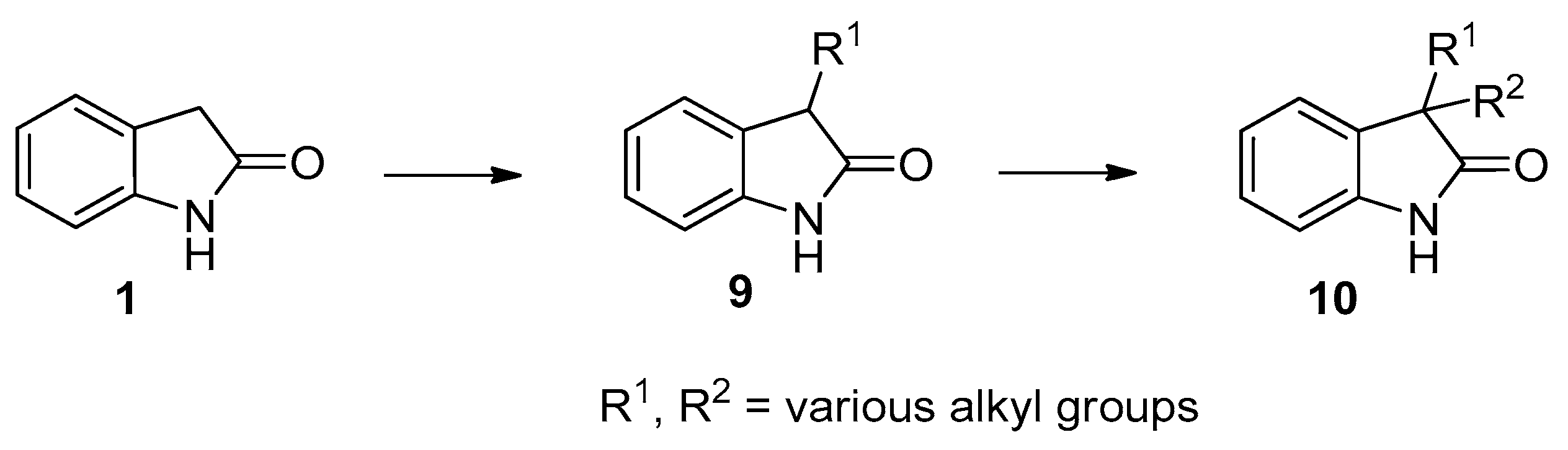
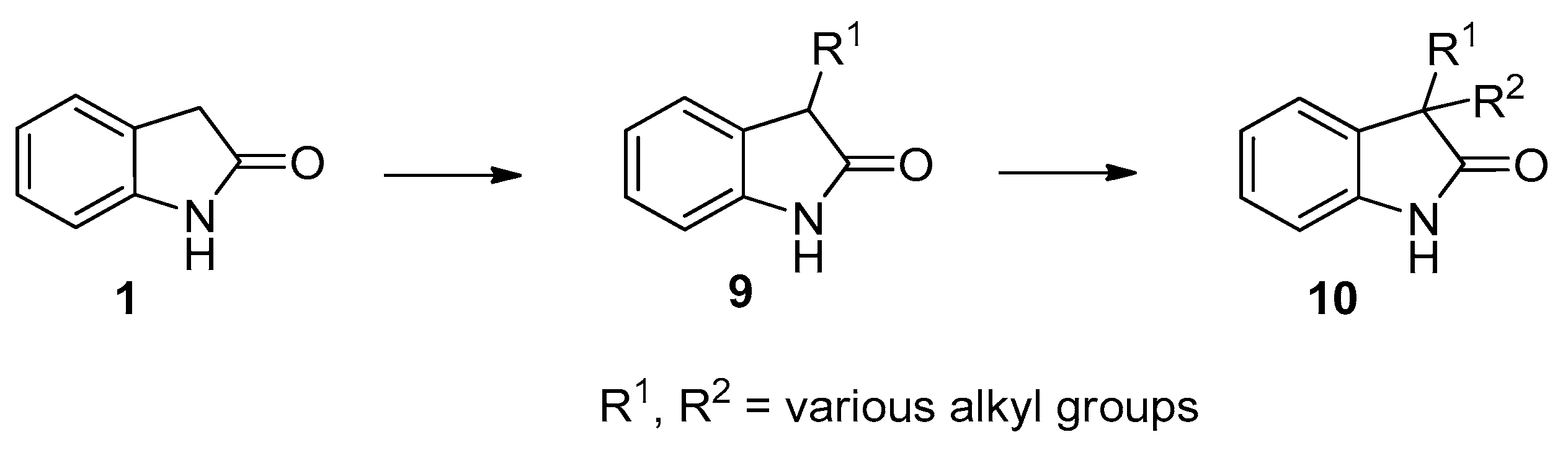
In continuation of our studies on the monoalkylation at the C(3) position of N-unprotected oxindole (1) [14,15,16] to give 3-alkyloxindoles 9 and on the selective introduction of the second alkyl group, without N-alkylation to afford N-unprotected 3,3-dialkyloxindoles 10 (Scheme 2) [17], we now aimed to elaborate a method for the introduction of two different alkyl groups into the structurally related carbonyl-adjacent C(5) site of N-unprotected 1,3-diazaoxindoles 6.
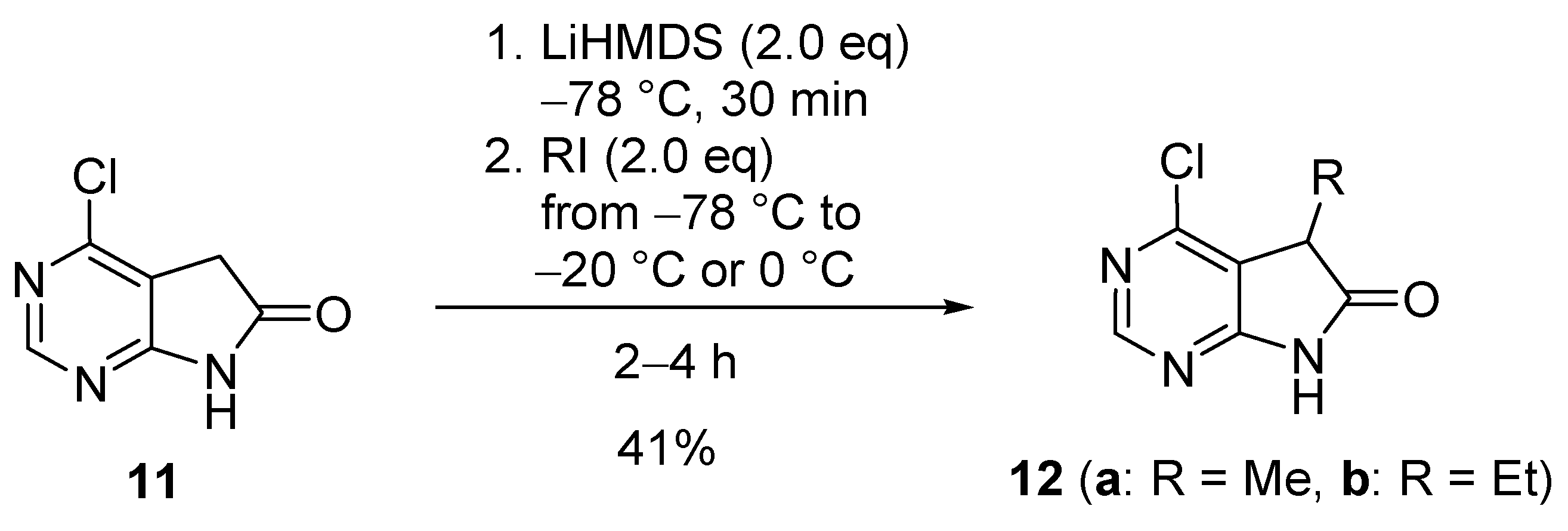
A comprehensive literature search revealed that no systematic study had been carried out on the C(5)-monoalkylation or C(5)-dialkylation of 1,3-diazaoxindoles 6. Alkylation of the parent compound 6a (Scheme 1) itself is unprecedented, and only some special examples are described in patent applications as discussed below. C(5)-Monoalkylation of 4-chloro-1,3-diazaoxindole (11, Scheme 3), unsubstituted at the C(5) and N(7) atoms, was performed by Shepherd et al. using lithium bis(trimethylsilyl)amide (LiHMDS, 2.0 eq.) as the base in tetrahydrofuran (THF), with methyl or ethyl iodide (MeI or EtI, 2.0 eq.) to give the 5-methyl and 5-ethyl derivatives 12a and 12b, respectively, in both cases with 41% yield [18].
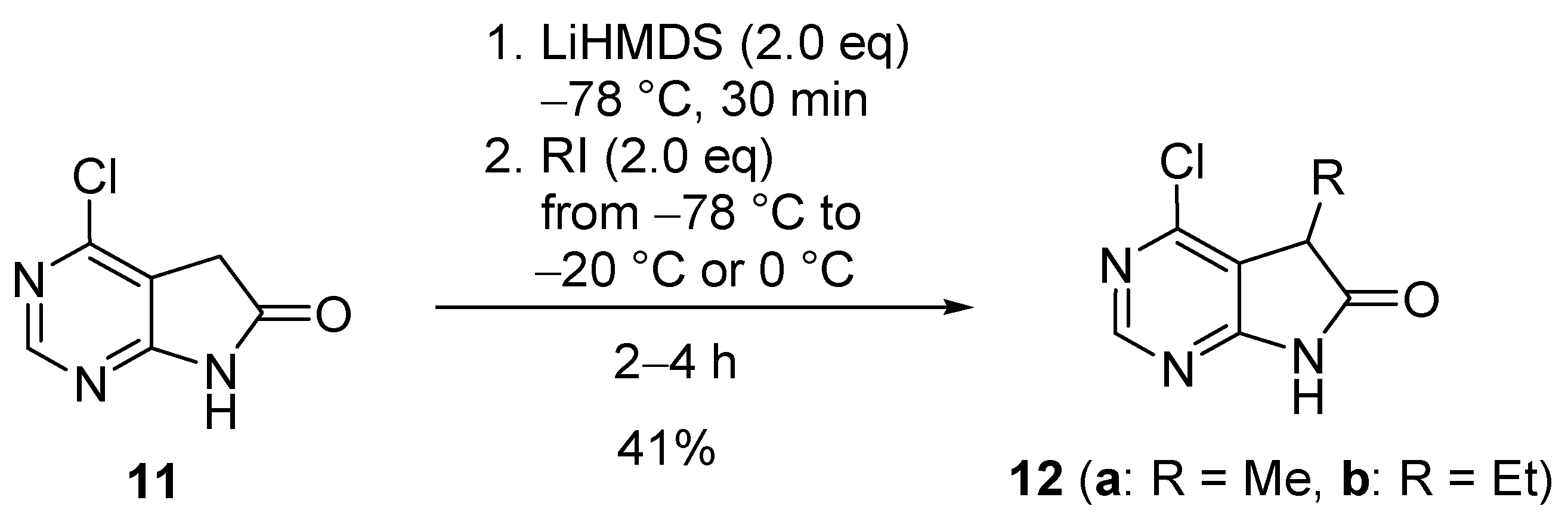
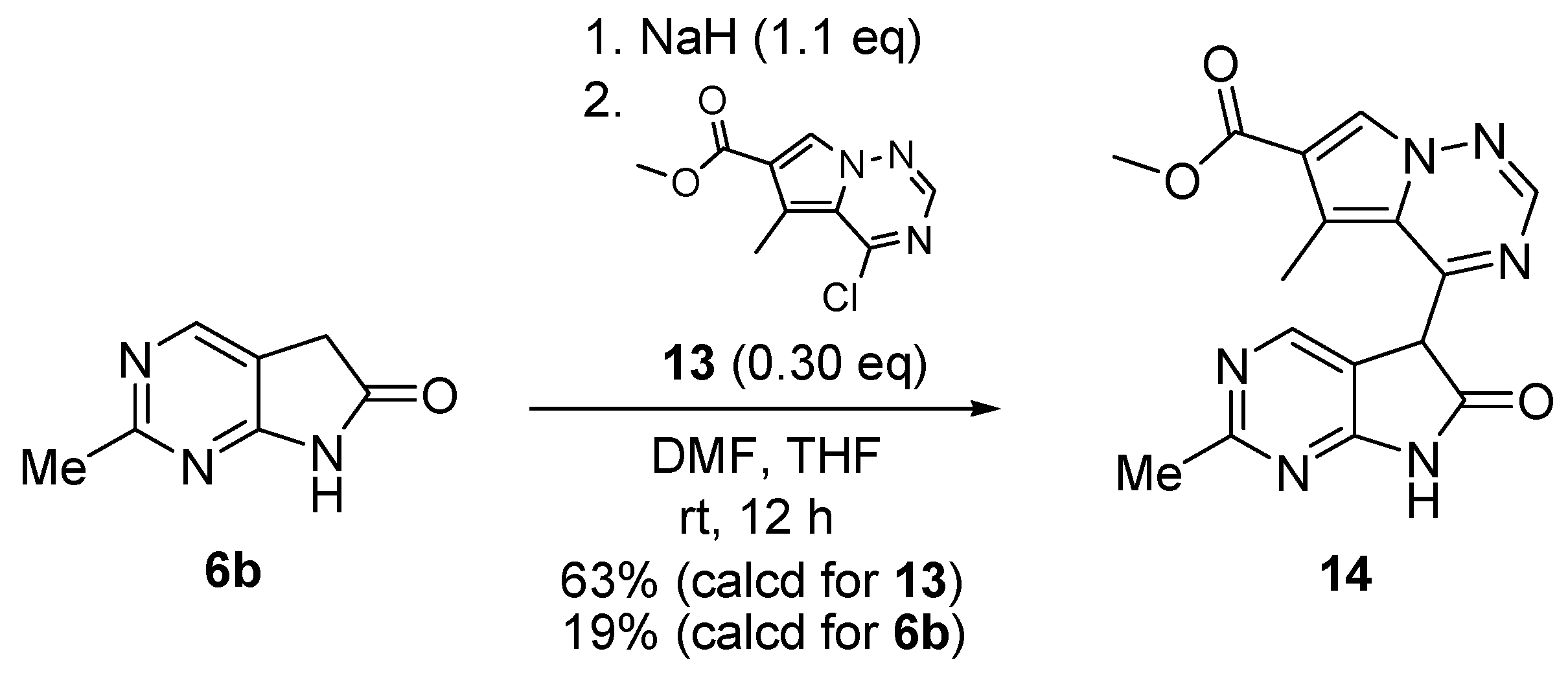
C(5)-Monosubstitution of 2-methyl-1,3-diazaoxindole (6b, Scheme 4) was carried out with chloro-substituted pyrrolotriazine 13 (0.30 eq.) in the presence of sodium hydride (1.1 eq.) to afford pyrrolopyrimidinyl-pyrrolotriazine 14 in 19% yield (calculated for 6b) [19].
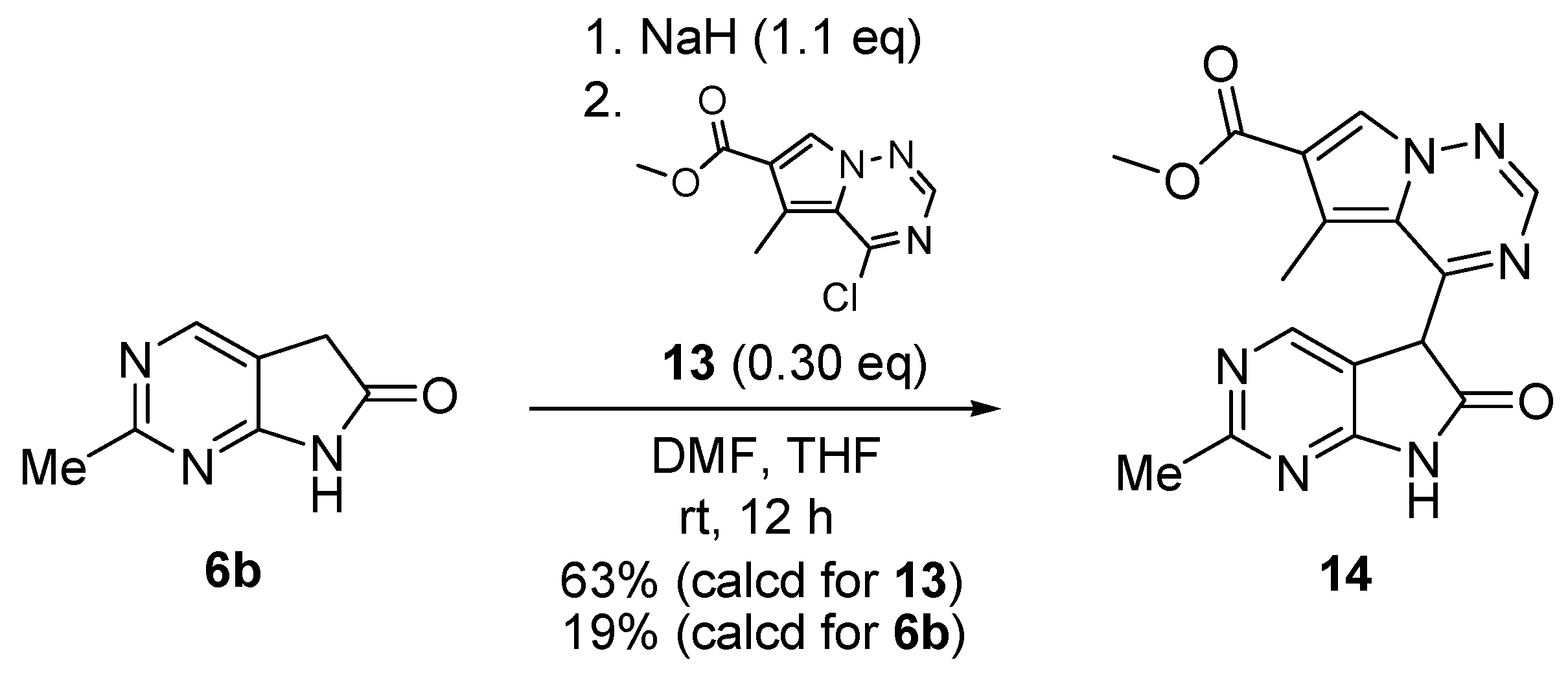
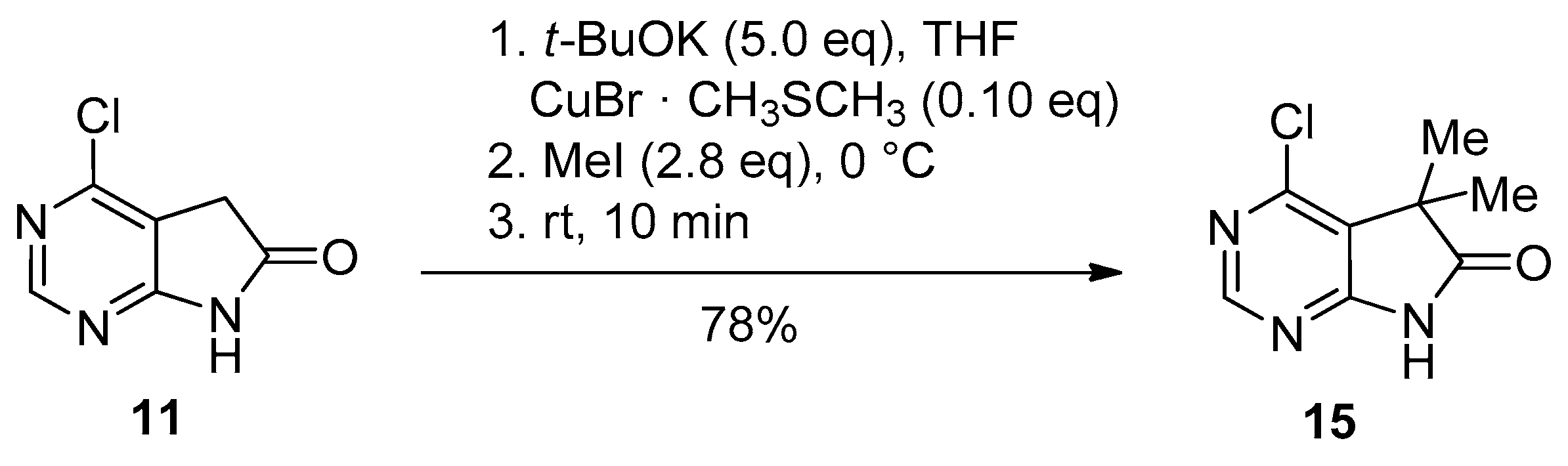
C(5)-Dialkylation of 4-chloro derivative 11 with potassium tert-butoxide (t-BuOK, 5.0 eq.) and MeI (2.8 eq.) in the presence of dimethyl sulfide and copper(I)bromide between 0 °C and ambient temperature gave 5,5-dimethyl derivative 15 in 78% yield (Scheme 5) [19].
