New Heterofunctional Supports Based on Glutaraldehyde-Activation: A Tool for Enzyme Immobilization at Neutral pH

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
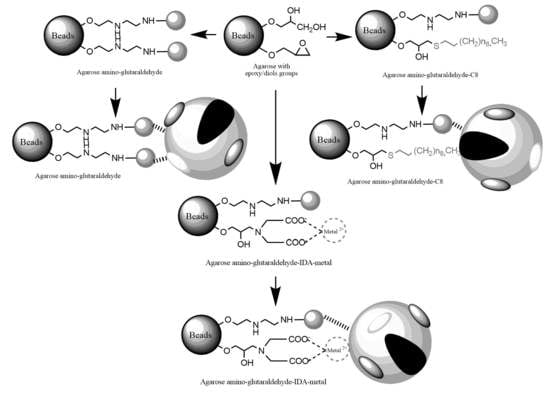
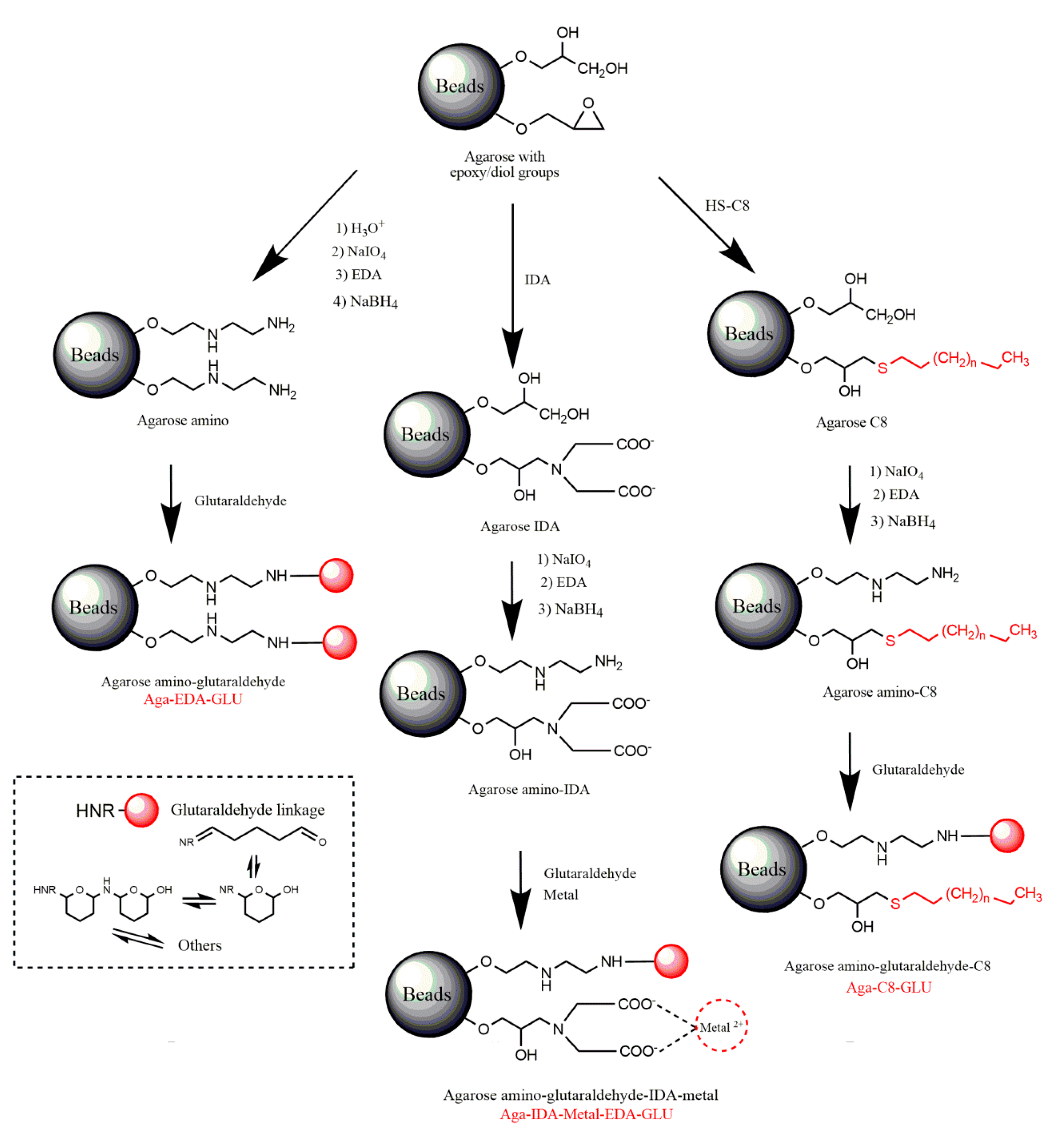
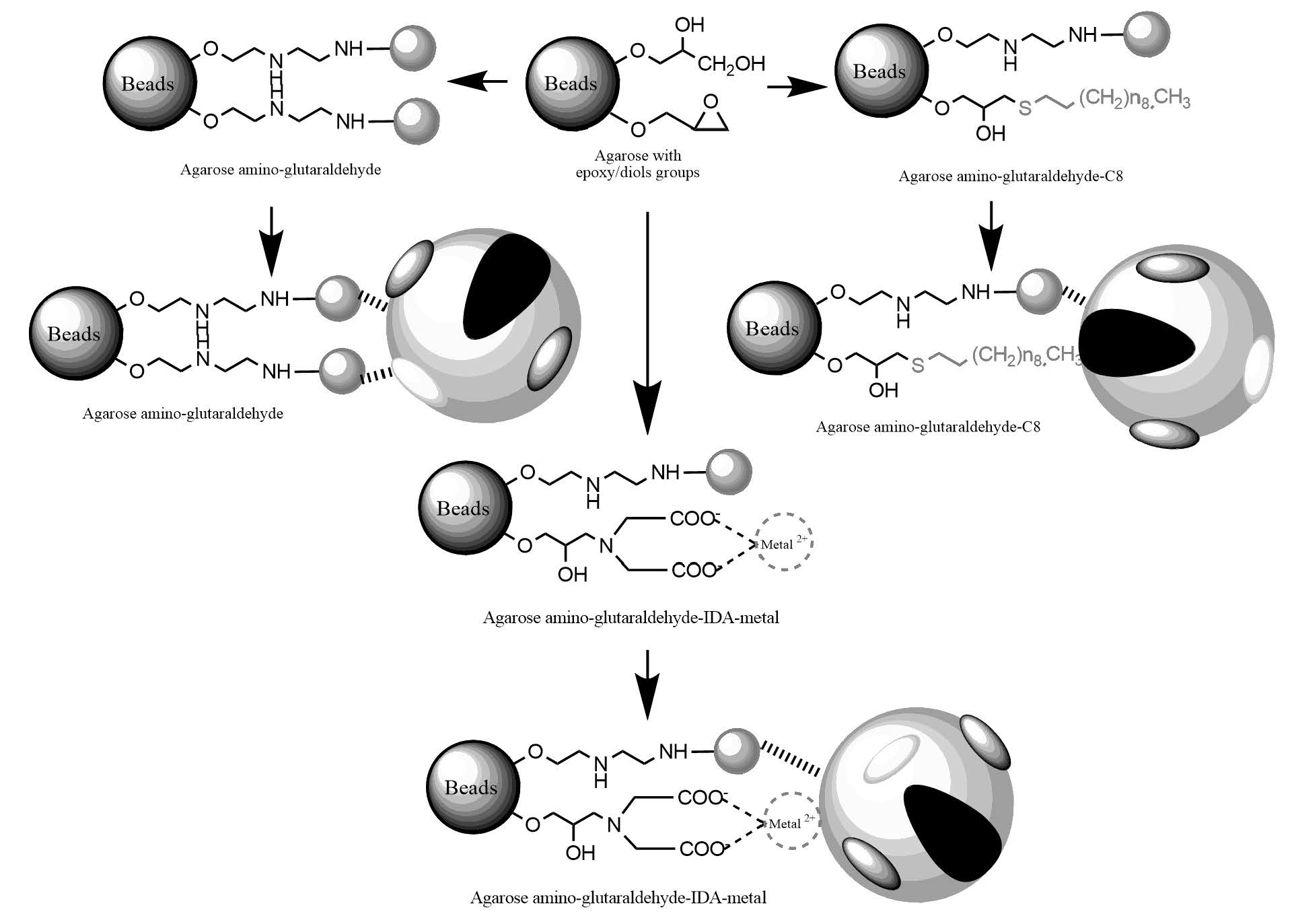
2.1. Construction and Analysis of New Heterofunctional Supports
2.2. Immobilization of Different Biocatalysts on the New Heterofunctional Supports
2.3. Thermal Stability
3. Materials and Methods
3.1. Materials
3.2. Standard Determination of Enzymatic Activities
3.3. Supports for Immobilization
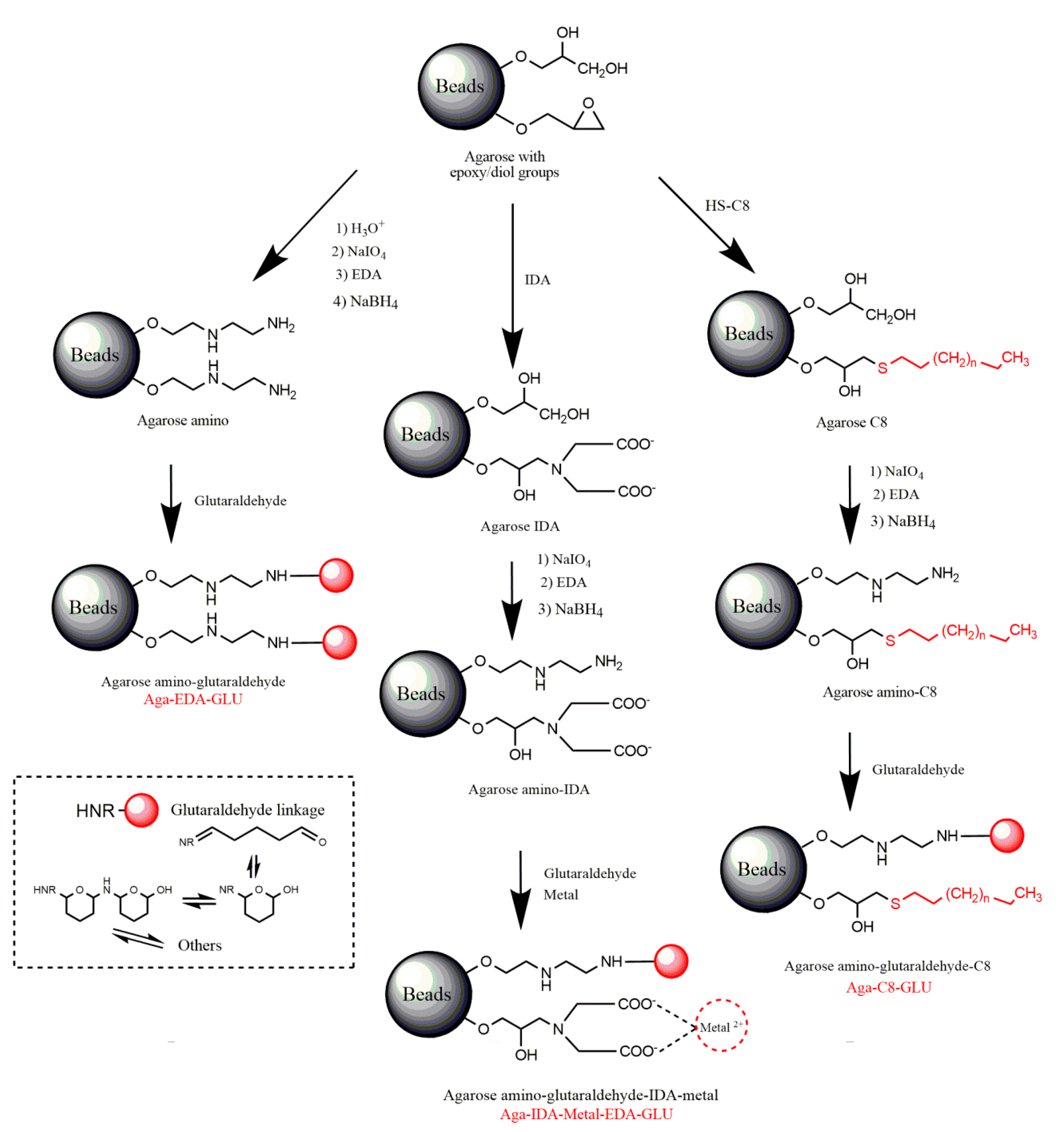
3.3.1. Activation of Agarose-Based Beads
3.3.2. Modification of the Epoxy-Activated Agarose with Different Reactive Groups
Cationic Supports
Support Activated with the Glutaraldehyde Group
Anionic Supports Activated with the Glutaraldehyde Group and Metal Chelate
Hydrophobic Supports Activated with the Glutaraldehyde Group
3.4. Immobilization of Enzymes
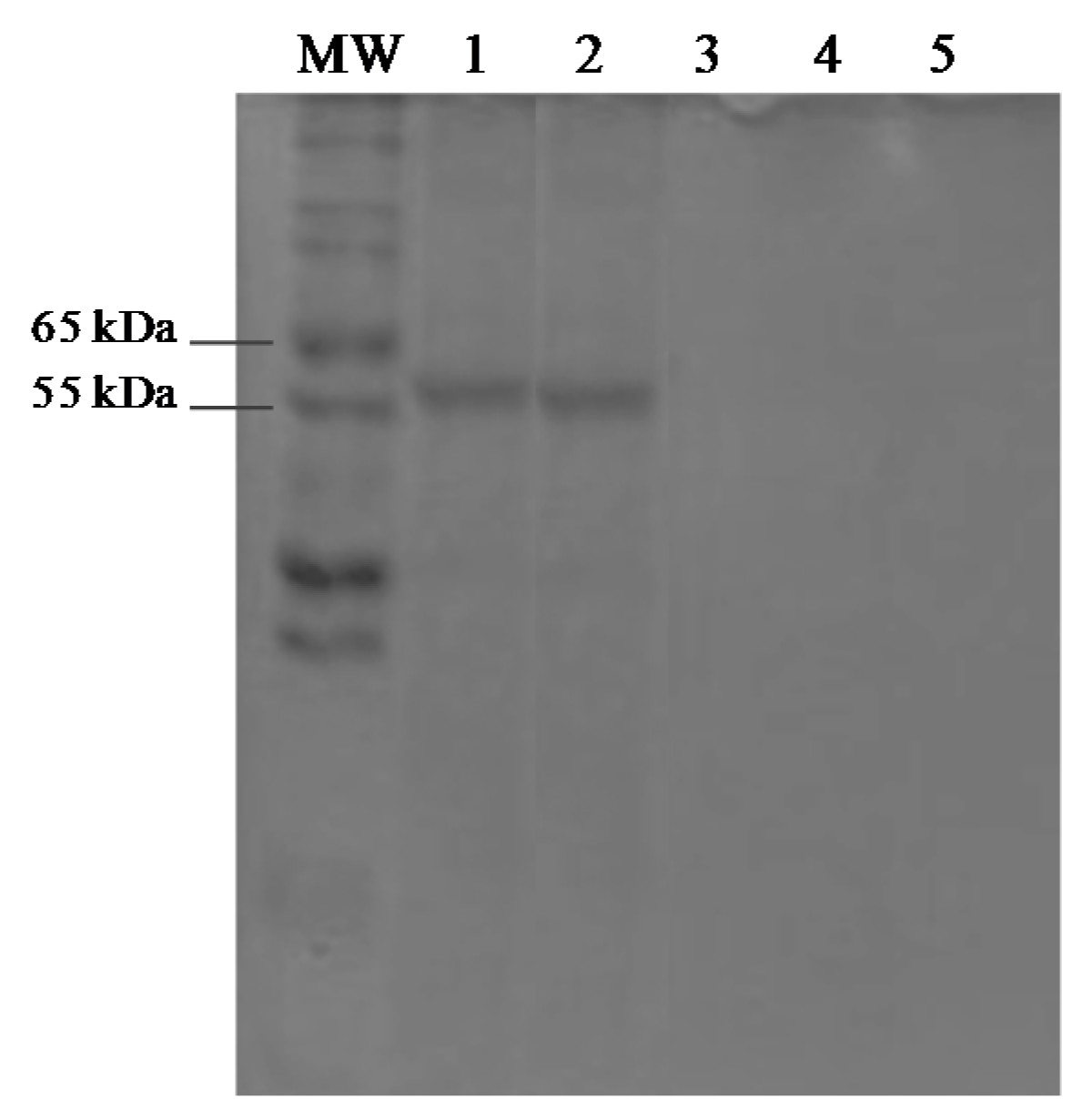
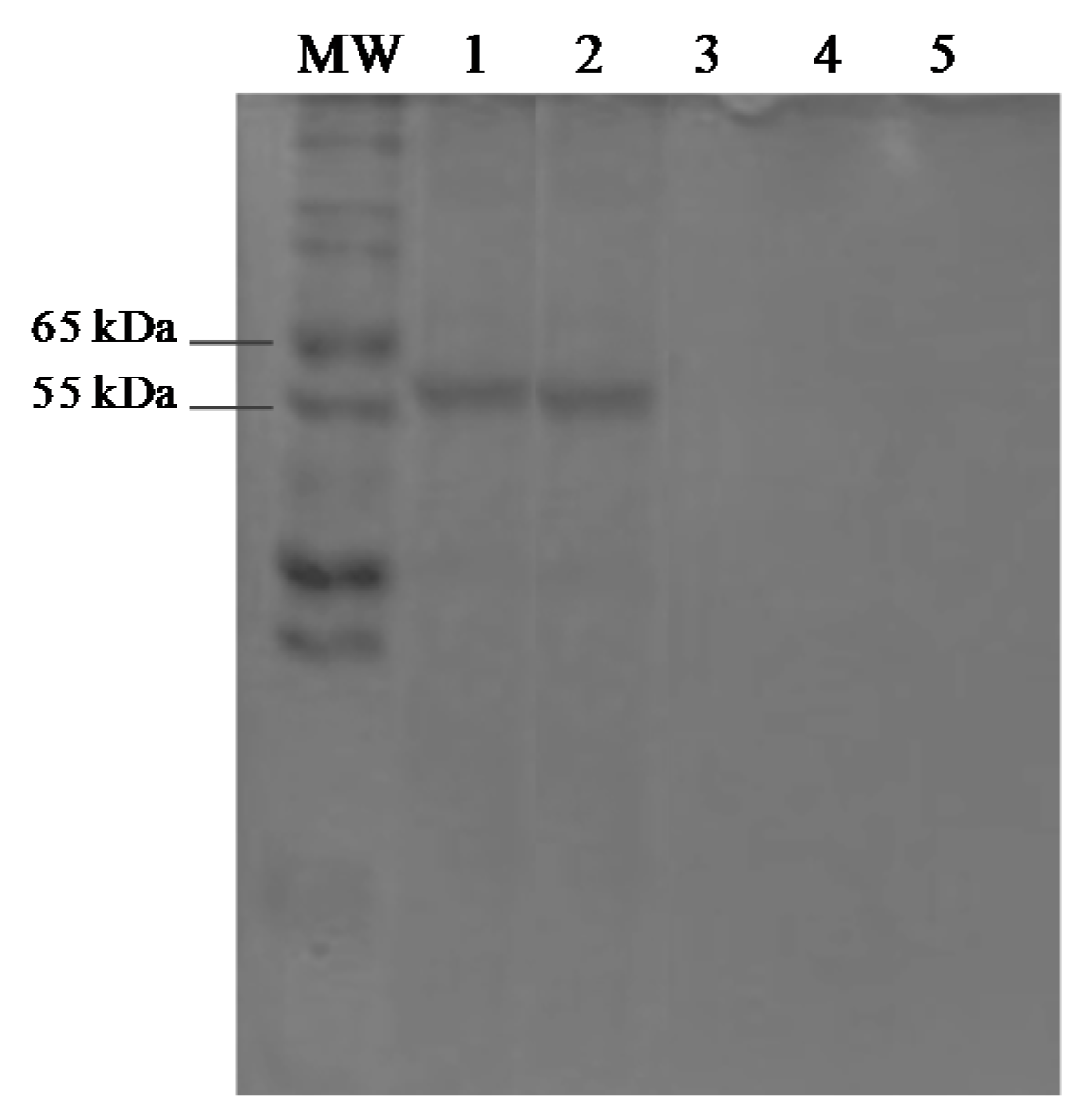
3.5. Protein Assays and SDS-PAGE
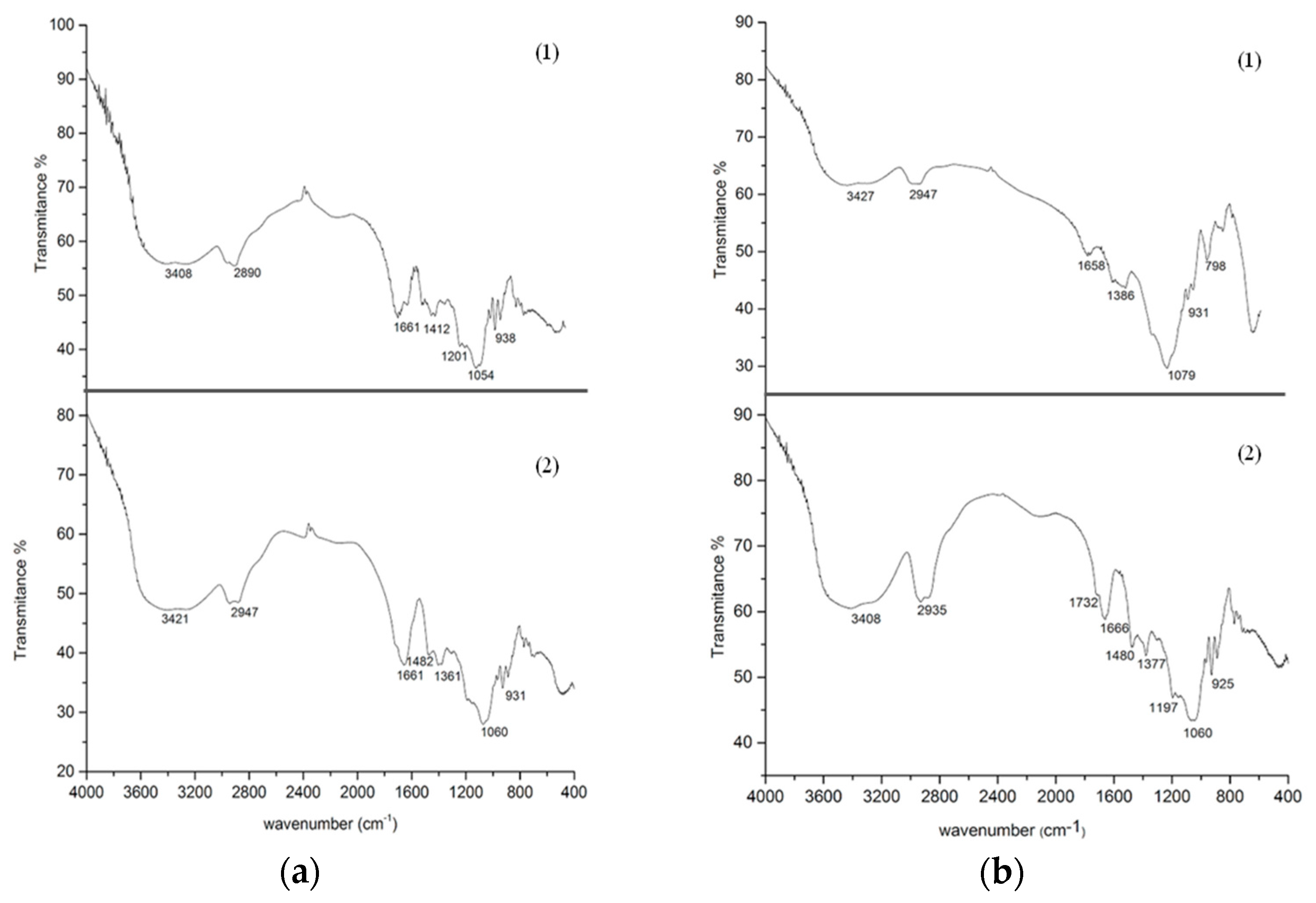
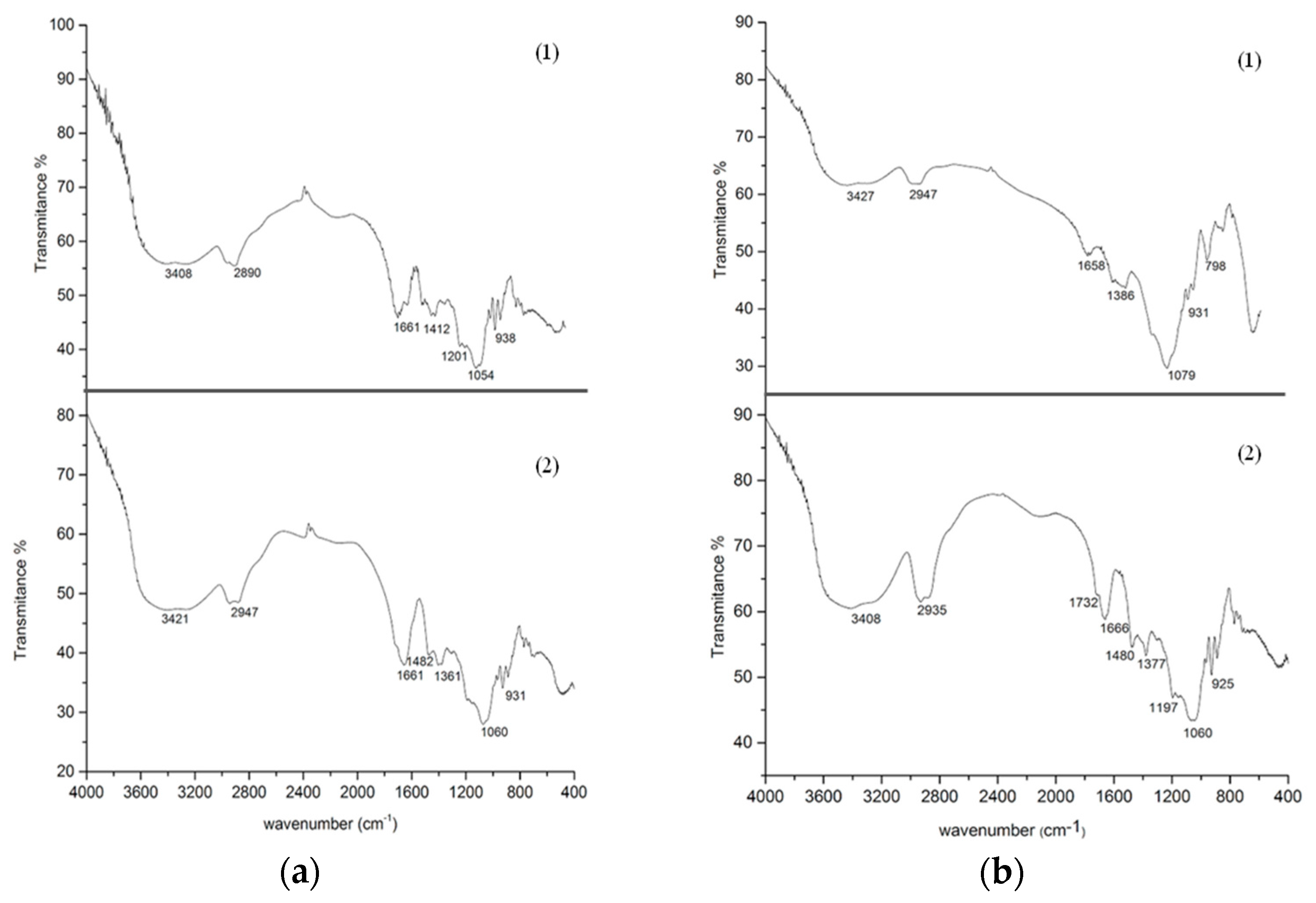
3.6. Fourier Transform Infrared (FTIR)
3.7. Desorption Process Analysis
3.8. Thermal Stability
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roy, J.J.; Abraham, T.E. Strategies in making cross-linked enzyme crystals. Chem. Rev. 2004, 104, 3705–3721. [Google Scholar] [CrossRef]
- Cao, L. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Eş, I.; Vieira, J.D.G.; Amaral, A.C. Principles, techniques, and applications of biocatalyst immobilization for industrial application. Appl. Microbiol. Biotechnol. 2015, 99, 2065–2082. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Bernedo, M.; Cuenca, E.; Fuentes, M.; Fernandez-Lorente, G.; Palomo, J.M.; Grazu, V.; Pessela, B.C.C.; Giacomini, C.; et al. Some special features of glyoxyl supports to immobilize proteins. Enzym. Microb. Technol. 2005, 37, 456–462. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.C.; Hidalgo, A.; Fernández-Lorente, G.; Fernández-Lafuente, R.; et al. Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins. Enzym. Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the support properties for immobilization or purification of enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Tural, B.; Tarhan, T.; Tural, S. Covalent immobilization of benzoylformate decarboxylase from Pseudomonas putida on magnetic epoxy support and its carboligation reactivity. J. Mol. Catal. B Enzym. 2014, 102, 188–194. [Google Scholar] [CrossRef]