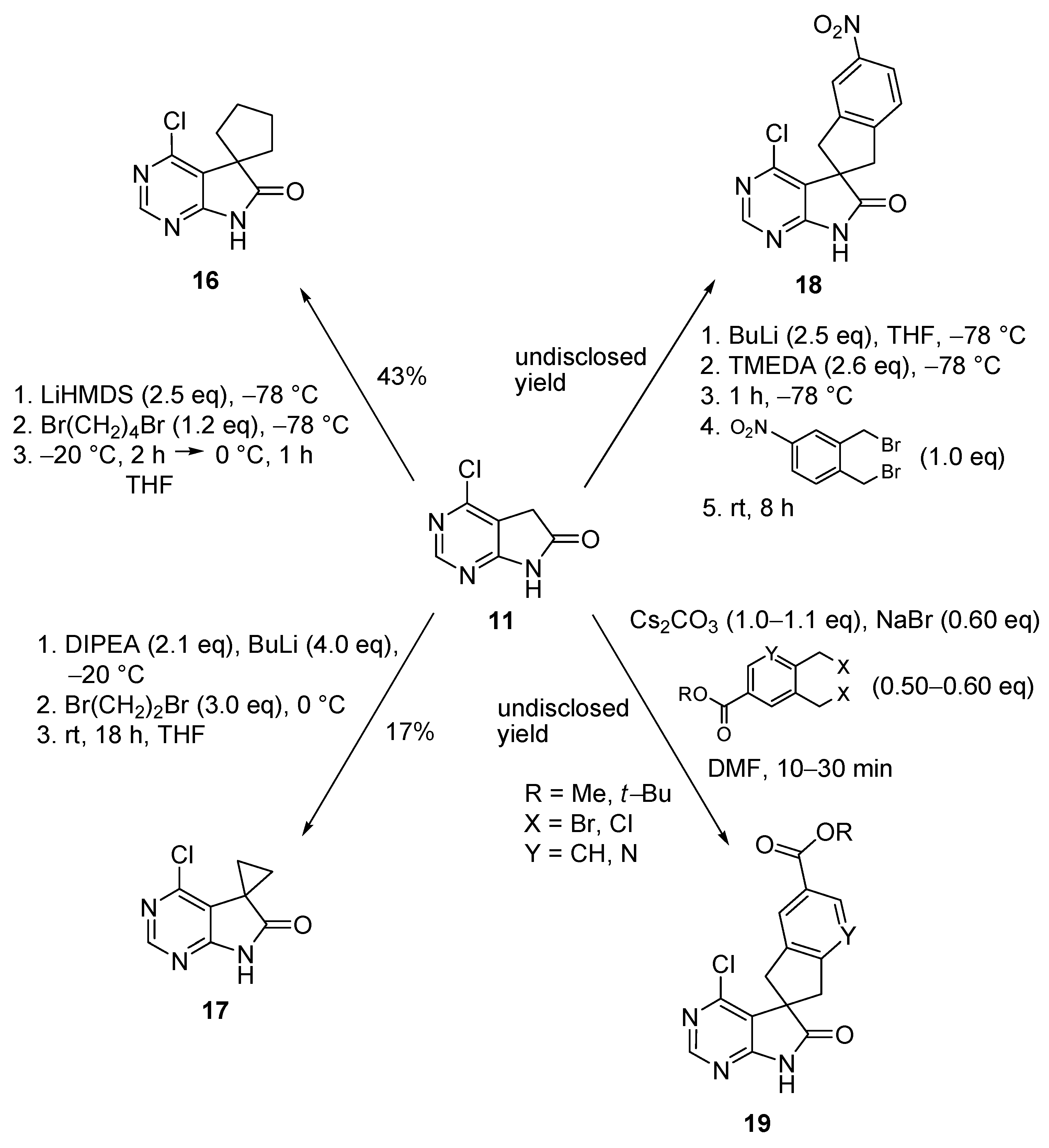
When the alkylation of 11 was performed with bis(haloalkyl) compounds, the corresponding spiro products 16–19 were obtained in low or undisclosed yields (Scheme 6): e.g., (i) with 1,4-dibromobutane (1.2 eq.) in the presence of LiHMDS (2.5 eq.) in THF [19]; (ii) with 1,2-dibromoethane (3.0 eq.) in the presence of butyllithium (BuLi, 4.0 eq.) and N,N-diisopropylethylamine (DIPEA) in THF [20]; (iii) with 1,2-bis(bromomethyl)-4-nitrobenzene (1.0 eq.) in the presence of BuLi (2.5 eq.) and tetramethylethylenediamine (TMEDA) in THF [21,22,23,24]; or (iv) with methyl 3,4-bis(bromomethyl)-benzoate (0.50 eq.) or tert-butyl 5,6-bis(chloromethyl)pyridine-3-carboxylate (0.60 eq.) in the presence of cesium carbonate (1.1 eq.) and sodium bromide in dimethyl formamide (DMF) [6,25,26].
All the above reactions led to the desired C(5)-alkyl or C(5)-dialkyl derivatives in low, moderate or undisclosed yields. The selectivity between the C(5) mono- and dialkyl products, and the formation of N(7)-alkylated by-products were not discussed.
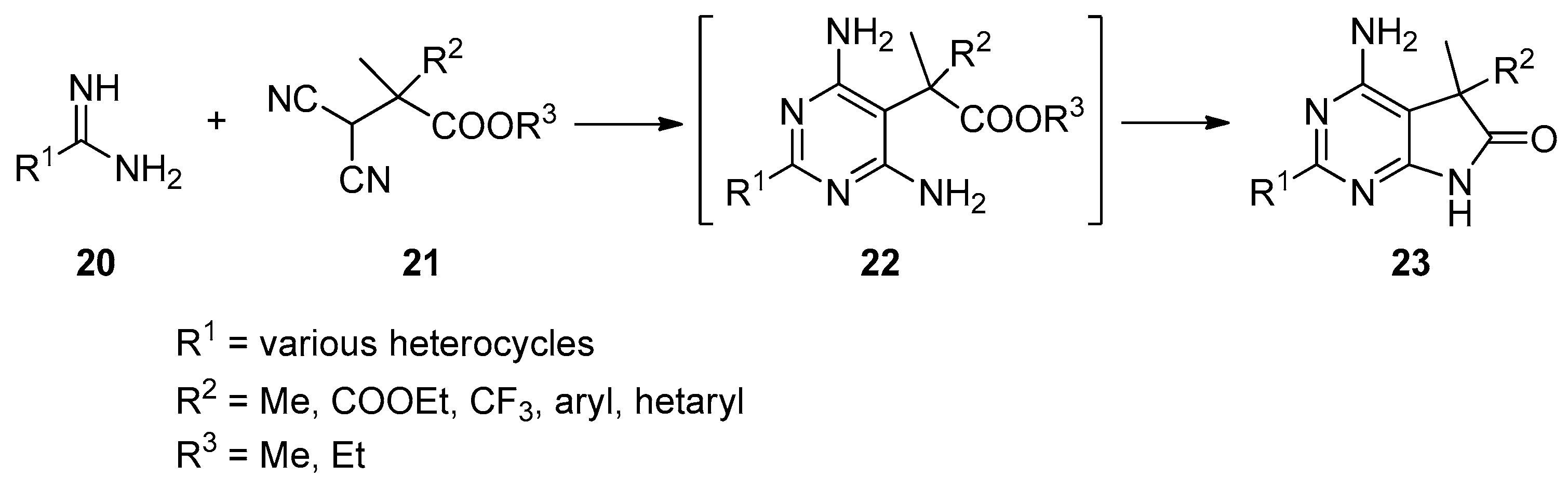
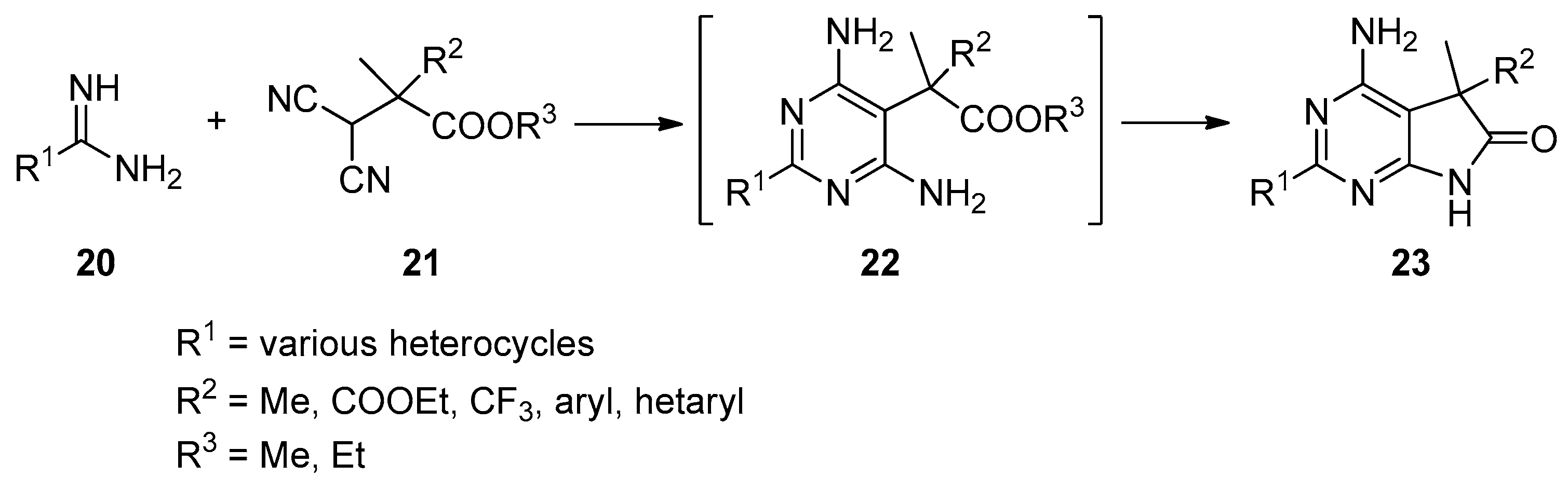
An alternative approach for the preparation of N(7)-unsubstituted-C(5)-mono- or C(5)-disubstituted 1,3-diazaoxindoles is also described [27,28,29,30,31]. Herein, the pyrrolo[2,3-d]pyrimidin-6-one scaffold is synthesized by a condensation of building blocks already containing the required substituents in the appropriate positions. This strategy is exemplified by the reaction sequence outlined in Scheme 7.
In the reaction of amidines 20 with the corresponding substituted malonitriles 21, C(5)-disubstituted 1,3-diazaoxindoles 23 were obtained, obviously via pyrimidines 22. The common drawback of these reactions is that the C(5) substituents of the target compounds have to be present in the starting materials 21, thereby limiting the versatility of this approach.
2. Results and Discussion
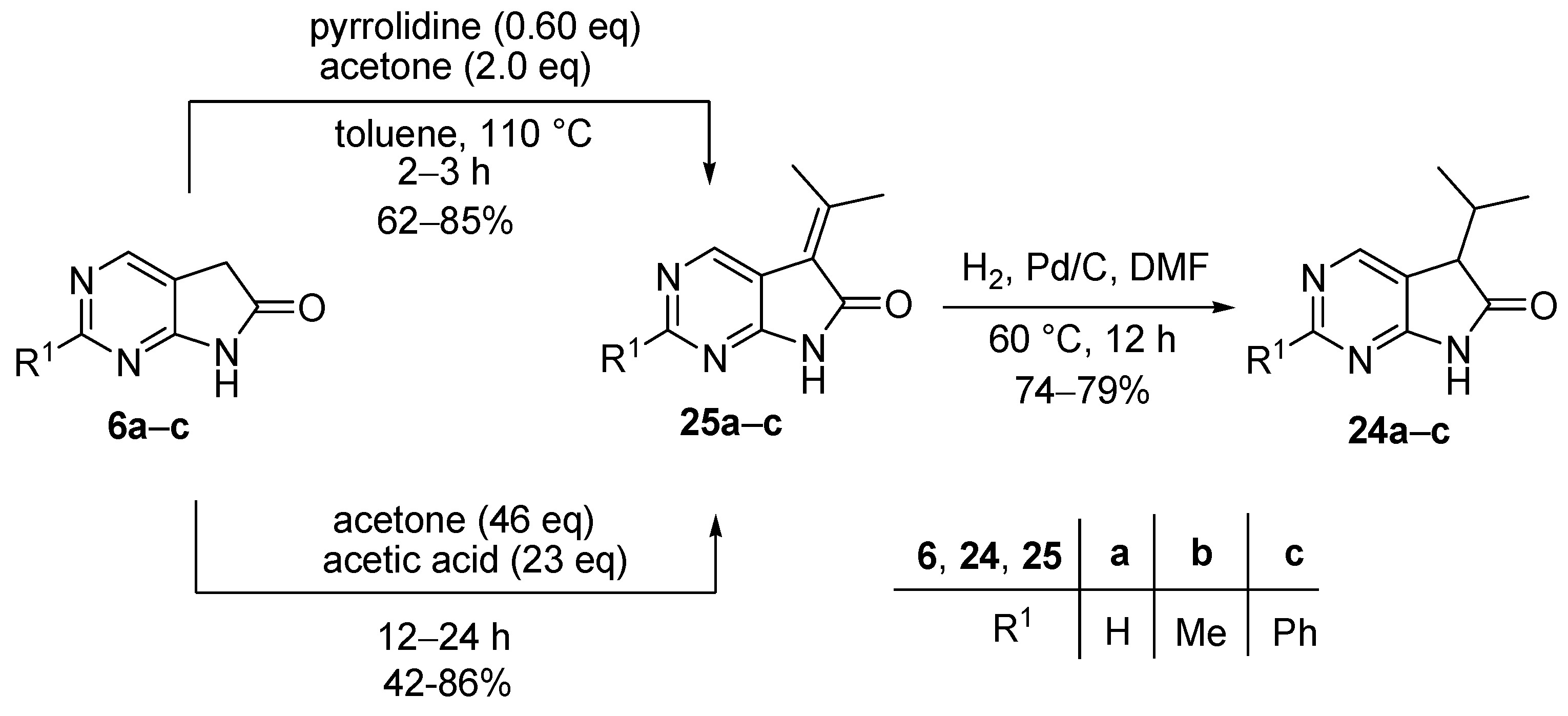
In the present study, we aimed to elaborate a general procedure for the introduction of two different alkyl groups into the C(5) position of N(7)-unprotected 1,3-diazaoxindoles 6. We decided to use the isopropyl group as the first alkyl substituent in position C(5), in order to increase steric hindrance in the second alkylation reaction. 5-Isopropyl-1,3-diazaoxindoles 24 can be prepared by using methods described in the literature for C(3)-alkylation of oxindoles [32,33,34]. Condensation of 1,3-diazaoxindoles 6 with acetone either under basic or acidic conditions afforded 5-isopropylidene-1,3-diazaoxindoles 25, which were reduced by catalytic hydrogenation to give 5-isopropyl-1,3-diazaoxindoles 24 (Scheme 8).
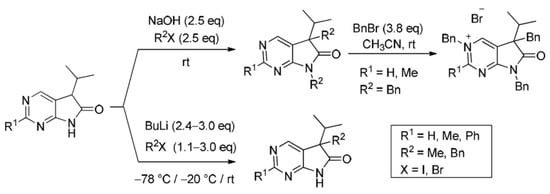
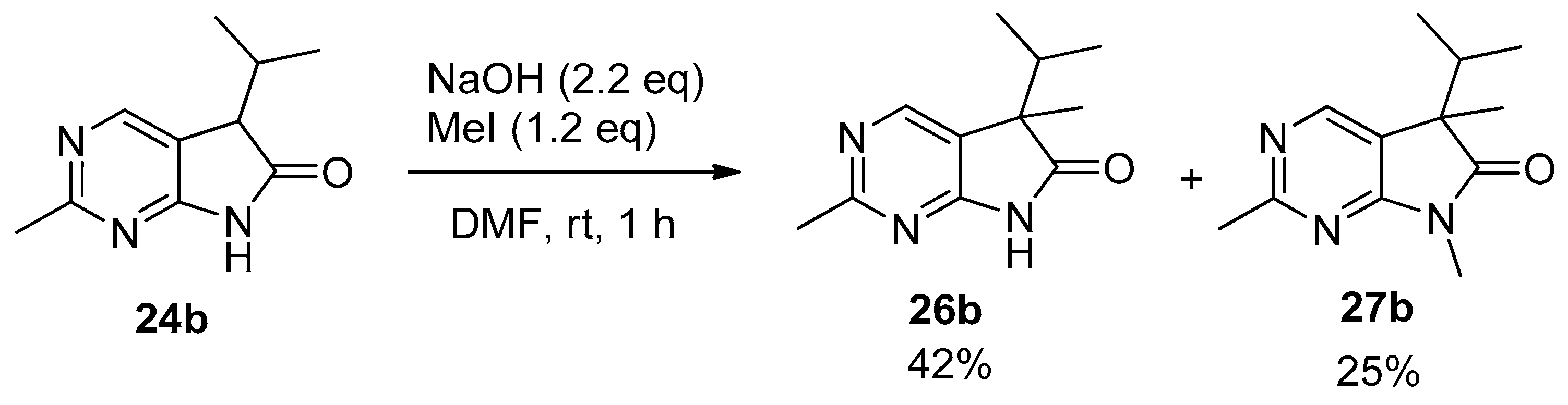
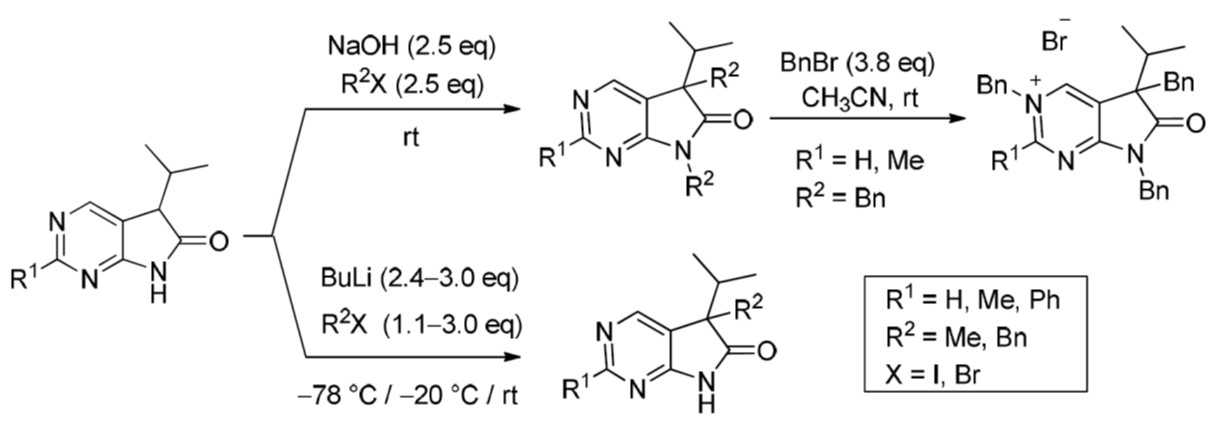
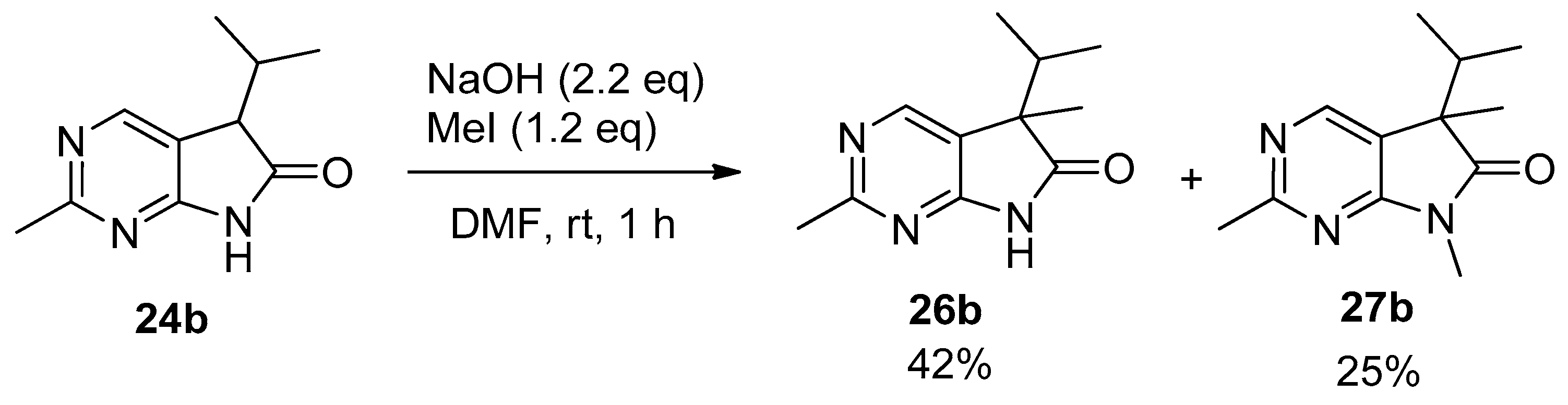
In order to study the introduction of a second alkyl group into the C(5) position of 5-isopropyl-1,3-diazaoxindoles 24, first we treated 2-methyl derivative 24b with MeI (1.2 eq.) in the presence of sodium hydroxide (NaOH, 2.2 eq.) in DMF at room temperature (Scheme 9). In addition to C(5)-methylated product 26b, a substantial amount of C(5),N(7)-dimethylated compound 27b was also formed (besides unidentified minor products), as detected by the LC-MS analysis of the product mixture. After purification by flash chromatography, products 26b and 27b were isolated in 42 and 25% yields, respectively. This finding is in agreement with the experience gained with oxindoles [35,36,37,38]: sodium bases seem to be unsuitable for the high-yielding selective introduction of a second substituent into position C(5) also in the case of 1,3-diaza-derivatives 24.
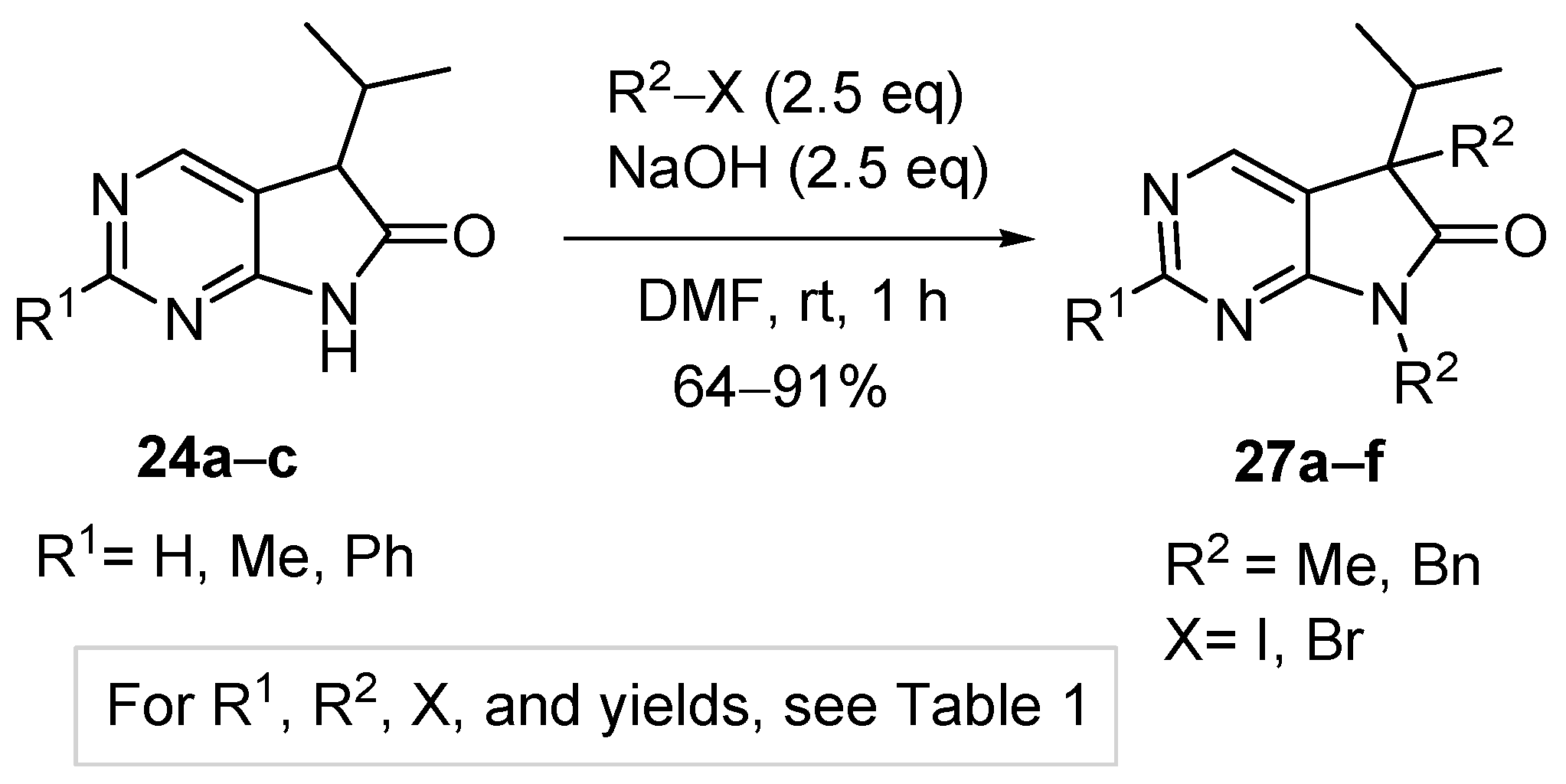
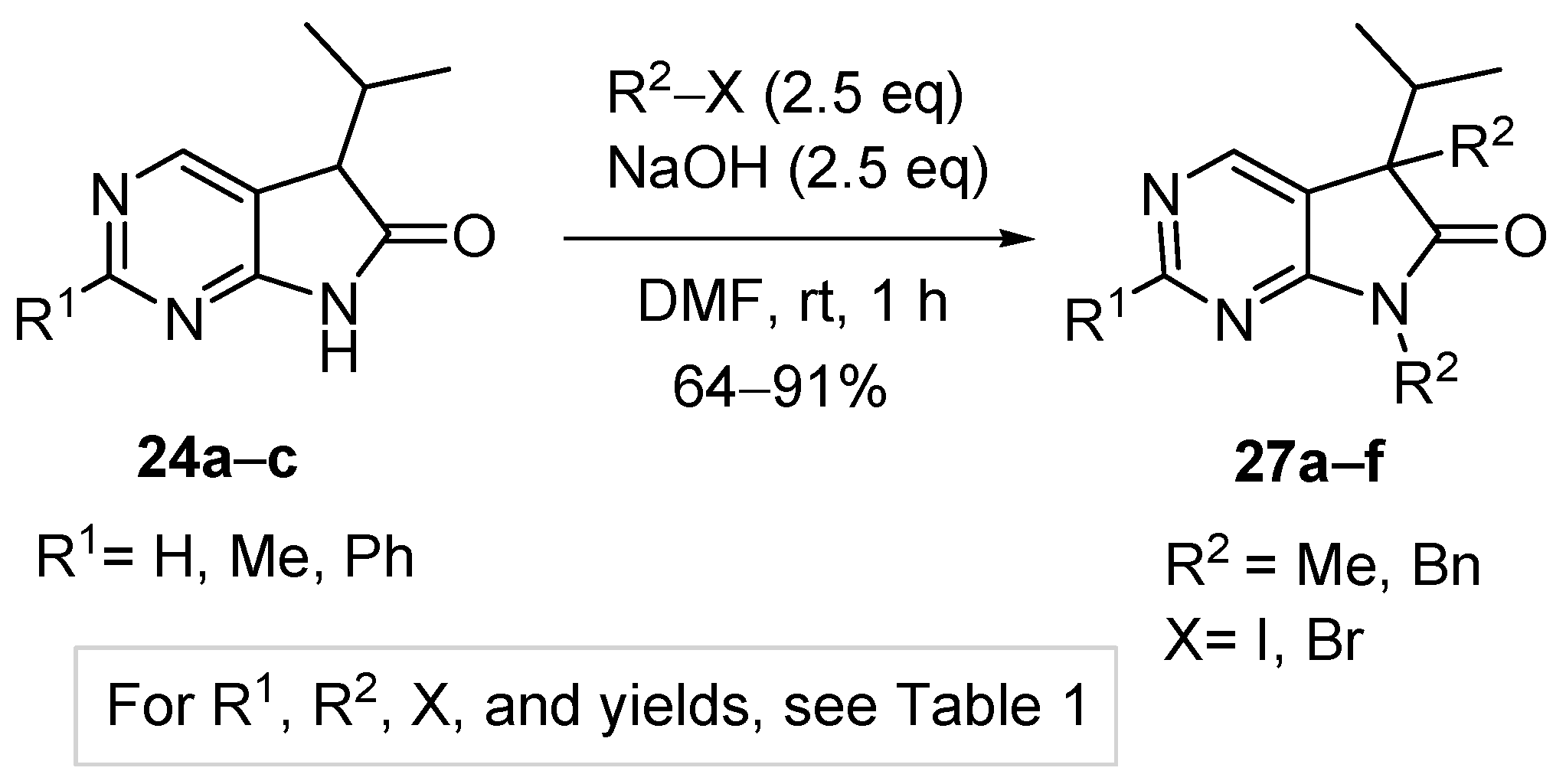
Aiming at the elaboration of an efficient method for the synthesis of C(5),N(7)-disubstituted derivatives 27, we carried out the alkylation of 2-unsubstituted (24a), 2-methyl- (24b) or 2-phenyl (24c) congeners with a higher excess of MeI (2.5 eq., Table 1, entries 1–3) or benzyl bromide (BnBr, 2.5 eq., entries 4–6) in the presence of NaOH (2.5 eq.) in DMF at ambient temperature. 5,7-Dimethyl(dibenzyl)-5-isopropyl-1,3-diazaoxindoles (27a–f) were obtained in good yields (Scheme 10).
As demonstrated by the reactions carried out using NaOH at our laboratory and by the procedures described in the literature using sodium, potassium or cesium bases for the deprotonation of 1,3-diazaoxindoles, selective alkylation at the C(5) position could not be achieved. Therefore, we continued our efforts by applying BuLi as the base in the alkylation reactions, which proved to be the optimal base in the analogous regioselective C(3)-alkylation reactions of N-unprotected oxindoles, particularly for the introduction of a second substituent into the carbonyl-adjacent C(3) position of N-unprotected 3-monoalkyloxindoles 9 (Scheme 2) [17,39].
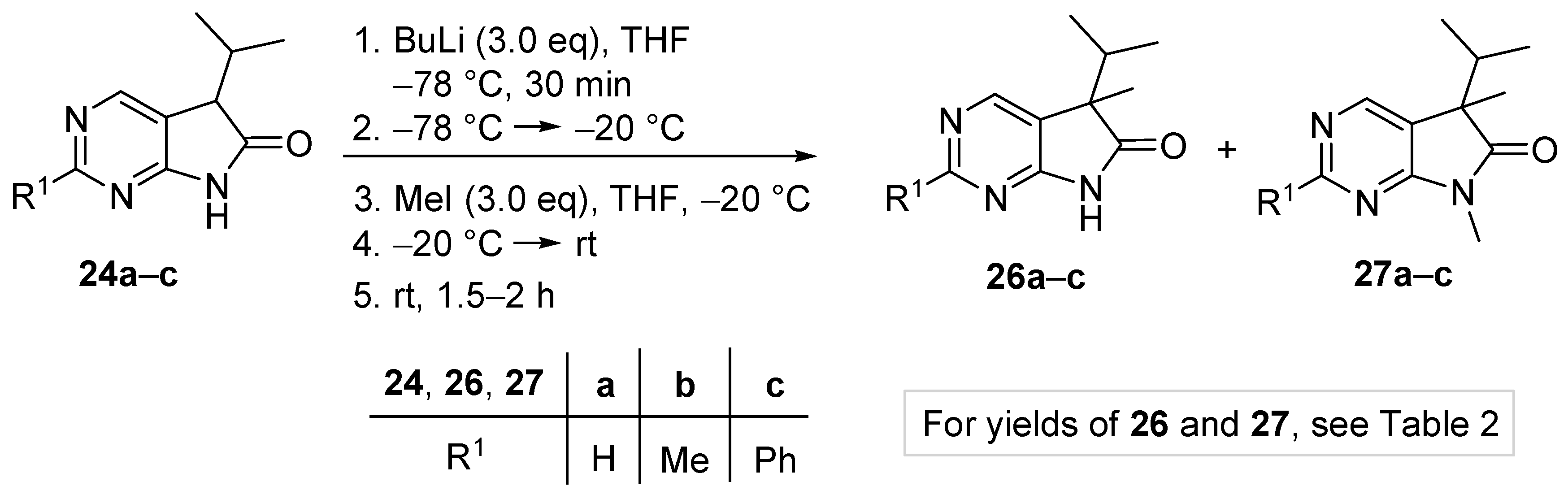
First we carried out reactions using a large excess of both BuLi and the alkylating agent in order to obtain a full picture of the regiochemistry of the alkylations. Methylation of compounds 24a–c with BuLi (3.0 eq.) and MeI (3.0 eq.) was performed as follows (Scheme 11): (i) a solution of compounds 24a–c in THF was added dropwise to the solution of BuLi in hexanes and THF −78 °C, and the reaction mixture was stirred at −78 °C for further 30 min; (ii) the reaction mixture was allowed to warm to −20 °C; (iii) a solution of MeI in THF was added dropwise at −20 °C; (iv) the mixture was allowed to warm to room temperature; (v) then it was stirred at this temperature for further 1.5–2 h. This protocol led to a mixture of 5-isopropyl-5-methyl- (compounds 26a–c) and 5-isopropyl-5,7-dimethyl-1,3-diazaoxindoles 27a–c. The yields of isolated products 26a–c and 27a–c after work-up and chromatographic purification can be found in Table 2 indicating that, despite the high excess of BuLi and MeI, the selectivity of the alkylation was quite high in the case of the 2-unsubstituted congener 24a (entry 1), while it was poor in the case of the 2-methyl (24b) and 2-phenyl (24c) derivatives (entries 2–3).
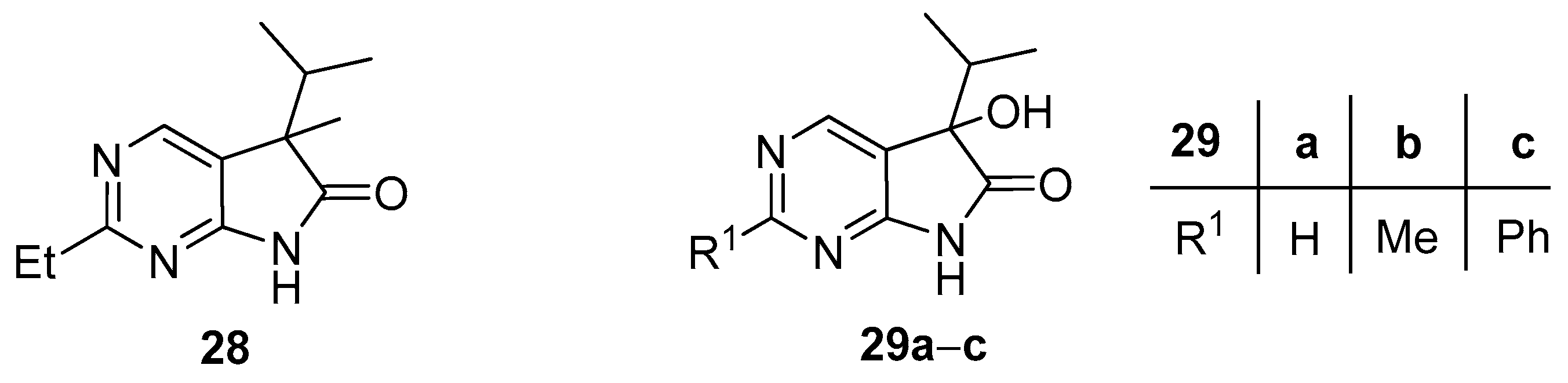
When deprotonation of 2-methyl derivative 24b with BuLi (3.0 eq.) was performed at −20 °C (instead of −78 °C), and the reaction mixture was allowed to warm to room temperature, finally MeI (3.0 eq.) was added at this temperature (instead of −20 °C), methylation of the 2-methyl moiety was also observed resulting in 2-ethyl derivative 28 (Figure 2) as a minor product in 11% yield after flash chromatography, besides 26b (30%) and 27b (17%). A similar reactivity of the methyl moiety of 2-methylpyrimidines was observed by other research groups [40,41].