- Alnoch, R.C.; Melo, R.R.D.; Palomo, J.M.; Souza, E.M.D.; Krieger, N.; Mateo, C. New tailor-made alkyl-aldehyde bifunctional supports for lipase immobilization. Catalyst 2016, 6, 191–204. [Google Scholar] [CrossRef]
- Garmroodi, M.; Mohammadi, M.; Ramazani, A.; Ashjari, M.; Mohammadi, J.; Sabour, B.; Yousefi, M. Covalent binding of hyper-activated Rhizomucor miehei lipase (RML) on hetero-functionalized siliceous supports. Int. J. Biol. Macromol. 2016, 86, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; Albuquerque, T.; Bartolome-Cabrero, R.; Fernandez-Lopez, L.; Torres, R.; Ortiz, C.; dos Santos, J.; Barbosa, O.; Fernandez-Lafuente, R. Reversible immobilization of lipases on heterofunctional octyl-amino agarose beads prevents enzyme desorption. Molecules 2016, 21, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, T.L.D.; Rueda, N.; dos Santos, J.C.S.; Barbosa, O.; Ortiz, C.; Binay, B.; Ozdemir, E.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Easy stabilization of interfacially activated lipases using heterofunctional divinyl sulfone activated-octyl agarose beads. Modulation of the immobilized enzymes by altering their nanoenvironment. Process Biochem. 2016, 51, 865–874. [Google Scholar] [CrossRef]
- Guajardo, N.; Bernal, C.; Wilson, L.; Cabrera, Z. Selectivity of R-α-monobenzoate glycerol synthesis catalyzed by Candida antarctica lipase B immobilized on heterofunctional supports. Process Biochem. 2015, 50, 1870–1877. [Google Scholar] [CrossRef]
- Hirata, D.B.; Albuquerque, T.L.; Rueda, N.; Virgen-ortíz, J.J.; Tacias-pascacio, V.G.; Fernandez-lafuente, R. Enzymatic evaluation of different immobilized lipases in transesterification reactions using tributyrin: Advantages of the heterofunctional octyl agarose beads. J. Mol. Catal. B Enzym. 2016, 133, 117–123. [Google Scholar] [CrossRef]
- Rueda, N.; Cleiton, S.; Daniela, M.; Albuquerque, T.L.; Barbosa, O.; Torres, R.; Ortiz, C.; Fernandez-lafuente, R. Enzymatic reversible immobilization of lipases on octyl-glutamic agarose beads: A mixed adsorption that reinforces enzyme immobilization. J. Mol. Catal. B Enzym. 2016, 128, 10–18. [Google Scholar] [CrossRef]
- Pessela, B.C.C.; Mateo, C.; Fuentes, M.; Vian, A.; Carrascosa, A.V.; Guisa, M. Stabilization of a multimeric β-galactosidase from Thermus sp. strain T2 by immobilization on novel heterofunctional epoxy supports plus aldehyde-dextran cross-linking. Biotechnol. Prog. 2004, 20, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, C.; Vera, C.; Serna, N.; Illanes, A. Immobilization of Aspergillus oryzae β-galactosidase in an agarose matrix functionalized by four different methods and application to the synthesis of lactulose. Bioresour. Technol. 2017, 232, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bernal, C.; Marciello, M.; Mesa, M.; Sierra, L.; Fernandez-lorente, G.; Mateo, C.; Guisan, J.M. Immobilisation and stabilisation of β-galactosidase from Kluyveromyces lactis using a glyoxyl support. Int. Dairy J. 2013, 28, 76–82. [Google Scholar] [CrossRef]
- Mateo, C.; Grazu, V.; Palomo, J.M.; Lopez-gallego, F. Fernandez-lafuente, R; Guisan, J.M. Immobilization of enzymes on heterofunctional epoxy supports. Nat. Protoc. 2007, 5, 1022–1033. [Google Scholar]
- Blanco, R.M.; Guistin, J.M. Stabilization of enzymes by multipoint covalent attachment to agarose-aldehyde gels. Borohydride reduction of trypsin-agarose derivatives. Enzym. Microb. Technol. 1989, 11, 360–366. [Google Scholar] [CrossRef]
- Tardioli, P.W.; Fernandez-Lafuente, R.; Guisan, J.M.; Giordano, R.L.C. Design of new immobilized-stabilized carboxypeptidase a derivative for production of aromatic free hydrolysates of proteins. Biotechnol. Prog. 2003, 19, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Bolivar, J.M.; Godoy, C.A.; Rocha-Martin, J.; Pessela, B.C.; Curiel, J.A.; Munoz, R.; Guisan, J.M.; Fernández-Lorente, G. Improvement of enzyme properties with a two-step immobilizaton process on novel heterofunctional supports. Biomacromolecules 2010, 11, 3112–3117. [Google Scholar] [CrossRef] [PubMed]
- Rivero, C.W.; Palomo, J.M. Covalent immobilization of Candida rugosa lipase at alkaline pH and their application in the regioselective deprotection of Per-O-acetylated thymidine. Catalyst 2016, 6, 115–126. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 2004, 37, 790–802. [Google Scholar] [PubMed]
- Barbosa, O.; Torres, R.; Ortiz, C.; Fernandez-lafuente, R. Versatility of glutaraldehyde to immobilize lipases: Effect of the immobilization protocol on the properties of lipase B from Candida antarctica. Process Biochem. 2012, 47, 1220–1227. [Google Scholar] [CrossRef]
- Fernández-Lorente, G.; Palomo, J.M.; Mateo, C.; Munilla, R.; Ortiz, C.; Cabrera, Z.; Guisán, J.M.; Fernández-Lafuente, R. Glutaraldehyde cross-linking of lipases adsorbed on aminated supports in the presence of detergents leads to improved performance. Biomacromolecules 2006, 7, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Bezbradica, D.I.; Mateo, C.; Guisan, J.M. Novel support for enzyme immobilization prepared by chemical activation with cysteine and glutaraldehyde. J. Mol. Catal. B Enzym. 2014, 102, 218–224. [Google Scholar] [CrossRef]