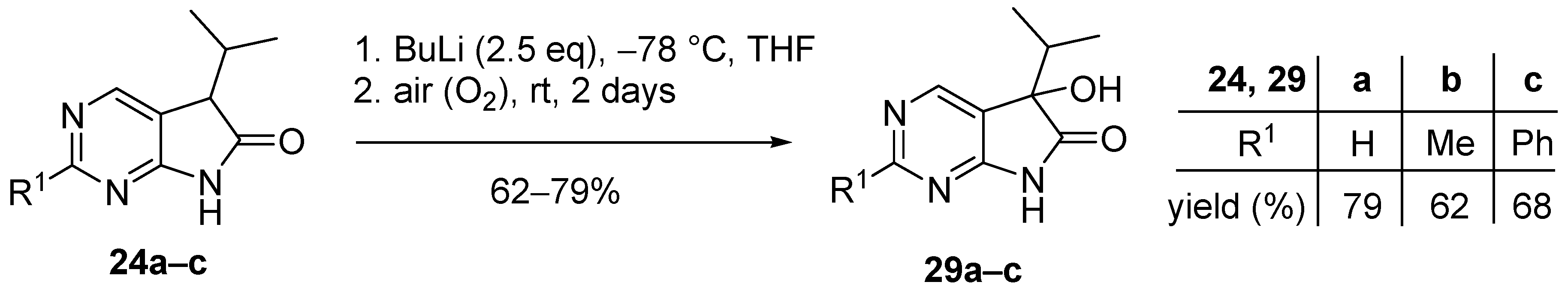
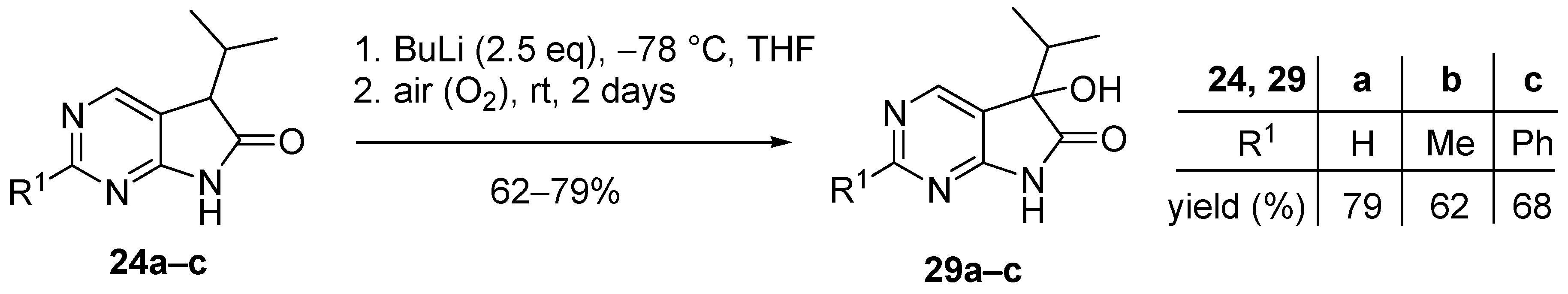
As the consequence of an insufficient pre-inertization with argon, 5-hydroxy byproducts 29a–c (Figure 2) were also isolated in some cases during our preliminary experiments. The targeted synthesis of the 5-hydroxy derivatives 29a–c was carried out with BuLi (2.5 eq.) without alkylating agent and under non-inert conditions with good yields (Scheme 12). The presence of the hydroxy moiety in products 29a–c renders further functionalizations possible thereby making these compounds valuable synthetic building blocks. An analogous oxidative side reaction via the corresponding hydroperoxide is also described in the oxindole family [42], and the corresponding 3-hydroxy derivatives in the oxindole series were also synthesised at our laboratory [17].
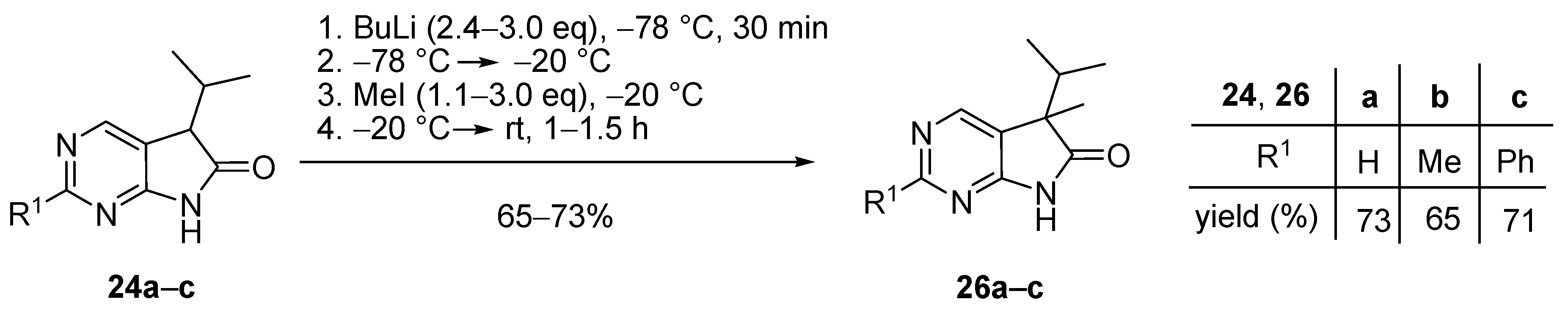
In order to achieve full conversion of the starting materials 24, to maximize the amount of monomethyl products 26 and to minimize that of dimethyl byproducts 27, various conditions were explored, including the excess of BuLi and MeI, different deprotonation temperatures and the temperature of MeI addition (Scheme 13). It was found that significantly different protocols had to be used for starting materials 24a, 24b and 24c. Deprotonation with BuLi leading to the formation of the corresponding dianions was performed at −78 °C in each case, although with different amounts of BuLi (2.4–3.0 eq.). Then, contrary to our earlier observations with 3-monoalkyloxindoles (9) where MeI was also added at −78 °C [17], for 1,3-diazaoxindoles 24a–c we found that addition of MeI had to be carried out at −20 °C so that starting materials 24a–c are fully consumed. Finally, the reaction mixture was allowed to warm to ambient temperature. When using 2-phenyl derivative 24c as the starting material, a significant amount (19%) of 5,7-dimethyl derivative 27c was formed even with 1.4 eq. of MeI, therefore the reaction was finally performed with a lower excess (1.1 eq.) of the alkylating agent. Using the optimized conditions, C(5)-methylated products 26a–c were obtained in good yields (65–73%).
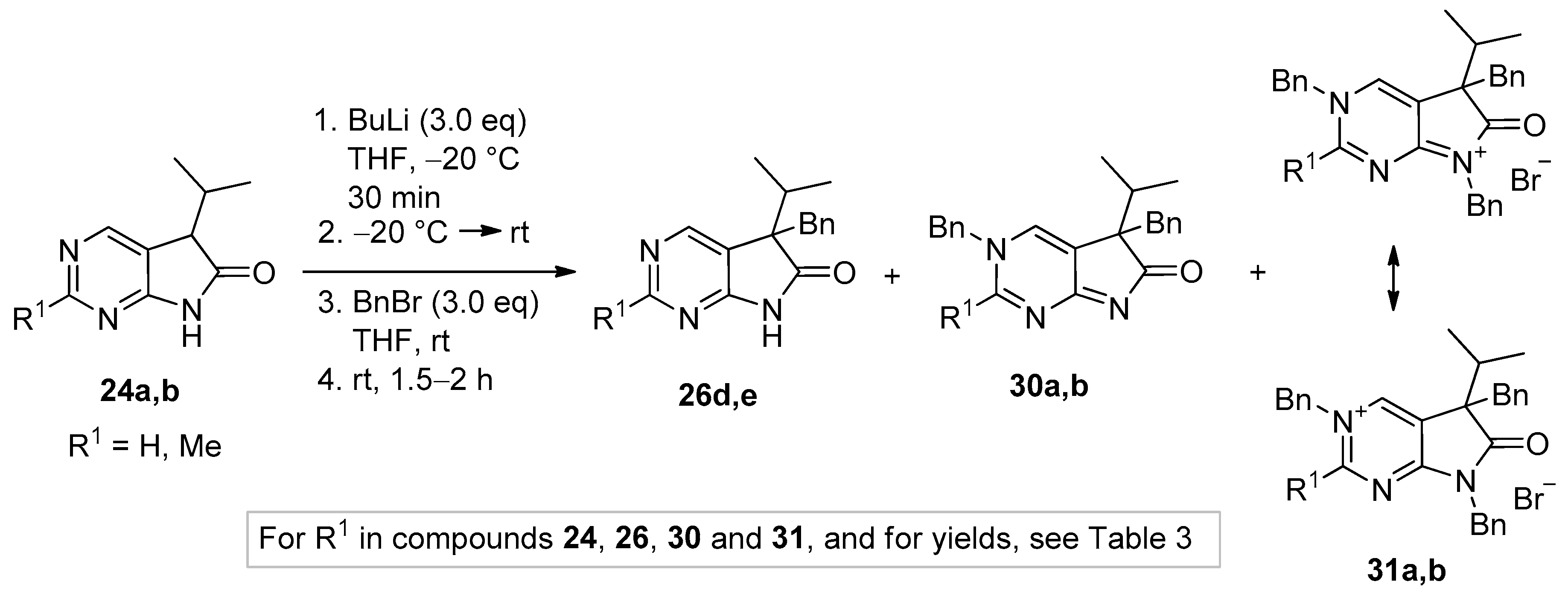
Next, alkylations with BnBr were attempted. When the deprotonation of 2-unsubstituted (24a) and 2-methyl-1,3-diazaoxindole (24b) with BuLi (3.0 eq.) was performed at −20 °C, then the addition of BnBr (3.0 eq.) at room temperature, surprising results were observed. After the work-up of the reaction mixture by flash chromatography, in addition to C(5)-benzylated derivatives 26d,e, N(3),C(5)-dibenzylated compounds 30a,b and N(3),C(5),N(7)-tribenzylated quaternary bromide salts 31a,b were also isolated (Scheme 14), with a product distribution quite different in the two reactions (compare entries 1 and 2 in Table 3) and also substantially different from those of the alkylations performed with 3.0 eq. MeI (Scheme 11). It is also noteworthy that benzylation of the 2-methyl moiety of 24b was not detected.
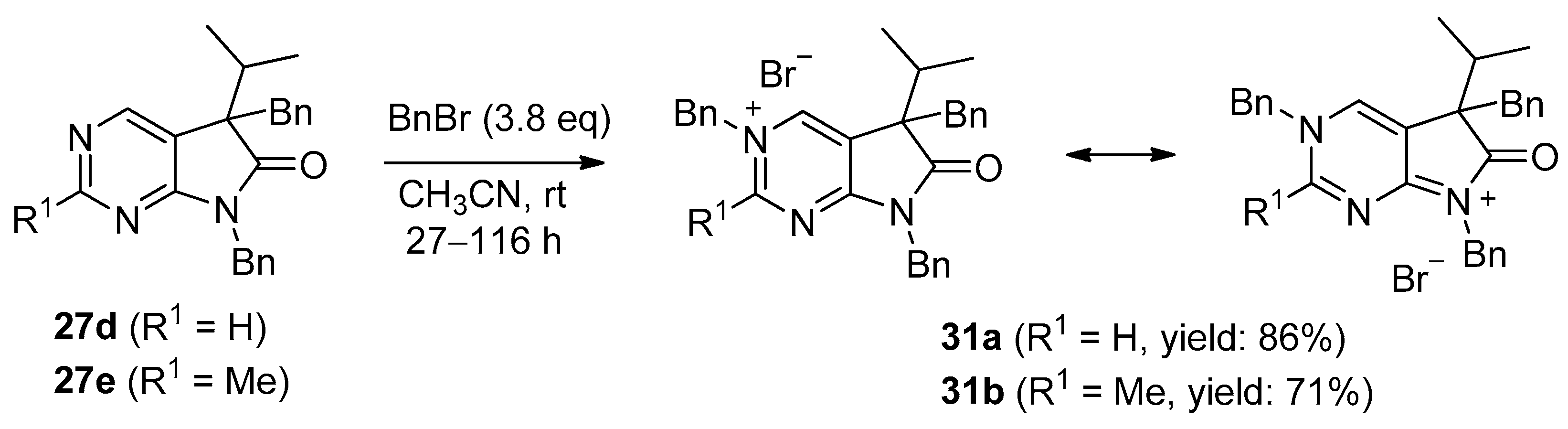
Tribenzylated quaternary bromide salts 31a,b were also prepared by benzylation of 5,7-dibenzyl-1,3-diazaoxindoles 27d,e with a high excess of BnBr (3.8 eq.) in acetonitrile at ambient temperature (Scheme 15). After long reaction times (27–116 h), quaternary salts 31a and 31b were obtained in good yields (86% and 71%, respectively).
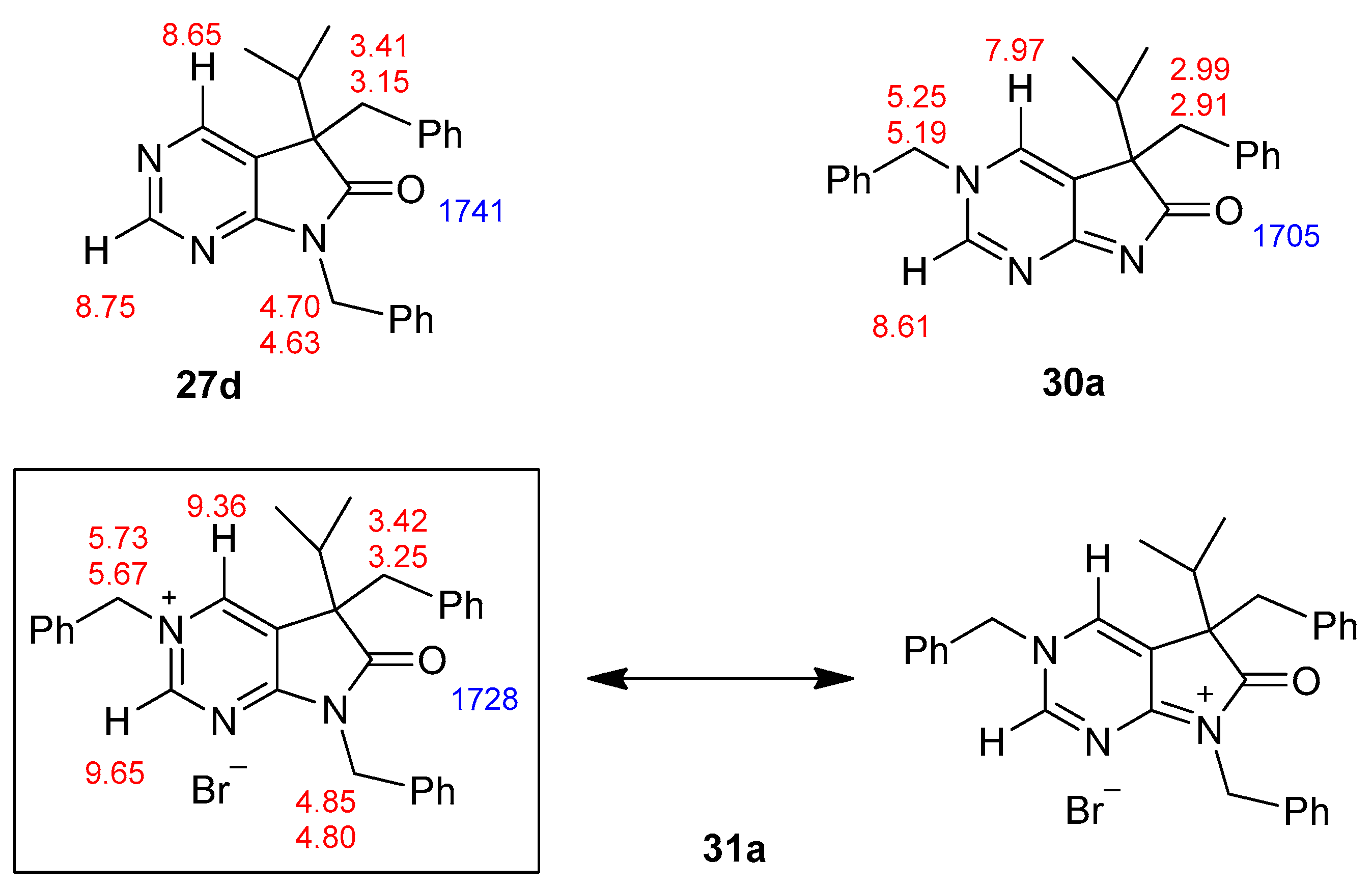
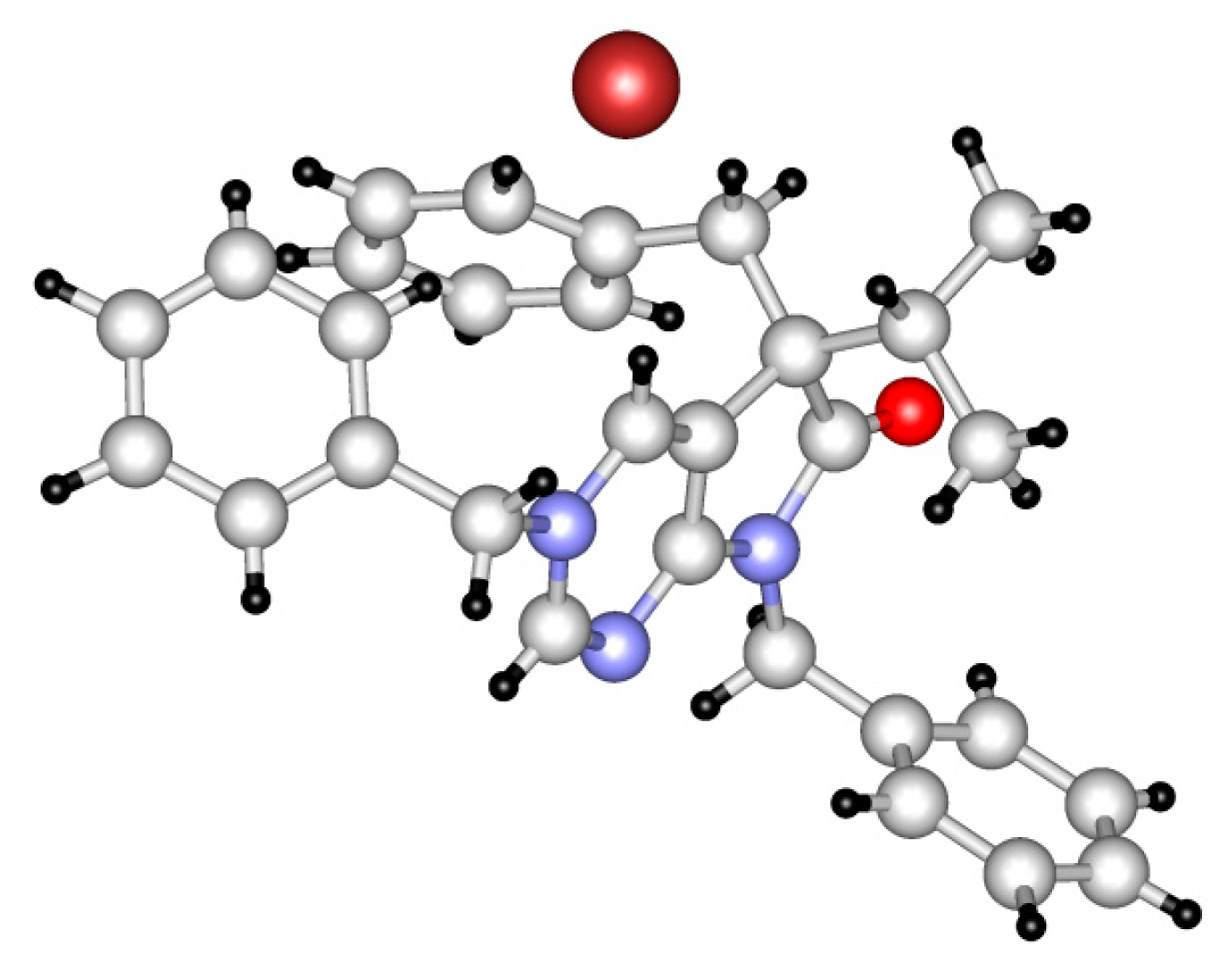
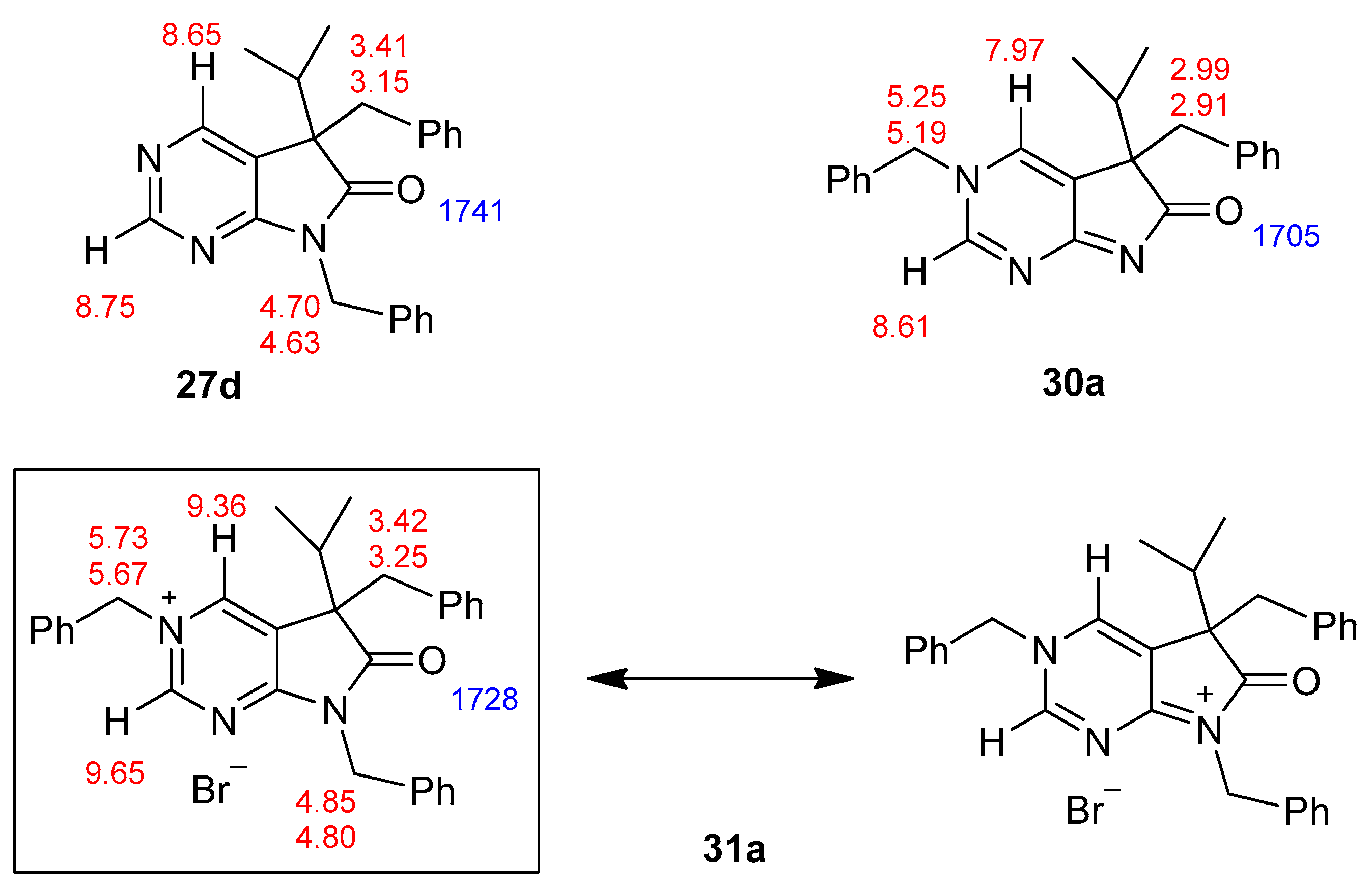
The structure of tribenzyl derivative 31a was also proven by single-crystal X-ray diffraction (Figure 3 and Supplementary Materials). A detailed analysis of the 1H-NMR and IR signals of 31a, compared to those of 5,7-dibenzyl (27d) and 3,5-dibenzyl (30a) derivatives, showed that the electron distribution of 31a is unambiguously closer to the framed resonance structure (Figure 4). The strong downfield shift of C(2)-H and C(4)-H signals in 31a in comparison to 27d and 30a indicate that the positive charge is mainly located in the pyrimidine ring. The N(3)-CH2 signal of 31a is also shifted by ca. 0.5 ppm when compared to the analogous signal in 30a, due to the positive charge of the N(3) atom in 31a, while the N(7)-CH2 signal remains practically unchanged when compared to 27d. The presence of a positively charged nitrogen atom in the pyrrole ring (as it is the case in the resonance structure of 31a on the right hand side) would shift the IR signal of the carbonyl moiety of 31a to a frequency higher than the carbonyl signal detected in 27d (1741 cm−1).