- Trivedi, T.J.; Srivastava, D.N.; Rogers, R.D.; Kumar, A. Agarose processing in protic and mixed protic-aprotic ionic liquids: Dissolution, regeneration and high conductivity, high strength ionogels. Green Chem. 2012, 14, 2831–2839. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and its derivatives as supports for enzyme immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Abian, O.; Fernandez-Lafuente, R.; Guisan, J.M. Increase in conformational stability of enzymes immobilized on epoxy-activated supports by favoring additional multipoint covalent attachment. Enzym. Microb. Technol. 2000, 26, 509–515. [Google Scholar] [CrossRef]
- Rodrigues, D.S.; Mendes, A.A.; Adriano, W.S.; Gonçalves, L.R.B.; Giordano, R.L.C. Multipoint covalent immobilization of microbial lipase on chitosan and agarose activated by different methods. J. Mol. Catal. B Enzym. 2008, 51, 100–109. [Google Scholar] [CrossRef]
- Betancor, L.; López-Gallego, F.; Hidalgo, A.; Alonso-Morales, N.; Dellamora-Ortiz, G.; Mateo, C.; Fernández-Lafuente, R.; Guisán, J.M. Different mechanisms of protein immobilization on glutaraldehyde activated supports: Effect of support activation and immobilization conditions. Enzym. Microb. Technol. 2006, 39, 877–882. [Google Scholar] [CrossRef]
- Soltani, N.; Reza, F.; Babak, P. Direct introduction of amine groups into cellulosic paper for covalent immobilization of tyrosinase: Support characterization and enzyme properties. Cellulose 2017, 24, 1407–1416. [Google Scholar] [CrossRef]
- Suescun, A.; Rueda, N.; Dos Santos, J.C.S.; Castillo, J.J.; Ortiz, C.; Torres, R.; Barbosa, O.; Fernandez-Lafuente, R. Immobilization of lipases on glyoxyl-octyl supports: Improved stability and reactivation strategies. Process Biochem. 2015, 50, 1211–1217. [Google Scholar] [CrossRef]
- Palomo, J.M.; Muoz, G.; Fernández-Lorente, G.; Mateo, C.; Fernández-Lafuente, R.; Guisán, J.M. Interfacial adsorption of lipases on very hydrophobic support (octadecyl-Sepabeads): Immobilization, hyperactivation and stabilization of the open form of lipases. J. Mol. Catal. B Enzym. 2002, 19, 279–286. [Google Scholar] [CrossRef]
- Balcão, V.M.; Vila, M.M.D.C. Structural and functional stabilization of protein entities: State-of-the-art. Adv. Drug Deliv. Rev. 2015, 93, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Glogauer, A.; Martini, V.P.; Faoro, H.; Couto, G.H.; Müller-Santos, M.; Monteiro, R.A.; Mitchell, D.A.; de Souza, E.M.; Pedrosa, F.O.; Krieger, N. Identification and characterization of a new true lipase isolated through metagenomic approach. Microb. Cell Fact. 2011, 10, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Rodríguez, Á.; Fernández-Leiro, R.; González-Siso, M.I.; Cerdán, M.E.; Becerra, M.; Sanz-Aparicio, J. Structural basis of specificity in tetrameric Kluyveromyces lactis β-galactosidase. J. Struct. Biol. 2012, 177, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Zanphorlin, L.M.; de Giuseppe, P.O.; Honorato, R.V.; Tonoli, C.C.C.; Fattori, J.; Crespim, E.; de Oliveira, P.S.L.; Ruller, R.; Murakami, M.T. Oligomerization as a strategy for cold adaptation: Structure and dynamics of the GH1 β-glucosidase from Exiguobacterium antarcticum B7. Sci. Rep. 2016, 6, 23776–23790. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzym. Microb. Technol. 2009, 45, 405–418. [Google Scholar] [CrossRef]
- Palomo, J.M.; Fuentes, M.; Fernández-Lorente, G.; Mateo, C.; Guisan, J.M.; Fernández-Lafuente, R. General trend of lipase to self-assemble giving bimolecular aggregates greatly modifies the enzyme functionality. Biomacromolecules 2003, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.M.; Segura, R.L.; Mateo, C.; Fernandez-Lafuente, R.; Guisan, J.M. Improving the activity of lipases from thermophilic organisms at mesophilic temperatures for biotechnology applications. Biomacromolecules 2004, 5, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Henley, J.P.; Sadana, A. Deactivation theory. Biotechnol. Bioeng. 1986, 28, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Crespim, E.; Zanphorlin, L.M.; de Souza, F.H.M.; Diogo, J.A.; Gazolla, A.C.; Machado, C.B.; Figueiredo, F.; Sousa, A.S.; Nóbrega, F.; Pellizari, V.H.; et al. A novel cold-adapted and glucose-tolerant GH1 β-glucosidase from Exiguobacterium antarcticum B7. Int. J. Biol. Macromol. 2016, 82, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Pessela, B.C.C.; Vian, A.; Mateo, C.; Fernandez-Lafuente, R.; Garcia, J.L.; Guisán, J.M.; Carrascosa, A.V. Overproduction of Thermus sp. strain T2 β-galactosidase in Escherichia coli and preparation by using tailor-made metal chelate supports. Appl. Environ. Microbiol. 2003, 69, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Whistler, R.L.; Wolfrom, M.L.; BeMiller, J. Methods in Carbohydrate Chemistry, 2nd ed.; Academic Press: New York, NY, USA; London, UK, 1963. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Addorisio, V.; Sannino, F.; Mateo, C.; Guisan, J.M. Oxidation of phenyl compounds using strongly stable immobilized-stabilized laccase from Trametes versicolor. Process Biochem. 2013, 48, 1174–1180. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
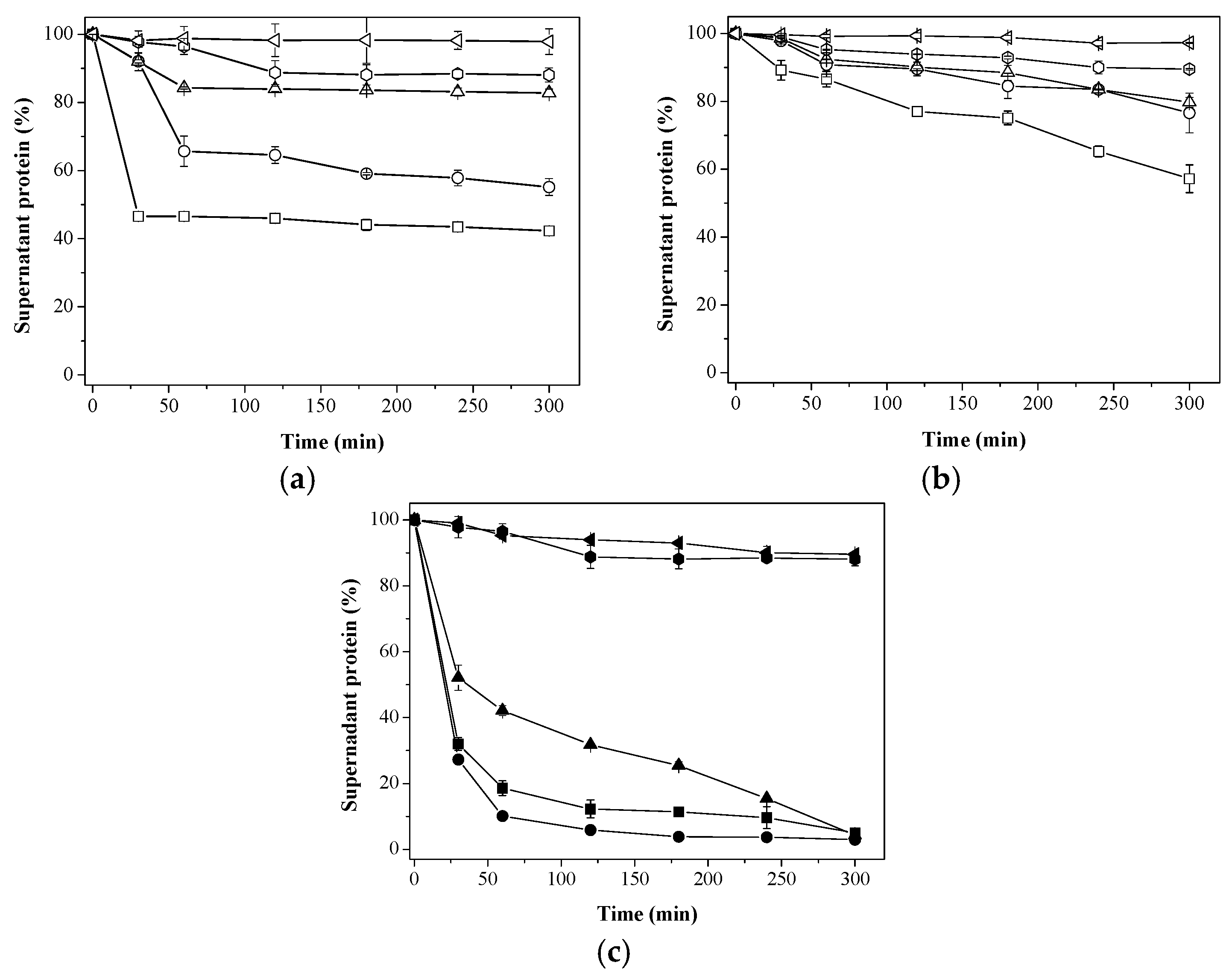
 25 mM;
25 mM;  50 mM;
50 mM;  100 mM;
100 mM;  200 mM; and
200 mM; and  500 mM sodium phosphate buffer at pH 7; (c) Immobilization on different supports using 200 mM sodium phosphate buffer at pH 7.