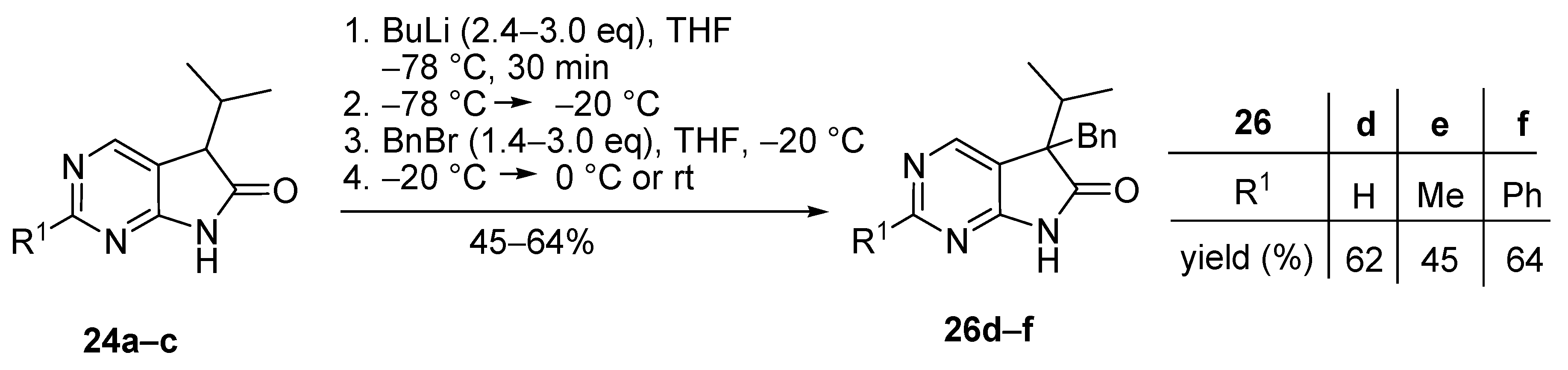
As the next step, the optimal conditions of C(5)-monobenzylation were sought. Similarly to the methylation reactions described above (Scheme 13), different protocols had to be elaborated for the benzylation of starting materials 24a, 24b and 24c (Scheme 16). BnBr was added at −20 °C in an excess of 1.4–3.0 eq. Use of lower excesses led in each case to significant amounts of the starting materials in the crude product, probably due to the higher steric hindrance of BnBr compared to MeI. By using the optimized conditions, C(5)-benzylated products 26d–f were obtained in acceptable yields (45–64%).
The different behavior of the 2-H (24a), 2-Me (24b) and 2-Ph (24c) derivatives observed in the C(5)-alkylation reactions can be attributed first of all to the different character of the C(2) substituents, on the other hand to the different alkylating agents, giving rise to various side reactions. Besides the 5,7-dialkylated 1,3-diazaoxindoles 27, the formation of C(2)-ethyl derivative 28 and products 30a,b and 31a,b benzylated at the N(3) position was observed, indicating that the selective alkylation at the C(5) position of N(7)-unsubstituted 5-isopropyl-1,3-diazaoxindoles 24 is an even more challenging task than the analogous selective C(3)-alkylation of oxindoles.
3. Experimental Section
3.1. General Information
Compounds 24a–c, 25a–c, 26a–f, 27a–f, 28, 29a–c, 30a,b, and 31a,b are new and characterized below. All melting points were determined on an OptiMelt Automated Melting Point System by Stanford Research Systems (Sunnyvale, CA, USA). IR spectra were obtained on a Bruker ALPHA FT-IR spectrometer (Bruker, Billerica, MA, USA). NMR spectra were recorded either on a Bruker AVANCE III 400 (1H and 13C frequencies were 400 and 100 MHz, respectively) or a Bruker AVANCE III HD 600 (1H and 13C frequencies were 600 and 150 MHz, respectively) spectrometer. CDCl3, DMSO-d6 or TFA-d was used as the solvent and tetramethylsilane (TMS) as the internal standard. Chemical shifts (δ) and coupling constants (J) are given in ppm and in Hz, respectively. Some representatives of compound families 26–31 were also investigated using special NMR measurements (1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC, selective NOESY). High-resolution mass spectra (HRMS spectra) were recorded either on a Micromass GCT (Waters, Milford, MA, USA) with EI (direct inlet) or on a Bruker Q-TOF MAXIS Impact mass spectrometer coupled to a Dionex Ultimate 3000 RS HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) with a diode array detector. The reactions were followed by analytical thin layer chromatography on silica gel 60 F254 (Merck, Darmstadt, Germany) and by HPLC-MS on a Shimadzu LC-20 HPLC equipment (Kyoto, Japan) with an SPD-M20A diode-array detector coupled with a LCMS-2020 mass spectrometer. All unspecified reagents were purchased from commercial sources.
3.2. General Procedure I for the Synthesis of Compounds 25a–c
A mixture of the corresponding 1,3-diazaoxindole (6a, 6b or 6c), pyrrolidine (0.60 eq.) and acetone (2.0 eq.) in toluene was stirred at reflux temperature for 2–4 h. The water was removed with a Dean-Stark apparatus. After the reaction was complete, the volatile components were removed in vacuo at 60 °C and the residue was triturated thoroughly with hexane. The precipitate was filtered off, washed with hexane four times and dried.
3.3. General Procedure II for the Synthesis of Compounds 25a–c
A mixture of the corresponding 1,3-diazaoxindole (6a, 6b or 6c), acetic acid (24 eq.) and acetone (46 eq.) was stirred at reflux temperature under argon atmosphere (Ar) for 12–48 h. After the reaction was complete, the volatile components were removed in vacuo at 40 °C and the residue was triturated thoroughly with diethyl ether (DEE) or water. The precipitate was filtered off and dried.
5-(1-Methylethylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (25a). Method A: This compound was prepared according to General Procedure I using 6a (500 mg, 3.7 mmol), pyrrolidine (0.18 mL, 0.16 g, 2.2 mmol), acetone (0.54 mL, 0.43 g, 7.4 mmol) in toluene (10 mL). The crude product was recrystallized from N,N-dimethylformamide (DMF) to give 25a (399 mg, 62%) as pale brown crystals, m.p. 200 °C (decomp., DMF). IR (KBr) ν 1718 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.52 (br s, 1H), 8.66 (s, 1H), 8.63 (s, 1H), 2.53 (s, 3H), 2.36 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 167.6, 160.40, 160.36, 156.1, 147.3, 119.1, 116.5, 25.7, 22.7. HRMS calcd. for C9H9N3O [M˙]+ 175.0746; found 175.0762. Method B: This compound was also prepared according to General Procedure II using 6a (3.0 g, 22.2 mmol), acetic acid (30 mL, 524 mmol), acetone (75 mL, 59.25 g, 1022 mmol). The crude product was recrystallized from DMF to give 25a (3.34 g, 86%) as pale brown crystals. Spectral data were identical with those of the product obtained using General Procedure I.
2-Methyl-5-(1-methylethylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (25b). Method A: This compound was prepared according to General Procedure I using 6b (15.00 g, 100 mmol), pyrrolidine (4.96 mL, 4.27 g, 60 mmol), acetone (14.68 mL, 11.60 g, 200 mmol) in toluene (150 mL). The crude product was recrystallized from DMF to give 25b (16.08 g, 85%) as yellow crystals, m.p. 303 °C (decomp., DMF). IR (KBr) ν 1709, 1434 cm−1. 1H-NMR (600 MHz, TFA-d) δ 11.71 (br s, 1H), 8.74 (s, 1H), 2.99 (s, 3H), 2.85 (s, 3H), 2.62 (s, 3H). 13C-NMR (150 MHz, TFA-d) δ 179.8, 171.1, 166.1, 165.6, 138.2, 118.4, 118.1, 27.9, 25.9, 23.1. HRMS calcd. for C10H11N3O [M˙]+ 189.0902; found 189.0891. Method B: This compound was also prepared according to General Procedure II using 6b (180 mg, 1.21 mmol), acetic acid (1.66 mL, 1.74 g, 29.04 mmol), acetone (4.09 mL, 3.23 g, 55.66 mmol). The product 25b was obtained as yellow crystals (98 mg, 42%). Spectral data were identical with those of the product obtained using General Procedure I.
2-Phenyl-5-(1-methylethylidene)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (25c). Method A: This compound was prepared according to General Procedure I using 6c (2.00 g, 9.50 mmol), pyrrolidine (0.47 mL, 0.41 g, 5.7 mmol), acetone (1.39 mL, 1.10 g, 19 mmol) in toluene (20 mL). The crude product was recrystallized from toluene–DMF to give 25c (1.74 g, 73%) as yellow crystals, m.p. 211 °C (decomp., toluene–DMF). IR (KBr) ν 1706, 1592 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.60 (br s, 1H), 8.78 (s, 1H), 8.37–8.34 (m, 2H), 7.52−7.50 (m, 3H), 2.54 (s, 3H), 2.38 (s, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 168.0, 161.1, 160.9, 159.4, 147.7, 137.5, 130.8, 128.8, 127.7, 119.3, 114.8, 25.7, 22.6. HRMS calcd. for C15H13N3O [M˙]+ 251.1059; found 251.1056. Method B: This compound was also prepared according to General Procedure II using 6c (120 mg, 0.57 mmol), acetic acid (0.78 mL, 0.82 g, 13.70 mmol), acetone (1.90 mL, 1.50 g, 26.20 mmol). The product 25c was obtained as orange crystals (89 mg, 62%). Spectral data were identical with those of the product obtained using General Procedure I.
3.4. General Procedure III for the Synthesis of Compounds 24a–c
Diazaoxindole derivatives 25a–c were hydrogenated at 60 °C under atmospheric pressure in DMF using activated palladium on charcoal (Pd/C, 10% Pd) catalyst for 12 h. After the reaction was completed, the catalyst was filtered off and washed with DMF three times. The filtrate and the washings were combined and the solvent was removed at 60 °C under reduced pressure. The residue was triturated thoroughly with water, the precipitate was filtered off and dried.
5-(1-Methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a). This compound was prepared according to General Procedure III using 25a (500 mg, 2.85 mmol) and Pd/C catalyst (140 mg) in DMF (50 mL). The crude product was recrystallized from toluene–DMF to give 25a (389 mg, 77%) as colorless crystals, m.p. 127–128 °C (toluene–DMF). IR (KBr) ν 1739, 1596 cm−1. 1H-NMR (600 MHz, CDCl3) δ 10.98 (br s, 1H), 8.87 (d, J = 0.8 Hz, 1H), 8.46 (d, J = 0.8 Hz, 1H), 3.57 (d, J = 4.0 Hz, 1H), 2.78 (m, 1H), 1.22 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 177.6, 164.3, 157.0, 149.3, 120.3, 50.3, 30.1, 20.2, 17.6. HRMS calcd. for C9H11N3O [M˙]+ 177.0902; found 177.0910.
2-Methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b). This compound was prepared according to General Procedure III using 25b (18.2 g, 96 mmol) and Pd/C catalyst (3.0 g) in DMF (300 mL). The crude product was recrystallized from acetonitrile to give 24b (13.57 g, 74%) as colorless crystals, m.p. 180–181 °C (acetonitrile). IR (KBr) ν 1738, 1618 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.84 (br s, 1H), 8.31 (s, 1H), 3.49 (d, J = 4.0 Hz, 1H), 2.75 (s, 3H), 2.54 (m, 1H), 1.20 (d, J = 6.8 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 177.6, 167.0, 164.5, 149.7, 117.0, 50.2, 30.1, 25.5, 20.4, 17.7. HRMS calcd. for C10H13N3O [M˙]+ 191.1059; found 191.1070.
2-Phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c). This compound was prepared according to General Procedure III using 25c (17.3 g, 68.80 mmol) and Pd/C catalyst (3.0 g) in DMF (250 mL). The crude product was recrystallized from toluene to give 24c (13.77 g, 79%) as colorless crystals, m.p. 178–180 °C (toluene). IR (KBr) ν 1738, 1620 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.56 (br s, 1H), 8.54 (s, 1H), 8.35–8.33 (m, 2H), 7.53–7.51 (m, 3H), 3.66 (d, J = 3.7 Hz, 1H), 2.40 (m, 1H), 1.10 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 178.0, 165.4, 162.3, 149.7, 137.5, 130.8, 128.8, 127.7, 118.6, 49.7, 29.6, 20.0, 18.0. HRMS calcd. for C15H15N3O [M˙]+ 253.1215; found 253.1220.
5-Methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26a). To a mixture of BuLi in hexane (5.63 mL, 9.00 mmol, 3.0 eq., 1.6 M) and THF (8 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 532 mg, 3.00 mmol) in THF (10 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.56 mL, 1.28 g, 9.00 mmol, 3.00 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the stirring was continued for further 1 h. The mixture was quenched with saturated ammonium chloride (NH4Cl) solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with ethyl acetate (EtOAc, 3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with diisopropyl ether (DIPE, 2 × 2 mL) and dried. The product 26a was obtained as yellow crystals (420 mg, 73%), m.p. 149–151 °C (hexane–EtOAc, colorless crystals). IR (KBr) ν 1732, 1603 cm−1. 1H-NMR (600 MHz, CDCl3) δ 9.85 (br s, 1H), 8.83 (s, 1H), 8.36 (s, 1H), 2.21 (sp, J = 6.9 Hz, 1H), 1.47 (s, 3H), 1.11 (d, J = 6.9 Hz, 3H), 0.81 (d, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 181.0, 163.0, 157.0, 149.1, 125.1, 51.6, 34.9, 20.9, 17.7, 17.2. HSQC: 8.83–157.0; 8.36–149.1; 2.21–34.9; 1.47–20.9; 1.11–17.2; 0.81–17.7. HMBC (Jlong-range = 8 Hz; characteristic cross-peaks): 8.83–163.0, 149.1; 8.36–163.0, 157.0, 125.1; 2.21–125.1, 51.6, 17.7, 17.2; 1.47–181.0, 125.1, 51.6, 34.9; 1.11–51.6, 34.9, 17.7; 0.81–51.6, 34.9, 17.2. Selective NOESY: 1.47–8.36, 2.21, 1.11, 0.81; 1.11–8.36, 2.21, 1.47, 0.81; 0.81–8.36, 2.21, 1.47, 1.11. HRMS calcd. for C10H13N3O [M˙]+ 191.1059; found 191.1075.
2,5-Dimethyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26b). To a mixture of BuLi in hexane (4.50 mL, 7.20 mmol, 2.4 eq., 1.6 M) and THF (5 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 574 mg, 3.00 mmol) in THF (15 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.26 mL, 0.60 g, 4.20 mmol, 1.4 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was allowed to warm to room temperature. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with DIPE (2 × 2 mL) and dried. The product 26b was obtained as yellow crystals (399 mg, 65%), m.p. 190–191 °C (hexane–EtOAc, colorless crystals). IR (KBr) ν 1744, 1621, 1429, 1190 cm−1. 1H-NMR (600 MHz, CDCl3) δ 11.36 (br s, 1H), 8.30 (s, 1H), 2.49 (s, 3H), 2.01 (sp, J = 6.8 Hz, 1H), 1.30 (s, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 181.5, 166.1, 163.9, 149.0, 121.4, 50.6, 34.3, 25.8, 20.7, 17.6, 17.2. HRMS calcd. for C11H15N3O [M˙]+ 205.1215; found 205.1219.
5-Methyl-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26c). To a mixture of BuLi in hexane (2.96 mL, 4.73 mmol, 2.4 eq., 1.6 M) and THF (3 mL), the solution of 2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c, 500 mg, 1.97 mmol) in THF (8 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to −20 °C, and MeI (0.13 mL, 0.31 g, 2.17 mmol, 1.1 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was stirred for further 1 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with DIPE (2 × 2 mL) and dried. The product 26c was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product 26c was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give colorless crystals (372 mg, 71%), m.p. 155–156 °C. IR (KBr) ν 1728, 1599, 1405, 1201 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.58 (br s, 1H), 8.43 (s, 1H), 8.37–8.35 (m, 2H), 7.49–7.48 (m, 3H), 2.23 (sp, J = 6.8 Hz, 1H), 1.49 (s, 3H), 1.13 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 181.1, 163.7, 163.0, 149.5, 137.1, 130.9, 128.6, 128.1, 122.3, 51.7, 35.0, 21.0, 17.8, 17.3. HRMS calcd. for C16H17N3O [M˙]+ 267.1372; found 267.1370.