500 mM sodium phosphate buffer at pH 7; (c) Immobilization on different supports using 200 mM sodium phosphate buffer at pH 7.  Aga-C8-GLU;
Aga-C8-GLU;  Aga-IDA-Ni2+-EDA-GLU;
Aga-IDA-Ni2+-EDA-GLU;  Aga-EDA-GLU;
Aga-EDA-GLU;  Aga-EDA (Control); and
Aga-EDA (Control); and  Aga-EDA-GLU-Control (Control). The results are expressed as the average of triplicate assays ± the standard error of the mean.
25 mM; 50 mM; 100 mM; 200 mM; and 500 mM sodium phosphate buffer at pH 7; (c) Immobilization on different supports using 200 mM sodium phosphate buffer at pH 7. Aga-C8-GLU; Aga-IDA-Ni2+-EDA-GLU; Aga-EDA-GLU; Aga-EDA (Control); and Aga-EDA-GLU-Control (Control). The results are expressed as the average of triplicate assays ± the standard error of the mean.
Aga-EDA-GLU-Control (Control). The results are expressed as the average of triplicate assays ± the standard error of the mean.
25 mM; 50 mM; 100 mM; 200 mM; and 500 mM sodium phosphate buffer at pH 7; (c) Immobilization on different supports using 200 mM sodium phosphate buffer at pH 7. Aga-C8-GLU; Aga-IDA-Ni2+-EDA-GLU; Aga-EDA-GLU; Aga-EDA (Control); and Aga-EDA-GLU-Control (Control). The results are expressed as the average of triplicate assays ± the standard error of the mean.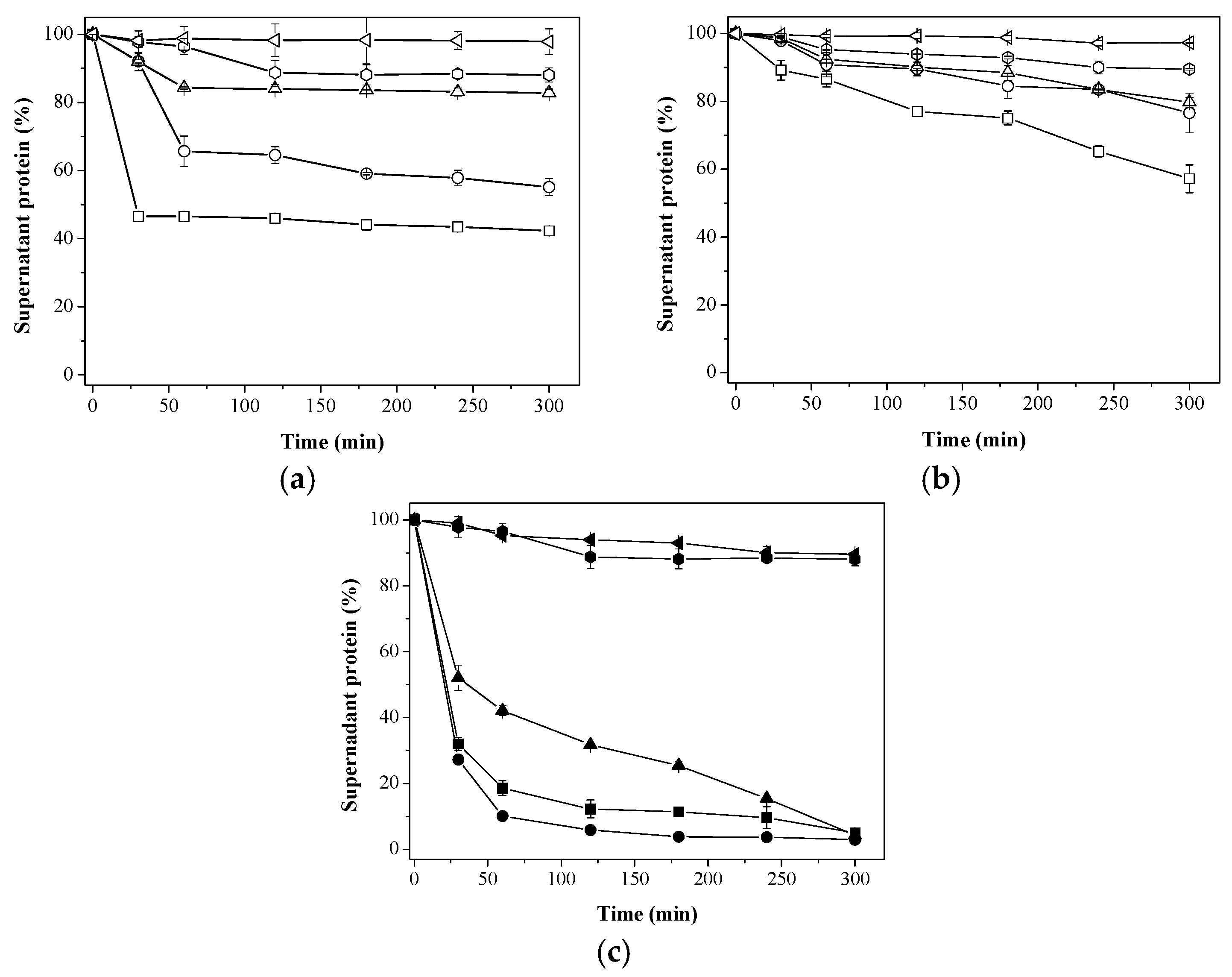
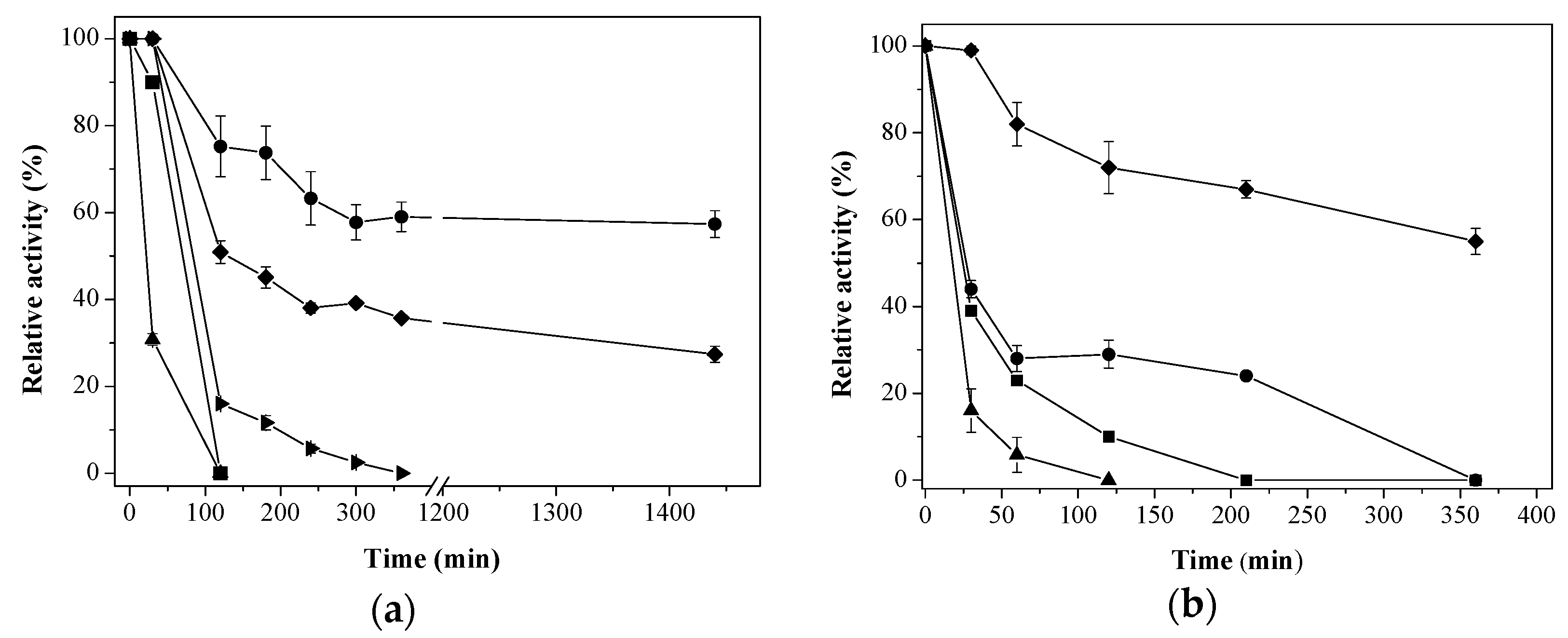
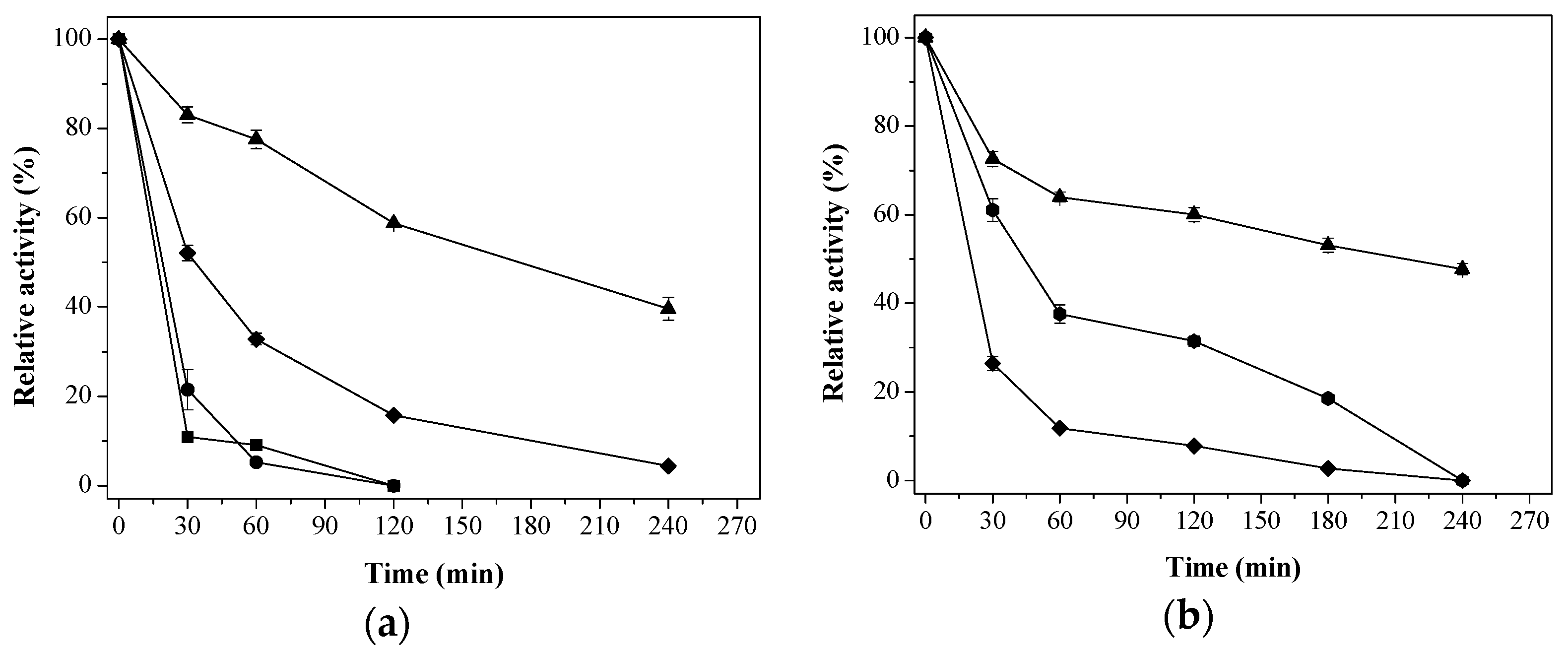
 ) Aga-IDA-Metal-EDA-GLU; (
) Aga-IDA-Metal-EDA-GLU; (  ) Aga-C8-GLU; (
) Aga-C8-GLU; (  ) Aga-EDA-GLU; (
) Aga-EDA-GLU; (  ) Octyl-Sepharose; and (
) Octyl-Sepharose; and (  ) free enzyme; (b) Lipase LipC12 at pH 7, 70 °C. (
) free enzyme; (b) Lipase LipC12 at pH 7, 70 °C. (  ) Aga-IDA-Metal-EDA-GLU; (
) Aga-IDA-Metal-EDA-GLU; (  ) Aga-C8-GLU; (
) Aga-C8-GLU; (  ) Aga-EDA-GLU; and (
) Aga-EDA-GLU; and (  ) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.
) Aga-IDA-Metal-EDA-GLU; ( ) Aga-C8-GLU; ( ) Aga-EDA-GLU; ( ) Octyl-Sepharose; and ( ) free enzyme; (b) Lipase LipC12 at pH 7, 70 °C. ( ) Aga-IDA-Metal-EDA-GLU; ( ) Aga-C8-GLU; ( ) Aga-EDA-GLU; and ( ) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.
) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.
) Aga-IDA-Metal-EDA-GLU; ( ) Aga-C8-GLU; ( ) Aga-EDA-GLU; ( ) Octyl-Sepharose; and ( ) free enzyme; (b) Lipase LipC12 at pH 7, 70 °C. ( ) Aga-IDA-Metal-EDA-GLU; ( ) Aga-C8-GLU; ( ) Aga-EDA-GLU; and ( ) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.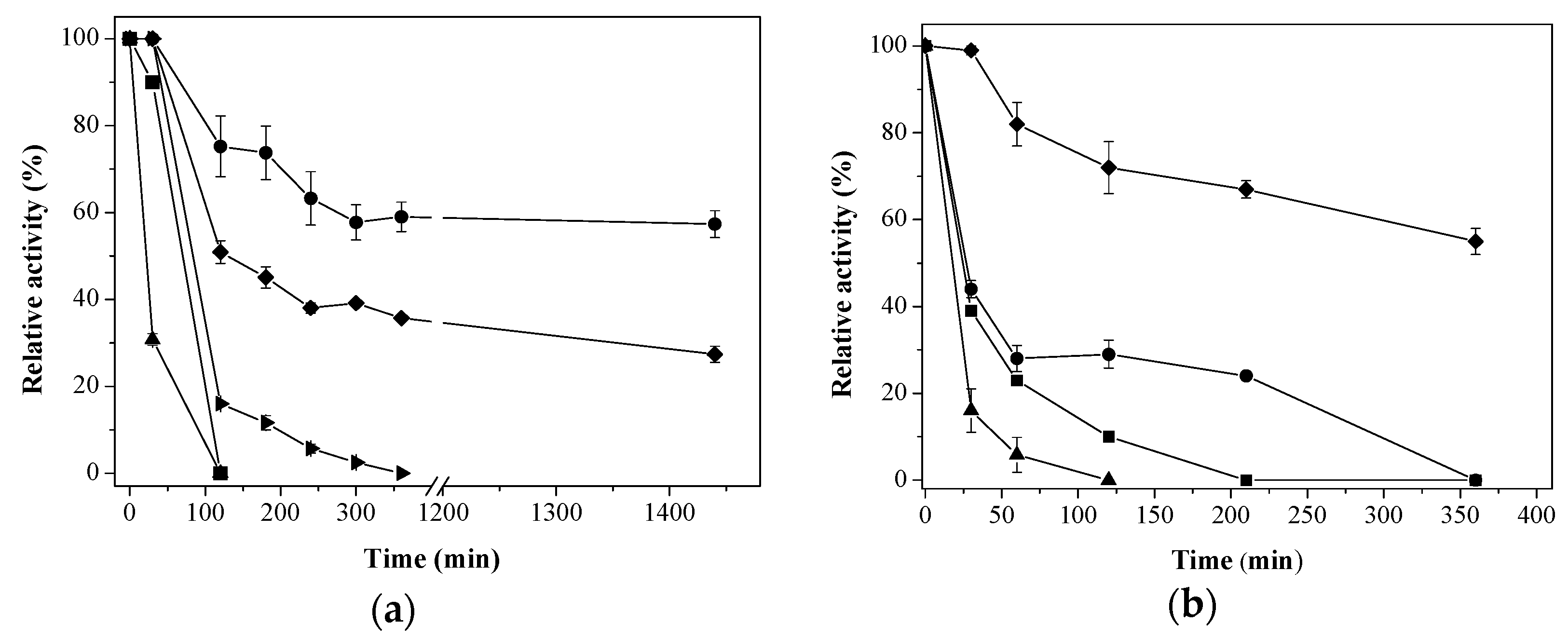
 ) Aga-C8-GLU; (
) Aga-C8-GLU; (  ) Ag-EDA-GLU; (
) Ag-EDA-GLU; (  ) Aga-IDA-Metal-EDA-GLU, and (
) Aga-IDA-Metal-EDA-GLU, and (  ) free enzyme; (b) β-glucosidase (EaglA) at pH 7, 40 °C. (
) free enzyme; (b) β-glucosidase (EaglA) at pH 7, 40 °C. (  ) Ag-EDA-GLU; (
) Ag-EDA-GLU; (  ) Aga-IDA-Metal-EDA-GLU, and (
) Aga-IDA-Metal-EDA-GLU, and (  ) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.