5-Benzyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26d). To a mixture of BuLi in hexane (2.81 mL, 4.50 mmol, 3.0 eq., 1.6 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 266 mg, 1.50 mmol) in THF (5 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to −20 °C and BnBr (0.53 mL, 0.77 g, 4.50 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was stirred for further 1 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), then it was filtered off, washed with DIPE (2 × 2 mL) and dried. The product 26d was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product 26d was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 26d (250 mg, 62%) as colorless crystals, m.p. 195–197 °C. IR (KBr) ν 1731, 1601 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.70 (br, 1H), 8.65 (s, 1H), 8.43 (s, 1H), 7.09–7.04 (m, 3H), 6.87 (m, 2H), 3.26 (d, J = 13.2 Hz, 1H), 3.21 (d, J = 13.2 Hz, 1H), 2.38 (sp, J = 6.8 Hz, 1H), 1.19 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 179.1, 163.0, 157.1, 149.8, 134.9, 129.7, 128.1, 127.0, 122.5, 58.5, 40.9, 35.0, 17.9, 17.6. HSQC: 8.65–157.1; 8.43–149.8, (7.09–7.04)–127.0, 128.1; 6.87–129.7; 3.26–40.9; 3.21–40.9; 2.38–35.0; 1.19–17.6; 0.86–17.9. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.65–163.0, 149.8; 8.43–163.0, 157.1, 122.5; (7.09–7.04)–134.9; 6.87–40.9; 3.26–135.0, 129.7, 58.5; 3.21–179.1, 135.0, 129.7, 58.5; 2.38–179.1, 122.5, 58.5, 17.9, 17.6; 1.19–58.5, 35.0, 17.9; 0.86–58.5, 35.0, 17.6. HRMS calcd. for C16H17N3O [M˙]+ 267.1372; found 267.1371.
5-Benzyl-2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26e). To a mixture of BuLi in hexane (2.36 mL, 3.66 mmol, 2.4 eq., 1.6 M) and THF (3 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 300 mg, 1.57 mmol) in THF (7 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to 0 °C and BnBr (0.26 mL, 0.38 g, 2.20 mmol, 1.4 eq.) in THF (1 mL) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for further 3 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane (5 mL), it was filtered off, washed with hexane (2 × 2 mL) and DIPE (2 mL), then dried. The crude product 26e was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. This product 26e was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 26e (199 mg, 45%) as colorless crystals, m.p. 185–187 °C. IR (KBr) ν 1740, 1620, 1426 cm−1. 1H-NMR (400 MHz, CDCl3) δ 9.79 (br s, 1H), 8.29 (s, 1H), 7.07–7.05 (m, 3H), 6.90 (m, 2H), 3.23 (d, J = 13.2 Hz, 1H), 3.19 (d, J = 13.2 Hz, 1H), 2.60 (s, 3H), 2.34 (sp, J = 6.8 Hz, 1H), 1.17 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 179.7, 166.8, 163.6, 149.8, 135.3, 129.8, 128.0, 126.8, 119.2, 58.1, 40.8, 34.9, 25.5, 17.9, 17.6. HRMS calcd. for C17H19N3O [M˙]+ 281.1528; found 281.1524.
5-Benzyl-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (26f). To a mixture of BuLi in hexane (1.89 mL, 4.73 mmol, 2.4 eq., 2.5 M) and THF (3 mL), the solution of 2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c, 500 mg, 1.97 mmol) in THF (6 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and BnBr (0.70 mL, 1.01 g, 5.91 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The acetone-dry ice bath was removed and the reaction mixture was allowed to warm to 0 °C. The stirring was continued for further 5 h at 0 °C. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane/DIPE = 3/1 (5 mL), then it was filtered off, washed with hexane/DIPE = 3/1 (2 × 2 mL) and dried. The product 26f was obtained as yellow crystals (435 mg, 64%), m.p. 192–193 °C (hexane–EtOAc, colorless crystals). IR (KBr) ν 1728, 1625, 1410 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.52 (s, 1H), 8.31–8.28 (m, 2H), 7.96 (br s, 1H), 7.46–7.44 (m, 3H), 7.07–7.05 (m, 3H), 6.95–6.93 (m, 2H), 3.27 (s, 2H), 2.40 (sp, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 179.3, 163.5, 163.2, 150.1, 137.0, 135.2, 130.9, 129.8, 128.6, 128.1, 128.0, 126.9, 119.8, 58.5, 41.0, 35.2, 18.0, 17.7. HRMS calcd. for C22H21N3O [M˙]+ 343.1685; found 343.1660.
3.5. General Procedure IV for the Synthesis of Compounds 27a–f
To a mixture of NaOH (2.5 eq.) in DMF, the solution of 5-isopropyl-1,3-diazaoxindoles 24a–c in DMF was added dropwise at room temperature, under Ar atmosphere and it was stirred for 30 min. Then the solution of the appropriate alkyl halide (2.5 eq.) in DMF was added dropwise at 15–20 °C. The stirring was continued for further 1 h. The reaction mixture was poured onto ice–water. The precipitate was filtered off or the aqueous layer was extracted with dichloromethane (DCM, 3 × 15 mL). The combined organic layer was washed with brine (5 × 50 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. Purification of the crude products was performed as described below.
5,7-Dimethyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27a). This compound was prepared according to General Procedure IV using 24a (275 mg, 1.55 mmol) in DMF (4 mL), NaOH (155 mg, 3.88 mmol) in DMF (2 mL) and MeI (0.24 mL, 0.55 g, 3.88 mmol) in DMF (1 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 27a (203 mg, 64%) as colorless crystals, m.p. 64–66 °C. IR (KBr) ν 1731, 1586, 1492 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.81 (s, 1H), 8.29 (s, 1H), 3.28 (s, 3H), 2.20 (sp, J = 7.0 Hz, 1H), 1.43 (s, 3H), 1.07 (d, J = 7.0 Hz, 3H), 0.71 (d, J = 7.0 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 180.2, 163.7, 157.5, 148.1, 124.5, 50.9, 35.0, 25.1, 20.8, 17.6, 17.3. HSQC: 8.81–157.5; 8.29–148.1; 3.28–25.1; 2.20–35.0; 1.43–20.8; 1.07–17.3; 0.71–17.6. HMBC (Jlong-range = 8 Hz; characteristic cross-peaks): 8.81–163.7, 148.1; 8.29–163.7, 157.5, 124.5; 3.28–180.2, 163.7; 1.43–180.2, 124.5, 50.9, 35.0; 1.07–50.9, 35.0, 17.6; 0.71–50.9, 35.0, 17.3. Selective NOESY: 1.43–8.29, 3.28, 2.20, 1.07, 0.71. HRMS calcd. for C11H15N3O [M˙]+ 205.1215; found 205.1223.
2,5,7-Trimethyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27b). This compound was prepared according to General Procedure IV using 24b (574 mg, 3.00 mmol) in DMF (7 mL), NaOH (300 mg, 7.5 mmol) in DMF (2 mL) and MeI (0.47 mL, 1.06 g, 7.5 mmol) in DMF (1 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents to give 27b (527 mg, 80%) as a colorless oil. IR (film) ν 1732, 1600, 1488 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 3.26 (s, 3H), 2.67 (s, 3H), 2.17 (sp, J = 6.9 Hz, 1H), 1.40 (s, 3H), 1.10 (d, J = 6.9 Hz, 3H), 0.69 (d, J = 6.9 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 180.6, 167.1, 164.0, 148.0, 121.1, 50.7, 34.9, 26.1, 25.1, 20.9, 17.6, 17.3. HRMS calcd. for C12H17N3O [M˙]+ 219.1372; found 219.1373.
5,7-Dimethyl-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27c). This compound was prepared according to General Procedure IV using 24c (507 mg, 2 mmol) in DMF (7 mL), NaOH (200 mg, 5.00 mmol) in DMF (2 mL) and MeI (0.31 mL, 0.71 g) in DMF (1 mL). The precipitate was filtered off, washed with water (3 × 5 mL) and dried. The product 27c was obtained as colorless crystals (512 mg, 91%). An analytical sample was obtained by recrystallization from hexane, m.p. 106–107 °C (hexane). IR (KBr) ν 1716, 1678 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.48–8.45 (m, 2H), 8.36 (s, 1H), 7.50–7.49 (m, 3H), 3.36 (s, 3H), 2.21 (sp, J = 6.8 Hz, 1H), 1.45 (s, 3H), 1.10 (d, J = 6.8 Hz, 3H), 0.73 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 180.7, 164.2, 163.6, 148.5, 137.4, 130.8, 128.5, 128.1, 122.1, 51.0, 35.1, 25.2, 20.9, 17.7, 17.3. HRMS calcd. for C17H19N3O [M˙]+ 281.1528; found 281.1522.
5,7-Dibenzyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27d). This compound was prepared according to General Procedure IV using 24a (250 mg, 1.41 mmol) in DMF (2 mL), NaOH (141 mg, 3.53 mmol) in DMF (2 mL) and BnBr (0.42 mL, 0.60 g, 3.53 mmol) in DMF (1 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 27d (353 mg, 70%) as colorless crystals, m.p. 65–66 °C. IR (KBr) ν 1741, 1496 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 8.75 (s, 1H), 8.65 (s, 1H), 7.20–7.16 (m, 3H), 7.07 (m, 1H), 6.99 (t, J = 7.6 Hz, 2H), 6.83 (m, 2H), 6.81 (m, 2H), 4.70 (d, J = 15.4 Hz, 1H), 4.63 (d, J = 15.4 Hz, 1H), 3.41 (d, J = 13.0 Hz, 1H), 3.15 (d, J = 13.0 Hz, 1H), 2.30 (sp, J = 6.8 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 178.2, 163.1, 157.4, 150.3, 135.8, 135.7, 129.8, 128.5, 128.0, 127.4, 127.2, 126.8, 121.6, 57.3, 41.8, 39.7, 35.2, 17.7, 17.6. COSY: (7.20–7.16)–6.83; 6.99–6.81; 3.41–3.15; 2.30–1.08, 0.68. HSQC: 8.75–150.3; 8.65–157.4; (7.20–7.16)–128.5, 127.4; 7.07–126.8; 6.99–128.0; 6.83–127.2; 6.81–129.8; 4.70–41.8; 4.63–41.8; 3.41–39.7; 3.15–39.7; 2.30–35.2; 1.08–17.7; 0.68–17.6. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.75–163.1, 157.4, 121.6; 6.83–41.8; 6.81–39.7; (7.20–7.16)–135.7; 6.99–135.8; 4.70–178.2, 163.1, 135.7, 127.2; 4.63–178.2, 163.1, 135.7, 127.2; 3.41–178.2, 135.8, 129.8, 57.3; 3.15–135.8, 129.8, 121.6, 57.3; 2.30–121.6, 57.3, 17.7, 17.6; 1.08–57.3, 35.2; 0.68–57.3, 35.2. COSY: (7.20–7.16)–6.83; 6.99–6.81; 3.41–3.15; 2.30–1.08, 0.68. HRMS calcd. for C23H23N3O [M˙]+ 357.1841; found 357.1829.
5,7-Dibenzyl-2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27e). This compound was prepared according to General Procedure IV using 24b (200 mg, 1.05 mmol) in DMF (2 mL), NaOH (105 mg, 2.63 mmol) in DMF (2 mL) and BnBr (0.31 mL, 0.45 g, 2.63 mmol) in DMF (1 mL). The precipitate was filtered off, washed with water (3 × 5 mL) and dried. The product 27e was obtained as orange crystals (320 mg, 82%). An analytical sample was obtained by recrystallization from a mixture of hexane and EtOAc to give 27e as colorless crystals, m.p. 84–86 °C (hexane–EtOAc). IR (KBr) ν 1736, 1478 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 8.61 (br s, 1H), 7.21–7.17 (m, 3H), 7.07 (t, J = 7.4 Hz, 1H), 7.00 (t, J = 7.4 Hz, 2H), 6.86–6.80 (m, 4H), 4.67 (d, J = 15.4 Hz, 1H), 4.61 (d, J = 15.4 Hz, 1H), 3.37 (d, J = 13.0 Hz, 1H), 3.12 (d, J = 13.0 Hz, 1H), 2.44 (s, 3H), 2.26 (sp, J = 6.8 Hz, 1H), 1.07 (d, J = 6.8 Hz, 3H), 0.64 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 178.5, 166.3, 163.4, 150.2, 135.90, 135.85, 129.8, 128.5, 128.0, 127.3, 127.1, 126.7, 118.1, 57.0, 41.7, 39.6, 35.2, 25.9, 17.604, 17.596. HRMS calcd. for C24H25N3O [M˙]+ 371.1998; found 371.1989.
5,7-Dibenzyl-5-(1-methylethyl)-2-phenyl-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (27f). This compound was prepared according to General Procedure IV using 24c (760 mg, 3.00 mmol) in DMF (10 mL), NaOH (300 mg, 7.50 mmol) in DMF (2 mL) and BnBr (0.89 mL, 1.28 g, 7.50 mmol) in DMF (2 mL). The residual oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents to give 27f (1062 mg, 82%) as a colorless oil. IR (film) ν 1732, 1596, 1475, 1398 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.49 (s, 1H), 8.38 (m, 2H), 7.47–7.44 (m, 3H), 7.21–7.17 (m, 3H), 7.13 (m, 2H), 6.98 (m, 1H), 6.90 (m, 2H), 6.82 (m, 2H), 4.80 (s, 2H), 3.27 (d, J = 13.2 Hz, 1H), 3.24 (d, J = 13.2 Hz, 1H), 2.40 (sp, J = 6.8 Hz, 1H), 1.19 (d, J = 6.8 Hz, 3H), 0.72 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 178.7, 164.1, 163.3, 149.3, 137.2, 135.7, 135.1, 130.8, 129.7, 128.5, 128.4, 128.3, 128.0 (signal overlapping), 127.4, 126.7, 119.5, 57.6, 42.5, 40.8, 35.4, 17.8, 17.7. HRMS calcd. for C29H27N3O [M˙]+ 433.2154; found 433.2102.
2-Ethyl-5-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (28). To a mixture of BuLi in hexane (2.51 mL, 6.27 mmol, 3.0 eq., 2.5 M) and THF (3 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 400 mg, 2.09 mmol) in THF (12 mL) was added dropwise at −20 °C under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then MeI (0.39 mL, 0.89 g, 6.27 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 2 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow crystals were purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 28 (50 mg, 11%) as colorless crystals, m.p. 149–151 °C. IR (KBr) ν 1743, 1620, 1460, 1193 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 11.37 (br s, 1H), 8.34 (s, 1H), 2.77 (q, J = 7.6 Hz, 2H), 2.01 (sp, J = 6.8 Hz, 1H), 1.30 (s, 3H), 1.24 (t, J = 7.6 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 181.5, 170.1, 164.0, 149.1, 121.6, 50.6, 34.4, 32.1, 20.7, 17.6, 17.2, 12.6. HSQC: 8.34–149.1; 2.77–32.1; 2.01–34.4; 1.30–20.7; 1.24–12.6; 0.98–17.3; 0.66–17.6. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 11.37–121.6; 8.34–170.1, 164.0, 121.6; 2.77–170.1; 1.30–181.5, 121.6; 1.24–170.1. HRMS calcd. for C12H17N3O [M˙]+ 219.1372; found 219.1387.
3.6. General Procedure V for the Synthesis of Compounds 29a–c
To a mixture of BuLi in hexane (1.50 mL, 3.75 mmol, 2.5 eq., 2.5 M) and THF (3 mL), the solution of the appropriate 2-substituted 5-isopropyl-1,3-diazaoxindole 24a–c (1.50 mmol) in THF (8 mL) was added dropwise at −78 °C, under argon atmosphere. After the addition was complete, the acetone–dry ice bath was removed, the reaction mixture was allowed to warm to room temperature and the apparatus was opened to the air. The stirring was continued for further 2 days. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the layers were separated and the aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic layer was dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The residue crystallized upon treatment with hexane/DIPE = 3/1 (7 mL), then it was filtered off, washed with hexane/DIPE = 3/1 (2 × 2 mL) and dried.
5-Hydroxy-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (29a). Method A: This compound was prepared according to General Procedure V. The product 29a was obtained as off-white crystals (228 mg, 79%), m.p. 175–176 °C. IR (KBr) ν 1760, 1159 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 11.42 (br s, 1H), 8.71 (s, 1H), 8.39 (s, 1H), 6.25 (s, 1H), 2.12 (sp, J = 6.9 Hz, 1H), 1.03 (d, J = 6.9 Hz, 3H), 0.64 (d, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 179.3, 164.1, 158.3, 149.8, 122.4, 78.1, 34.4, 16.3, 16.0. HSQC: 8.71–158.3; 8.39–149.8; 2.12–34.4; 1.03–16.0; 16.3. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.71–164.0, 149.8; 8.39–164.0, 158.3, 122.4; 6.25–122.4, 78.1, 34.4; 1.03–78.1, 34.4, 16.3; 0.64–78.1, 34.4, 16.0. HRMS calcd. for C9H11N3O2 [M + H]+ 193.0851; found 193.0850. Method B: To a mixture of BuLi in hexane (3.80 mL, 9.44 mmol, 3.3 eq., 2.5 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 500 mg, 2.86 mmol) in THF (8 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.59 mL, 1.33 g, 9.44 mmol, 3.3 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (20 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (2 mL), washed with DIPE (2 × 1 mL) and dried to give 29a (50 mg, 9%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
5-Hydroxy-2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (29b). Method A: This compound was prepared according to General Procedure V. The product 29b was obtained as off-white crystals (194 mg, 62%), m.p. 182–184 °C. IR (KBr) ν 1624, 1432 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.28 (br s, 1H), 8.26 (s, 1H), 6.16 (s, 1H), 2.49 (s, 3H), 2.10 (sp, J = 6.8 Hz, 1H), 1.03 (d, J = 6.8 Hz, 3H), 0.62 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 179.5, 167.2, 164.3, 149.8, 119.1, 78.0, 34.3, 25.9, 16.4, 16.0. HRMS calcd. for C10H13N3O2 [M˙]+ 207.1008; found 207.1026. Method B: To a mixture of BuLi in hexane (4.13 mL, 6.60 mmol, 2.2 eq., 1.6 M) and THF (5 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 574 mg, 3.00 mmol) in THF (14 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then MeI (0.41 mL, 0.94 g, 6.60 mmol, 2.2 eq.) in THF (1 mL) was added dropwise. The temperature was allowed to warm to room temperature and stirring was continued for further 5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining orange oil was purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (2 mL), washed with DIPE (2 × 1 mL) and dried to give 29b (100 mg, 16%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
5-Hydroxy-2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (29c). Method A: This compound was prepared according to General Procedure V. The product 29c was obtained as off-white crystals (273 mg, 68%), m.p. 184–185 °C. IR (KBr) ν 1730, 1621, 1167 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 11.49 (br s, 1H), 8.51 (s, 1H), 8.35–8.33 (m, 2H), 7.54–7.51 (m, 3H), 6.24 (s, 1H), 2.16 (sp, J = 6.8 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, DMSO-d6) δ 179.6, 164.8, 163.2, 150.2, 137.3, 131.1, 128.9, 127.9, 120.5, 78.1, 34.4, 16.4, 16.1. HRMS calcd. for C15H15N3O2 [M˙]+ 269.1164; found 269.1150. Method B: To a mixture of BuLi in hexane (4.13 mL, 6.60 mmol, 2.2 eq., 1.6 M) and THF (15 mL), the solution of 2-phenyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24c, 760 mg, 3.00 mmol) in THF (12 mL) was added dropwise at −78 °C under Ar atmosphere. The mixture was stirred for 30 min, then the temperature was allowed to warm to –20 °C and MeI (0.22 mL, 0.51 g, 3.60 mmol, 1.2 eq.) in THF (1 mL) was added dropwise. The temperature was allowed to warm to 0 °C and stirring was continued for further 4 h. The mixture was quenched with saturated NH4Cl solution (10 mL), and water (5 mL) was added. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow crystals were purified by gradient elution column chromatography using hexane and EtOAc as the eluents. The product was triturated in hexane (2 mL), washed with DIPE (2 × 1 mL) and dried to give 29c (64 mg, 8%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
3,5-Dibenzyl-5-(1-methylethyl)-3,5-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (30a). To a mixture of BuLi in hexane (3.20 mL, 5.07 mmol, 3.0 eq., 1.6 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 300 mg, 1.69 mmol) in THF (6 mL) was added dropwise at −20 °C under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.60 mL, 0.87 g, 5.07 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then the mixture was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 30a (95 mg, 16%) as colorless crystals, m.p. 271–272 °C. IR (KBr) ν 1705, 1656, 1464 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 8.61 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 2.0 Hz, 1H), 7.50 (m, 2H), 7.44 (m, 1H), 7.39 (m, 2H), 6.99 (m, 1H), 6.90 (m, 2H), 6.74 (m, 2H), 5.25 (d, J = 14.8 Hz, 1H), 5.19 (d, J = 14.8 Hz, 1H), 2.99 (d, J = 12.7 Hz, 1H), 2.91 (d, J = 12.7 Hz, 1H), 2.02 (sp, J = 6.8 Hz, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.67 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 194.5, 182.0, 153.6, 136.8, 136.1, 132.9, 130.0, 129.2, 128.8, 128.0, 127.5, 127.0, 126.2, 58.8, 57.0, 39.4, 34.3, 17.74, 17.73. COSY (characteristic cross-peaks): 7.50–7.44, 7.39; 2.02–1.00, 0.67. HSQC: 8.61–153.6; 7.97–132.9; 7.50–129.2; 7.44–128.8; 7.39–128.0; 6.99–127.0; 6.90–127.5; 6.74–130.0; 5.25–57.0; 5.19–57.0; 2.99–39.5; 2.91–39.5; 2.02–34.3; 1.00, 0.67–17.74, 17.73. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 8.61–182.0, 132.9, 57.0; 7.97–182.0, 153.6, 127.0; 7.39–57.0; 6.74–39.5; 5.25, 5.19–153.6, 136.1, 132.9, 128.0; 2.99–136.8, 130.0; 2.91–136.8, 130.0, 127.0; 2.02–194.5. HRMS calcd. for C23H23N3O [M˙]+ 357.1841; found 357.1851.