) Aga-C8-GLU; ( ) Ag-EDA-GLU; ( ) Aga-IDA-Metal-EDA-GLU, and ( ) free enzyme; (b) β-glucosidase (EaglA) at pH 7, 40 °C. ( ) Ag-EDA-GLU; ( ) Aga-IDA-Metal-EDA-GLU, and ( ) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.
) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.
) Aga-C8-GLU; ( ) Ag-EDA-GLU; ( ) Aga-IDA-Metal-EDA-GLU, and ( ) free enzyme; (b) β-glucosidase (EaglA) at pH 7, 40 °C. ( ) Ag-EDA-GLU; ( ) Aga-IDA-Metal-EDA-GLU, and ( ) free enzyme. The results are expressed as the average of triplicate assays ± the standard error of the mean.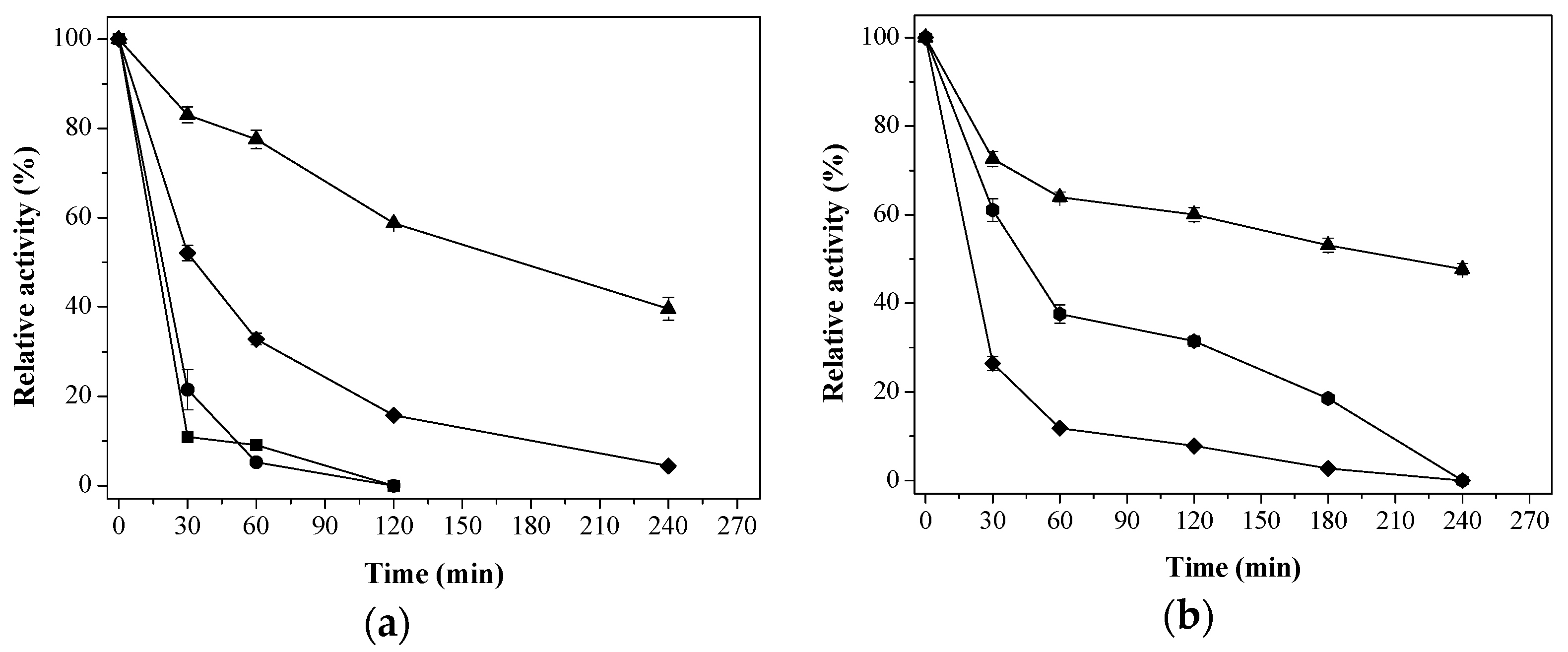

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Support | Immobilization Efficiency a (%), IE | Recovered Activity after Immobilization at pH 7 b (%), R |
|---|---|---|---|
| Aga-C8-GLU | 100 | 210 | |
| CRL | Aga-IDA-Ni2+-EDA-GLU | 95 | 54 |
| Aga-EDA-GLU | 100 | 95 | |
| Octyl-Sepharose | 100 | 225 | |
| Aga-C8-GLU | 100 | 269 | |
| LipC12 | Aga-IDA-Ni2+-EDA-GLU | 100 | 100 |
| Aga-EDA-GLU | 100 | 42 |
| Support | Immobilization Efficiency a (%), IE | Recovered Activity after Incubation b (%), R | Activity in the Supernatant after Incubation (%) |
|---|---|---|---|
| Aga-EDA-GLU * | 100 | 97 | 0 |
| Aga-C8-GLU ** | 100 | 90 | 0 |
| Octyl-Sepharose ** | 100 | 12 | 81 |
| Aga-IDA-Ni2+-EDA-GLU *** | 100 | 98 | 0 |
| Enzyme | Support | Immobilization Efficiency a (%), IE | Recovered Activity after Immobilization at pH 7 b (%), R |
|---|---|---|---|
| Aga-C8-GLU | 100 | 81 | |
| KlBgal | Aga-IDA-Co2+-EDA-GLU | >94 | 78 |
| Ag-EDA-GLU | 100 | 70 | |
| Aga-C8-GLU | 100 | 0 | |
| EaBglA | Aga-IDA-Co2+-EDA-GLU | 100 | 64 |
| Aga-EDA-GLU | 100 | 50 |
| Enzyme | Support | Half-Life (T1/2, Minutes) | Stability Factor |
|---|---|---|---|
| CRL a | Free form | 21 | |
| Aga-C8-GLU | 120 | 5.7 | |
| Aga-IDA-Ni2+-EDA-GLU | 60 | 2.8 | |
| Aga-EDA-GLU | 1560 | 74.3 | |
| Octyl-Sepharose | 76 | 3.6 | |
| LipC12 b | Free form | 13 | |
| Aga-C8-GLU | 420 | 32.3 | |
| Aga-IDA-Ni2+-EDA-GLU | 23 | 1.8 | |
| Aga-EDA-GLU | 76 | 5.8 | |
| KlBgal c | Free form | 33 | |
| Aga-C8-GLU | 17 | 0.52 | |
| Aga-IDA-Co2+-EDA-GLU | 160 | 4.8 | |
| Aga-EDA-GLU | 12 | 0.36 | |
| EaBglA c | Free form | 21 | |
| Aga-C8-GLU | - | - | |
| Aga-IDA-Co2+-EDA-GLU | 230 | 10.9 | |
| Aga-EDA-GLU | 39 | 1.8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melo, R.R.d.; Alnoch, R.C.; Vilela, A.F.L.; Souza, E.M.d.; Krieger, N.; Ruller, R.; Sato, H.H.; Mateo, C. New Heterofunctional Supports Based on Glutaraldehyde-Activation: A Tool for Enzyme Immobilization at Neutral pH. Molecules 2017, 22, 1088. https://doi.org/10.3390/molecules22071088
Melo RRd, Alnoch RC, Vilela AFL, Souza EMd, Krieger N, Ruller R, Sato HH, Mateo C. New Heterofunctional Supports Based on Glutaraldehyde-Activation: A Tool for Enzyme Immobilization at Neutral pH. Molecules. 2017; 22(7):1088. https://doi.org/10.3390/molecules22071088
Chicago/Turabian StyleMelo, Ricardo Rodrigues de, Robson Carlos Alnoch, Adriana Ferreira Lopes Vilela, Emanuel Maltempi de Souza, Nadia Krieger, Roberto Ruller, Hélia Harumi Sato, and Cesar Mateo. 2017. "New Heterofunctional Supports Based on Glutaraldehyde-Activation: A Tool for Enzyme Immobilization at Neutral pH" Molecules 22, no. 7: 1088. https://doi.org/10.3390/molecules22071088