3,5-Dibenzyl-2-methyl-5-(1-methylethyl)-3,5-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (30b). To a mixture of BuLi in hexane (1.26 mL, 3.14 mmol, 3.0 eq., 2.5 M) and THF (2 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 200 mg, 1.05 mmol) in THF (6 mL) was added dropwise at −20 °C under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.37 mL, 0.54 g, 3.14 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 2 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then it was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining orange oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 30b (215 mg, 56%) as off-white crystals, m.p. 250–252 °C. IR (KBr) ν 1757, 1653 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 9.11 (br s, 1H), 7.53 (m, 2H), 7.46 (m, 1H), 7.29 (m, 2H), 7.15 (m, 1H), 7.12 (m, 2H), 6.98 (m, 2H), 5.71 (d, J = 16.0 Hz, 1H), 5.64 (d, J = 16.0 Hz, 1H), 3.32 (d, J = 13.1 Hz, 1H), 3.18 (d, J = 13.1 Hz, 1H), 2.61 (s, 3H), 2.27 (sp, J = 6.8 Hz, 1H), 1.10 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 183.4 (br), 170.9 (br), 165.2, 142.8 (br), 135.7, 134.1, 129.9, 129.4, 128.9, 128.2, 127.4, 127.1, 122.8, 58.7, 58.5, 39.4, 34.9, 23.1, 17.6, 17.5. HSQC: 9.11–142.8; 7.53–129.4; 7.46–128.9; 7.29–127.4; 7.15–127.2; 7.12–128.2; 6.98–129.9; 5.71–58.5; 5.64–58.5; 3.32–39.4; 3.18–39.4; 2.61–23.1; 2.27–34.9; 1.10–17.6; 0.85–17.5. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 9.11–170.9, 165.2, 122.8, 58.5; 7.53–134.1; 7.29–58.5; 7.12–135.7; 5.71, 5.64–165.2, 142.8, 134.1; 3.32–183.4; 3.18–183.4, 122.8; 2.61–165.2. HRMS calcd. for C24H25N3O [M˙]+ 371.1998; found 371.1994.
3,5,7-Tribenzyl-5-(1-methylethyl)-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-3-ium bromide (31a). Method A: A mixture of 27d (340 mg, 0.95 mmol), BnBr (0.43 mL, 0.62 g, 3.80 mmol) and acetonitrile (3 mL) was stirred for 27 h at room temperature. The solvent was removed at 40 °C in vacuo and the crystals was triturated with hexane (5 mL), filtered off, washed with DIPE (3 × 2 mL) and dried. The product 31a was obtained as colorless crystals (431 mg, 86%), m.p. 220–222 °C (i-PrOH). IR (KBr) ν 1728, 1625, 1410 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 9.65 (d, J = 1.6 Hz, 1H), 9.36 (d, J = 1.6 Hz, 1H), 7.59 (m, 2H), 7.56 (m, 2H), 7.51 (m, 1H), 7.26 (m, 3H), 7.02 (m, 1H), 6.94 (m, 2H), 6.80 (m, 2H), 6.63 (m, 2H), 5.73 (d, J = 14.4 Hz, 1H), 5.67 (d, J = 14.4 Hz, 1H), 4.85 (d, J = 15.3 Hz, 1H), 4.80 (d, J = 15.3 Hz, 1H), 3.42 (d, J = 13.2 Hz, 1H), 3.25 (d, J = 13.2 Hz, 1H), 2.41 (sp, J = 6.9 Hz, 1H), 1.04 (d, J = 6.9 Hz, 3H), 0.86 (d, J = 6.9 Hz, 3H). 13C-NMR (150 MHz, DMSO-d6) δ 178.0, 166.1, 156.3, 143.0, 134.5, 134.4, 134.2, 129.7, 129.6, 129.4, 128.9, 128.8, 128.3, 128.1, 127.7, 127.2, 123.8, 59.9, 58.2, 43.1, 39.2, 35.4, 17.7, 17.2. COSY (characteristic cross-peaks): 7.26–6.94; 7.02–6.80; 6.80–6.63; 3.42–3.25; 2.41–1.04, 0.86. HSQC: 9.65–156.3; 9.36–143.0; 7.59–128.9; 7.56–129.5; 7.51–129.6; 7.26–128.8, 128.1; 7.02–127.2; 6.94–127.7; 6.63–129.7; 5.73–59.9; 5.67–59.9; 4.85–43.1; 4.80–43.1; 3.42–39.2; 3.25–39.2; 2.41–35.4; 1.04–17.7; 0.86–17.2. HMBC (Jlong-range = 7 Hz; characteristic cross-peaks): 9.65–166.1, 143.0, 59.9; 9.36–166.1, 156.3, 123.8, 59.9; 7.59–59.9; 7.56–134.4; 7.26–134.2; 6.94–43.1; 6.80–134.5; 6.63–39.2; 2.41–123.8, 178.0; 1.04–58.2; 0.86–58.2. COSY: 7.26–6.94; 7.02–6.80; 6.80–6.63; 3.42–3.25; 2.41–1.04, 0.86. HSQC: 9.65–156.3; 9.36–143.0; 7.59–128.9; 7.56–129.5; 7.51–129.6; 7.26–128.8, 128.1; 7.02–127.2; 6.94–127.7; 6.63–129.7; 5.73–59.9; 5.67–59.9; 4.85–43.1; 4.80–43.1; 3.42–39.2; 3.25–39.2; 2.41–35.4; 1.04–17.7; 0.86–17.2. Selective NOESY: 9.36–7.59, 6.63, 5.73, 5.67, 3.42, 1.04; 9.65–7.59, 5.73, 5.67. HRMS calcd. for C30H30N3O [M]+ 448.238; found 448.238. Elementary analysis of C30H30BrN3O calcd. for Br: 15.12%; found: 14.80%. Method B: To a mixture of BuLi in hexane (3.20 mL, 5.07 mmol, 3.0 eq., 1.6 M) and THF (3 mL), the solution of 5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24a, 300 mg, 1.69 mmol) in THF (6 mL) was added dropwise at −20 °C, under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.60 mL, 0.87 g, 5.07 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 1.5 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then it was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining yellow oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 30a (110 mg, 12%) as colorless crystals. Spectral data were identical with those of the product obtained using Method A.
3,5,7-Tribenzyl-2-methyl-5-(1-methylethyl)-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-3-ium bromide (31b). Method A: A mixture of 27e (371 mg, 1.00 mmol), BnBr (0.45 mL, 0.65 g, 3.80 mmol) and acetonitrile (3 mL) was stirred for 116 h at room temperature. The solvent was removed at 40 °C in vacuo and the crystals was triturated with hexane (5 mL), filtered off, washed with DIPE (3 × 2 mL) and dried. The product 31b was obtained as off-white crystals (386 mg, 71%), m.p. 104–106 °C (i-PrOH). IR (KBr) ν 1756, 1651 cm−1. 1H-NMR (600 MHz, DMSO-d6) δ 9.36 (s, 1H), 7.55 (t, J = 7.4 Hz, 2H), 7.48 (t, J = 7.4 Hz, 1H), 7.36 (d, J = 7.4 Hz, 2H), 7.27–7.26 (m, 3H), 7.07 (t, J = 7.4 Hz, 1H), 7.02–6.99 (m, 4H), 6.86 (d, J = 7.2 Hz, 2H), 5.79 (d, J = 15.8 Hz, 1H), 5.71 (d, J = 15.8 Hz, 1H), 4.84 (d, J = 15.2 Hz, 1H), 4.79 (d, J = 15.2 Hz, 1H), 3.44 (d, J = 13.3 Hz, 1H), 3.29 (d, J = 13.3 Hz, 1H), 2.77 (s, 3H), 2.39 (sp, J = 6.8 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H). 13C-NMR (150 MHz, CDCl3) δ 178.3, 166.6, 165.8, 145.0, 134.8, 134.3, 133.4, 129.7, 129.4, 129.2, 128.8, 128.5, 128.1, 127.9, 127.8, 127.4, 121.5, 59.3, 58.0, 43.0, 39.2, 35.6, 23.5, 17.6, 17.3. HRMS calcd. for C31H32N3O [M]+ 462.254; found 462.253. Elementary analysis of C31H32BrN3O calcd. for Br: 14.73%; found: 14.80%. Method B: To a mixture of BuLi in hexane (1.26 mL, 3.14 mmol, 3.0 eq., 2.5 M) and THF (2 mL), the solution of 2-methyl-5-(1-methylethyl)-5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one (24b, 200 mg, 1.05 mmol) in THF (6 mL) was added dropwise at −20 °C, under Ar atmosphere. The temperature was allowed to warm to room temperature and the mixture was stirred for 30 min, then BnBr (0.37 mL, 0.54 g, 3.14 mmol, 3.0 eq.) in THF (1 mL) was added dropwise. The stirring was continued for further 2 h. The mixture was quenched with saturated NH4Cl solution (10 mL), water (5 mL) was added, then it was stirred for 15 min under Ar atmosphere. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered and the solvent was removed in vacuo at 40 °C. The remaining orange oil was purified by gradient elution column chromatography using DCM and DCM/MeOH = 9/1 as the eluents. The product was triturated in hexane (3 mL), washed with DIPE (2 × 1 mL) and dried to give 31b (108 mg, 19%) as off-white crystals. Spectral data were identical with those of the product obtained using Method A.
4. Conclusions
The alkylation reactions of N(7)-unsubstituted 1,3-diazaoxindoles have been investigated using the 5-isopropyl derivatives as the model compounds. Alkylation of 5-isopropyl derivatives 24 (bearing various substituents at the C(2) position of the pyrimidine ring) with MeI or BnBr in the presence of NaOH at room temperature led to the C(5),N(7)-disubstituted analogues 27. When BuLi was used as the base and the alkylating agent was added at −20 °C, a selective C(5)-alkylation could be carried out resulting in derivatives 26. Some interesting by-products were also identified in the above reactions. When methylation of the lithium salt of 2-methyl derivative 24b was performed at ambient temperature, methylation of the 2-methyl moiety was also observed giving rise to the formation of 2-ethyl derivative 28. The insufficient pre-inertization of the lithiation reactions led to 5-hydroxy by-products 29. During the benzylation of 2-unsubstituted (24a) and 2-methyl-1,3-diazaoxindole (24b) with BuLi and BnBr at room temperature, in addition to C(5)-benzylated derivatives, the unexpected N(3),C(5)-dibenzylated compounds 30 and N(3),C(5),N(7)-tribenzylated quaternary bromide salts 31 were also isolated. The structure of the latter was proven by single-crystal X-ray diffraction and a detailed spectroscopic analysis. The present study may help other research groups perform various alkylation reactions of 1,3-diazaoxindoles.
Supplementary Materials
CCDC 1532352 contains the supplementary crystallographic data for this paper (X-Ray structure, conditions of the single-crystal X-ray measurements and CIF structure file for compound 31a). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
Author Contributions
E.K. performed the experiments and compiled the Experimental Section; J.N., G.S. and B. V. conceived and designed the experiments, and wrote the further chapters of the paper; J.H. and A.D. carried out and evaluated the analytical measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Costall, B.; Domeney, A.M.; Eden, R.J.; Gerrard, P.A.; Harvey, C.A.; Kelly, M.E. The antiparkinson potential of SK&F 101468-A revealed in rodent and primate test. Br. J. Pharmacol. 1989, 96, 90. [Google Scholar]
- Murayama, S.; Narabayashi, H.; Furukazu, H.; Yanagisawa, N.; Mizuno, Y.; Nakajima, M.; Kanazawa, I. Preclinical and clinical studies of ropinirole hydrochloride (SK&F 101468). Clinical evaluation of ropinirole hydrochloride (SK&F 101468) in patients with Parkinson’s disease: A double-blind comparative study vs bromocriptine mesilate. Jpn. Pharmacol. Ther. 1996, 24 (Suppl. 11), S1939–S2007. [Google Scholar]
- Prakash, C.; Kamel, A.; Gummerus, J.; Wilner, K. Metabolism and excretion of a new antipsychotic drug, ziprasidone, in humans. Drug Metab. Dispos. 1997, 25, 863–872. [Google Scholar] [PubMed]
- O’Farrell, A.-M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003, 101, 3597–3605. [Google Scholar] [CrossRef] [PubMed]
- Dallinger, C.; Trommeshauser, D.; Marzin, K.; Liesener, A.; Kaiser, R.; Stopfer, P. Pharmacokinetic properties of nintedanib in healthy volunteers and patients with advanced cancer. J. Clin. Pharmacol. 2016, 56, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Bell, I.M.; Fraley, M.E.; Gallicchio, S.N.; Ginnetti, A.; Mitchell, H.J.; Paone, D.V.; Staas, D.D.; Wang, C.; Zartman, C.B.; Stevenson, H.E. Preparation of Piperidinonylcarboxamideazaindane Derivatives for Use as CGRP Receptor Antagonists. WO Patent 2,012,064,910, 18 May 2012. [Google Scholar]
- Hewitt, D.J.; Aurora, S.K.; Dodick, D.W.; Goadsby, P.J.; Ge, Y.J.; Bachman, R.; Taraborelli, D.; Fan, X.; Assaid, C.; Lines, C. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 2011, 31, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Bell, I.M.; Gallicchio, S.N.; Wood, M.R.; Quigley, A.G.; Stump, C.A.; Zartman, C.B.; Fay, J.F.; Li, C.-C.; Lynch, J.J.; Moore, E.L.; et al. Discovery of MK-3207: A highly potent, orally bioavailable CGRP receptor antagonist. ACS Med. Chem. Lett. 2010, 1, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.R.; Gallicchio, S.N.; Selnick, H.G.; Zartman, C.B.; Bell, I.M.; Stump, C.A. Substituted Spiro [Indane-Azaindolinone] Derivatives as CGRP Receptor Antagonists and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of CGRP-Associated Diseases Such as Headaches. U.S. Patent 20,070,265,225, 15 November 2007. [Google Scholar]
- Savchuk, N.F.; Tkachenko, S.Y.; Nazarenkov, D.Y. Incretin Secretagogues, Methods for Preparing and Applying Them. WO Patent 2,012,161,622, 29 November 2012. [Google Scholar]
- Humphries, A.C.; Gancia, E.; Gilligan, M.T.; Goodacre, S.; Hallett, D.; Merchant, K.J.; Thomas, S.R. 8-Fluoroimidazo[1,2-a]pyridine: Synthesis, physicochemical properties and evaluation as a bioisosteric replacement for imidazo[1,2-a]pyrimidine in an allosteric modulator ligand of the GABAA receptor. Bioorg. Med. Chem. Lett. 2006, 16, 1518–1522. [Google Scholar] [CrossRef] [PubMed]
- Gundisch, D.; Kampchen, T.; Schwarz, S.; Seitz, G.; Siegl, J.; Wegge, T. Syntheses and evaluation of pyridazine and pyrimidine containing bioisosteres of (±)-pyrido[3.4-b]homotropane and pyrido-[3.4-b]tropane as novel nAChR ligands. Bioorg. Med. Chem. 2002, 10, 1–9. [Google Scholar] [CrossRef]
- Kókai, E.; Nagy, J.; Tóth, T.; Kupai, J.; Huszthy, P.; Simig, G.; Volk, B. Convenient synthesis of 2-substituted 5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-ones. Monatsh. Chem. Chem. Mon. 2016, 147, 767–773. [Google Scholar] [CrossRef]
- Volk, B.; Mezei, T.; Simig, G. Raney nickel-induced 3-alkylation of oxindole with alcohols and diols. Synthesis 2002, 2002, 595–597. [Google Scholar] [CrossRef]
- Volk, B.; Simig, G. New one-pot synthesis of 3-alkyl-and 3-(ω-hydroxyalkyl)oxindoles from isatins. Eur. J. Org. Chem. 2003, 2003, 3991–3996. [Google Scholar] [CrossRef]
- Porcs-Makkay, M.; Volk, B.; Kapiller-Dezsöfi, R.; Mezei, T.; Simig, G. New routes to oxindole derivatives. Monatsh. Chem. Chem. Mon. 2004, 135, 697–711. [Google Scholar] [CrossRef]
- Kókai, E.; Simig, G.; Volk, B. Literature survey and further studies on the 3-Alkylation of N-unprotected 3-monosubstituted oxindoles. Practical synthesis of N-unprotected 3,3-disubstituted oxindoles and subsequent transformations on the aromatic ring. Molecules 2017, 22, 24–41. [Google Scholar]
- Shepherd, T.A.; Dally, R.D.; Joseph, S. Preparation of Pyrrolopyrimidinone Derivatives as AKT and p70S6 Kinase Inhibitors. U.S. Patent 20,100,120,801, 13 May 2010. [Google Scholar]
- Leftheris, K.; Barrish, J.; Hynes, J.; Wrobleski, S.T. Preparation of Pyrrolo[2,1-f][1,2,4]Triazine Carboxylic Acid Derivatives for Use in Treating p38 Kinase-Associated Conditions. WO Patent 2,002,040,486, 23 May 2002. [Google Scholar]
- Jakob, F.; Konetzki, I.; Craan, T.; Nardi, A.; Hesslinger, C. Preparation of Substituted Condensed Pyrimidine Compounds as PDE4 Inhibitors. WO Patent 2,015,018,534, 12 February 2015. [Google Scholar]
- Bell, I.M.; Selnick, H.G.; Stump, C.A.; Theberge, C.R.; Zartman, C.B. Preparation of Spiro-Hydantoin Aryl CGRP Receptor Antagonists. WO Patent 2,007,061,696, 31 May 2007. [Google Scholar]
- Bell, I.M.; Selnick, H.G.; Wood, M.R.; Theberge, C.R.; Stump, C.A.; Gallicchio, S.N.; Zartman, C.B. Preparation of Spiro-Hydantoin Aryl CGRP Receptor Antagonists. WO Patent 2,007,061,677, 31 May 2007. [Google Scholar]
- Bell, I.M.; Stump, C.A.; Theberge, C.R.; Gallicchio, S.N.; Zartman, C.B.; Selnick, H.G. Preparation of Spiro-Hydantoin Aryl CGRP Receptor Antagonists. WO Patent 2,007,061,692, 31 May 2007. [Google Scholar]
- Bell, I.M.; Stump, C.A. Preparation of Monocyclic Anilide Spirolactam CGRP Receptor Antagonists Useful Against Headache, Migraine and Cluster Headache. WO Patent 2,006,029,153, 16 May 2006. [Google Scholar]
- Bell, I.M.; Fraley, M.; Biftu, T.; Zhu, C.; Nair, A. Preparation of Heterocyclyl Spirolactams as CGRP Receptor Antagonists. WO Patent 2,013,169,563, 14 November 2013. [Google Scholar]
- Bell, I.M.; Fraley, M.F.; Gallicchio, S.N.; Ginnetti, A.; Mitchell, H.J.; Paone, D.V.; Staas, D.D.; Wang, C.; Zartman, B.C.; Stevenson, H.E. Preparation of Piperidinone Carboxamide Indane as CGRP Receptor Antagonists for Treatment of Migraine. WO Patent 2,012,064,911, 18 May 2012. [Google Scholar]
- Rennie, G.R.; Perl, N.; Mermerian, A.; Jung, J.; Jia, J.; Iyengar, R.R.; Im, G.-Y.J.; Barden, T.C.; Sheppeck, J.E.; Renhowe, P.A.; et al. Preparation of Oxazolylpyrazole Compounds as Soluble Guanylate Cyclase (sGC) Stimulators. WO Patent 2,016,044,446, 24 March 2016. [Google Scholar]
- Follmann, M.; Stasch, J.-P.; Redlich, G.; Griebenow, N.; Lang, D.; Wunder, F.; Paulsen, H.; Huebsch, W. Preparation of Fused Imidazole and Pyrazole Derivatives Useful in the Prevention and/or Treatment of Cardiovascular Diseases. DE Patent 102,012,200,352, 11 July 2013. [Google Scholar]
- Follmann, M.; Stasch, J.-P.; Redlich, G.; Griebenow, N.; Lang, D.; Wunder, F.; Paulsen, H.; Huebsch, W. Substituted, Annulated Imidazoles and Pyrazoles, and Use Thereof. WO Patent 2,013,104,598, 18 July 2013. [Google Scholar]
- Follmann, M.; Stasch, J.-P.; Redlich, G.; Griebenow, N.; Lang, D.; Wunder, F. Heteroaryl-Substituted Pyrazolopyridines and Use Thereof as Soluble Guanylate Cyclase Stimulators and Their Preparation. WO Patent 2,013,004,785, 10 January 2013. [Google Scholar]
- Berger, R.; Dong, G.; Raghavan, S.; Yang, Z. Preparation of Triazolo-Pyrazinyl Derivatives Useful as Soluble Guanylate Cyclase Activators. WO Patent 2,016,081,668, 26 May 2016. [Google Scholar]
- Nozoye, T.; Nakai, T.; Kubo, A. Reactions of indole-related compounds. III. (E)-and (Z)-2-Oxoindolin-3-ethylidenes. Chem. Pharm. Bull. 1977, 25, 196–198. [Google Scholar] [CrossRef]
- Mertens, A.; Müller-Beckmann, B.; Kampe, W.; Hölck, J.-P.; von der Saal, W. Nonsteroidal cardiotonics. 1. 2-Pyridyl-6,7-dihydro-3H,5H-pyrrolo[2,3-f]benzimidazol-6-ones, a novel class of cardiotonic agents. J. Med. Chem. 1987, 30, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Tacconi, G.; Maggi, L.D.; Righetti, P.; Desimoni, G.; Azzolina, O.; Ghislandi, V. (Z)-and (E)-3-Alkylidene-1,3-dihydroindol-2-ones: Influence of configuration on the transmission of the inductive effect to the carbonyl group. J. Chem. Soc. Perkin Trans. 1976, 2, 150–154. [Google Scholar] [CrossRef]
- Gruda, I. Formation of N- and C-substituted derivatives during alkylation of indol-2(3H)-one. Can. J. Chem. 1972, 50, 18–23. [Google Scholar] [CrossRef]
- Shaun, J.; Tetsuro, K.; Katsura, T.; Tsuyoshi, H.; Yasufumi, U. Novel Compounds. U.S. Patent 2,011,195,954, 11 August 2011. [Google Scholar]
- Karig, G.; Ford, M.J.; Siegel, K.; Schnatterer, S. Method for Producing N-Sulfonyl-Substituted Oxindoles. U.S. Patent 2,015,133,660, 14 May 2015. [Google Scholar]
- Cook, B.N.; Huber, J.D.; Hughes, R.O.; Kirrane, T.M., Jr.; Lasota, C.; Li, X.; Liang, S.; Mugge, I.A.; Zhang, Q. Preparation of Bicyclic Compounds as Modulators of Retinoid-Related Orphan Receptor γt (RORγt or RORc). WO Patent 2,015,035,032, 12 March 2015. [Google Scholar]
- Kende, A.S.; Hodges, J.C. Regioselective C-3 alkylations of oxindole dianion. Synth. Commun. 1982, 12, 1–10. [Google Scholar] [CrossRef]
- Li, B.-F.; Moree, W.J.; Yu, J.; Coon, T.; Zamani-Kord, S.; Malany, S.; Jalali, K.; Wen, J.; Wang, H.; Yang, C.; et al. Selectivity profiling of novel indene H1-antihistamines for the treatment of insomnia. Bioorg. Med. Chem. Lett. 2010, 20, 2629–2633. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Thaisrivongs, D.A.; Hartwig, J. Palladium-catalyzed asymmetric allylic alkylations of polynitrogen-containing aromatic heterocycles. J. Am. Chem. Soc. 2011, 133, 12439–12441. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; You, Y.; Chen, Y.-Z.; Xiang, G.-Y.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. An unprecedented protocol for the synthesis of 3-hydroxy-3-phenacyloxindole derivatives with indolin-2-ones and α-substituted ketones. Org. Biomol. Chem. 2016, 14, 1395–1401, and references cited therein. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds 24a−c, 25a−c, 26a−f, 27a−f, 28, 29a−c, 30a,b, and 31a,b are available from the authors. |
Figure 1.
Oxindole 1, 7-azaoxindole 2, and drug candidates with a 7-azaoxindole skeletons: ubrogepant (3, MK-1602), MK-8031 4, MK-3207 5; and 1,3-diazaoxindole 6a.
Figure 1.
Oxindole 1, 7-azaoxindole 2, and drug candidates with a 7-azaoxindole skeletons: ubrogepant (3, MK-1602), MK-8031 4, MK-3207 5; and 1,3-diazaoxindole 6a.
Scheme 1.
Synthesis of 5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-ones (6, 1,3-diazaoxindoles).
Scheme 2.
Alkylation of oxindole (1) to 3-monoalkyloxindoles 9 and subsequent alkylation to give 3,3-dialkyloxindoles 10.
Scheme 2.
Alkylation of oxindole (1) to 3-monoalkyloxindoles 9 and subsequent alkylation to give 3,3-dialkyloxindoles 10.
Scheme 3.
C(5)-Monoalkylation of 4-chloro-1,3-diazaoxindole (11).
Scheme 4.
C(5)-Monoalkylation of 2-methyl-1,3-diazaoxindole (6b).
Scheme 5.
C(5)-Dimethylation of 4-chloro-1,3-diazaoxindole (11).
Scheme 6.
C(5)-Alkylation of 4-chloro-1,3-diazaoxindole (11) leading to spiro products 16–19.

Scheme 7.
Examples for the ring-closure based approaches for the synthesis of C(5)-disubstituted 4-amino-1,3-diazaoxindoles 23.
Scheme 7.
Examples for the ring-closure based approaches for the synthesis of C(5)-disubstituted 4-amino-1,3-diazaoxindoles 23.
Scheme 8.
Two-step synthesis of 5-isopropyl-1,3-diazaoxindoles 24.
Scheme 9.
Formation of a product mixture during the alkylation of 24b in the presence of NaOH.
Scheme 10.
5,7-Dialkylation of 5-isopropyl-1,3-diazaoxindoles 24a–c in the presence of NaOH.
Scheme 11.
Methylation of 5-isopropyl-1,3-diazaoxindoles 24a–c in the presence of a large excess of BuLi and MeI.
Scheme 11.
Methylation of 5-isopropyl-1,3-diazaoxindoles 24a–c in the presence of a large excess of BuLi and MeI.

Figure 2.
By-products of the methylation reactions of 5-isopropyl-1,3-diazaoxindoles 24.

Scheme 12.
5-Hydroxylation of 5-isopropyl-1,3-diazaoxindoles 24a–c.
Scheme 13.
C(5)-Methylation of 5-isopropyl-1,3-diazaoxindoles 24a–c.
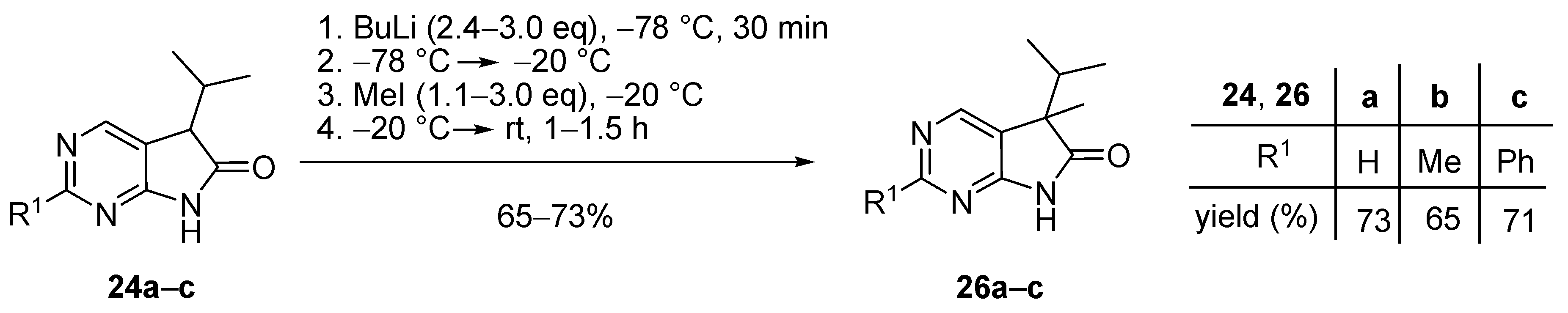
Scheme 14.
Benzylation of 5-isopropyl-1,3-diazaoxindoles 24a,b in the presence of a large excess of BuLi and BnBr.
Scheme 14.
Benzylation of 5-isopropyl-1,3-diazaoxindoles 24a,b in the presence of a large excess of BuLi and BnBr.
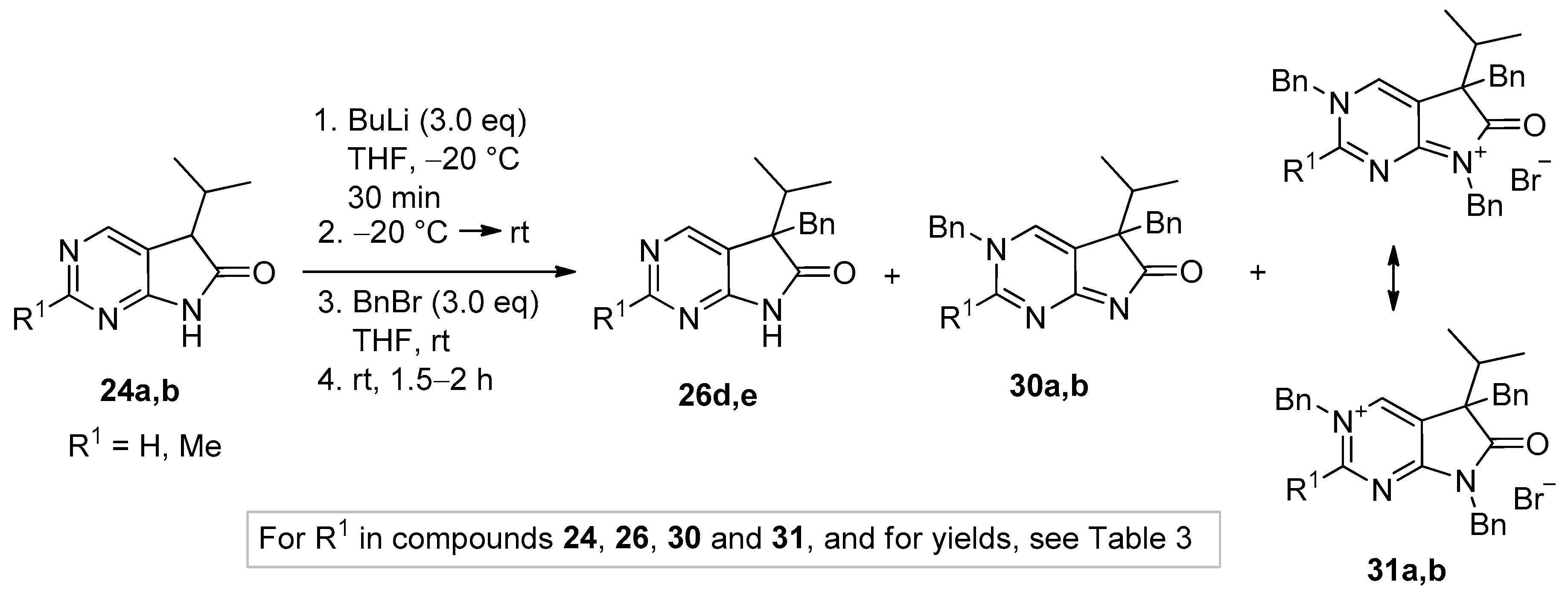
Scheme 15.
Targeted synthesis of 3,5,7-tribenzyl derivatives 31a,b starting from 5,7-dibenzyl derivatives 27d,e.
Scheme 15.
Targeted synthesis of 3,5,7-tribenzyl derivatives 31a,b starting from 5,7-dibenzyl derivatives 27d,e.
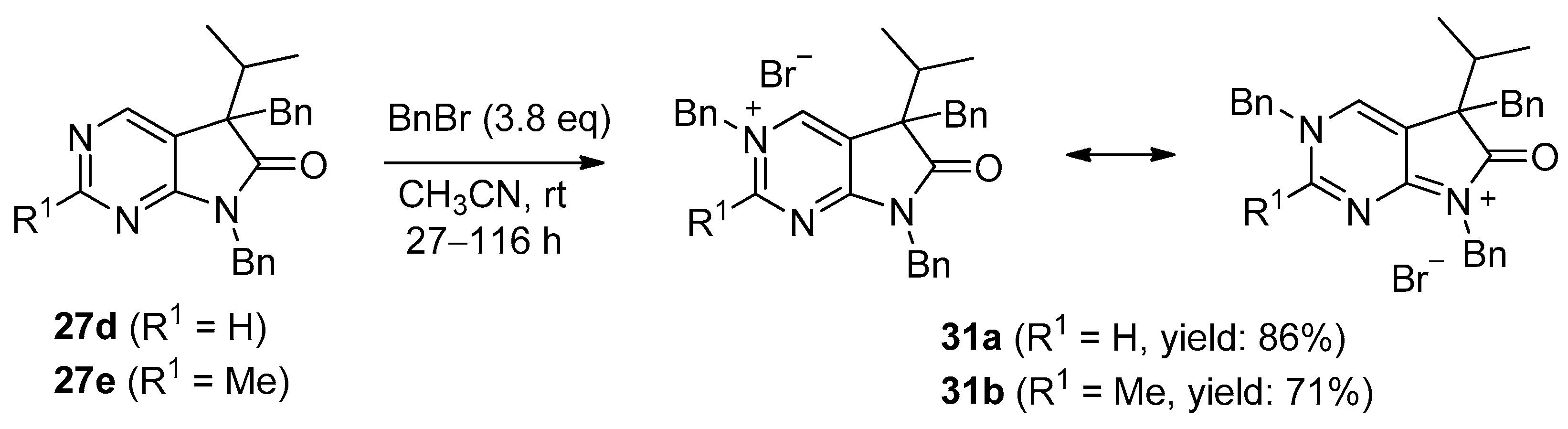
Figure 3.
Perspective view of bromide salt 31a.
Figure 4.
Characteristic 1H-NMR (red, ppm) and IR (blue, cm−1) signals of 5,7-dibenzyl- (27d), 3,5-dibenzyl- (30a) and 3,5,7-tribenzyl- (31a) derivatives.
Figure 4.
Characteristic 1H-NMR (red, ppm) and IR (blue, cm−1) signals of 5,7-dibenzyl- (27d), 3,5-dibenzyl- (30a) and 3,5,7-tribenzyl- (31a) derivatives.
Scheme 16.
C(5)-Benzylation of 5-isopropyl-1,3-diazaoxindoles 24a–c.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Yields of the 5,7-dialkylation reactions of 5-isopropyl-1,3-diazaoxindoles 24a–c.
| Entry | 24 | R1 | R2 | X | 27 | Yield of 27 (%) |
|---|---|---|---|---|---|---|
| 1 | a | H | Me | I | a | 64 |
| 2 | b | Me | Me | I | b | 80 |
| 3 | c | Ph | Me | I | c | 91 |
| 4 | a | H | Bn | Br | d | 70 |
| 5 | b | Me | Bn | Br | e | 82 |
| 6 | c | Ph | Bn | Br | f | 82 |
Table 2.
Isolated products of the methylation reactions of 5-isopropyl-1,3-diazaoxindoles 24 in the presence of a large excess of BuLi and MeI.
Table 2.
Isolated products of the methylation reactions of 5-isopropyl-1,3-diazaoxindoles 24 in the presence of a large excess of BuLi and MeI.
| Entry | 24 | R1 | Yield of 26 (%) | Yield of 27 (%) |
|---|---|---|---|---|
| 1 | a | H | 75 | 2 |
| 2 | b | Me | 42 | 22 |
| 3 | c | Ph | 56 | 31 |
Table 3.
Isolated products of the benzylation reactions of 5-isopropyl-1,3-diazaoxindoles 24a,b in the presence of a large excess of BuLi and BnBr.
Table 3.
Isolated products of the benzylation reactions of 5-isopropyl-1,3-diazaoxindoles 24a,b in the presence of a large excess of BuLi and BnBr.
| Entry | 24 | R1 | 26 | Yield of 26 (%) | 30 | Yield of 30 (%) | 31 | Yield of 31 (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | a | H | d | 36 | a | 16 | a | 12 |
| 2 | b | Me | e | 5 | b | 56 | b | 19 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kókai, E.; Halász, J.; Dancsó, A.; Nagy, J.; Simig, G.; Volk, B. Study on the Alkylation Reactions of N(7)-Unsubstituted 1,3-Diazaoxindoles. Molecules 2017, 22, 846. https://doi.org/10.3390/molecules22050846
AMA Style
Kókai E, Halász J, Dancsó A, Nagy J, Simig G, Volk B. Study on the Alkylation Reactions of N(7)-Unsubstituted 1,3-Diazaoxindoles. Molecules. 2017; 22(5):846. https://doi.org/10.3390/molecules22050846
Chicago/Turabian StyleKókai, Eszter, Judit Halász, András Dancsó, József Nagy, Gyula Simig, and Balázs Volk. 2017. "Study on the Alkylation Reactions of N(7)-Unsubstituted 1,3-Diazaoxindoles" Molecules 22, no. 5: 846. https://doi.org/10.3390/molecules22050846