Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein?


 , , , ,
, , , , 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
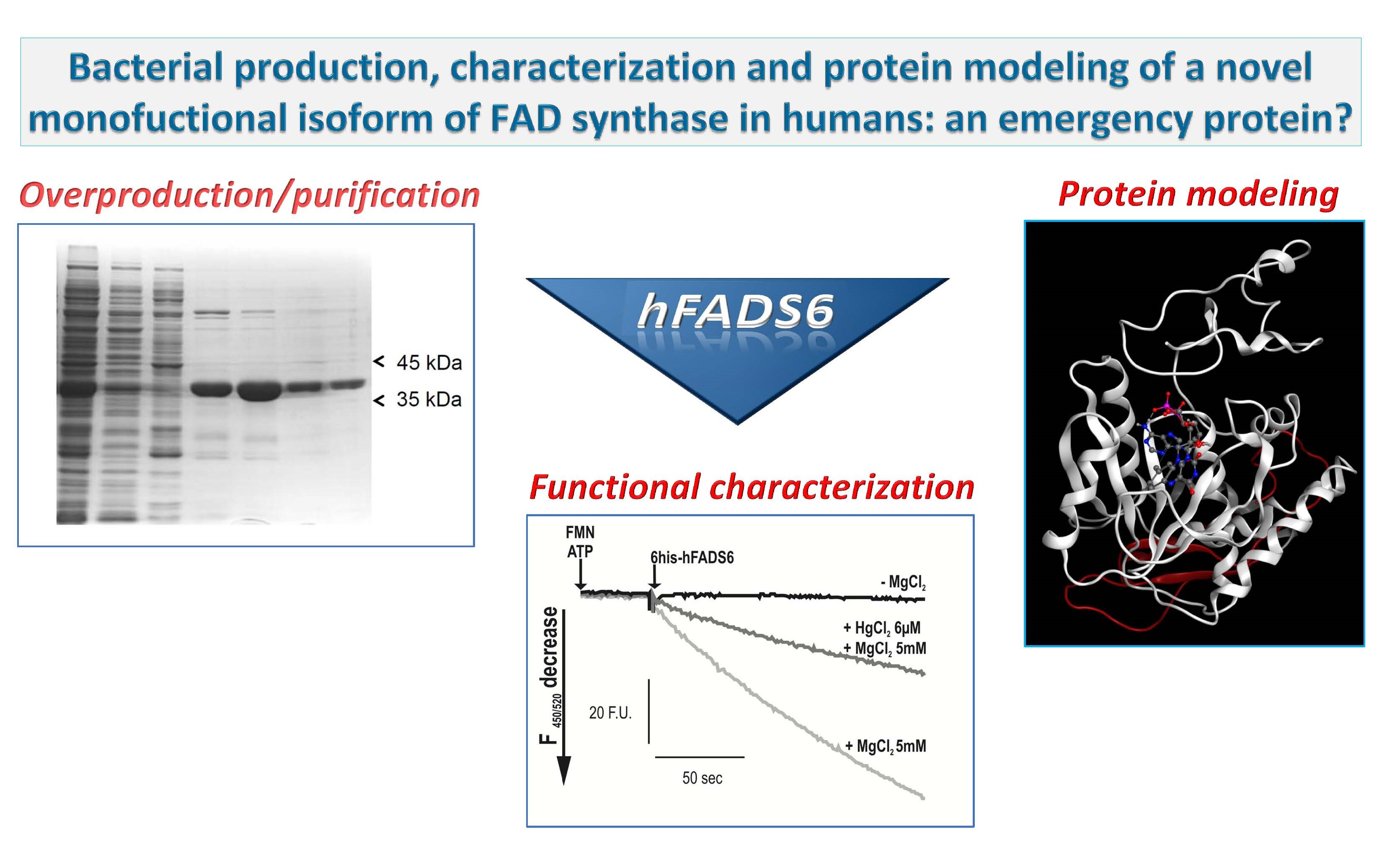
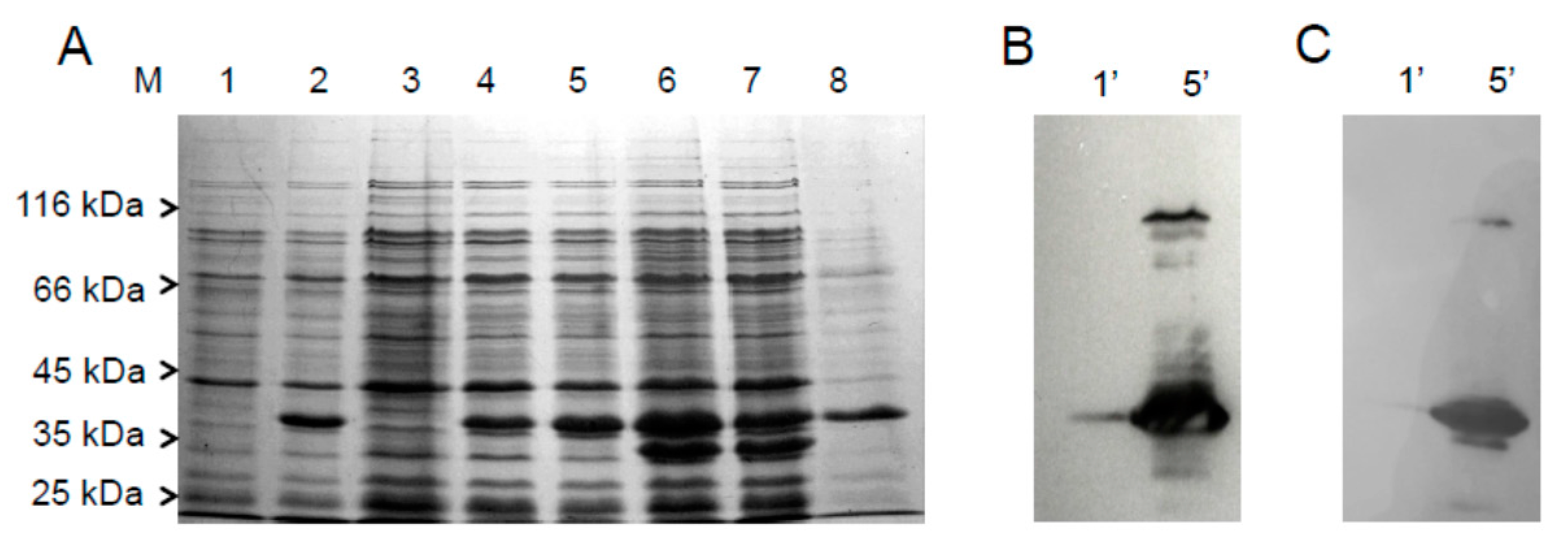
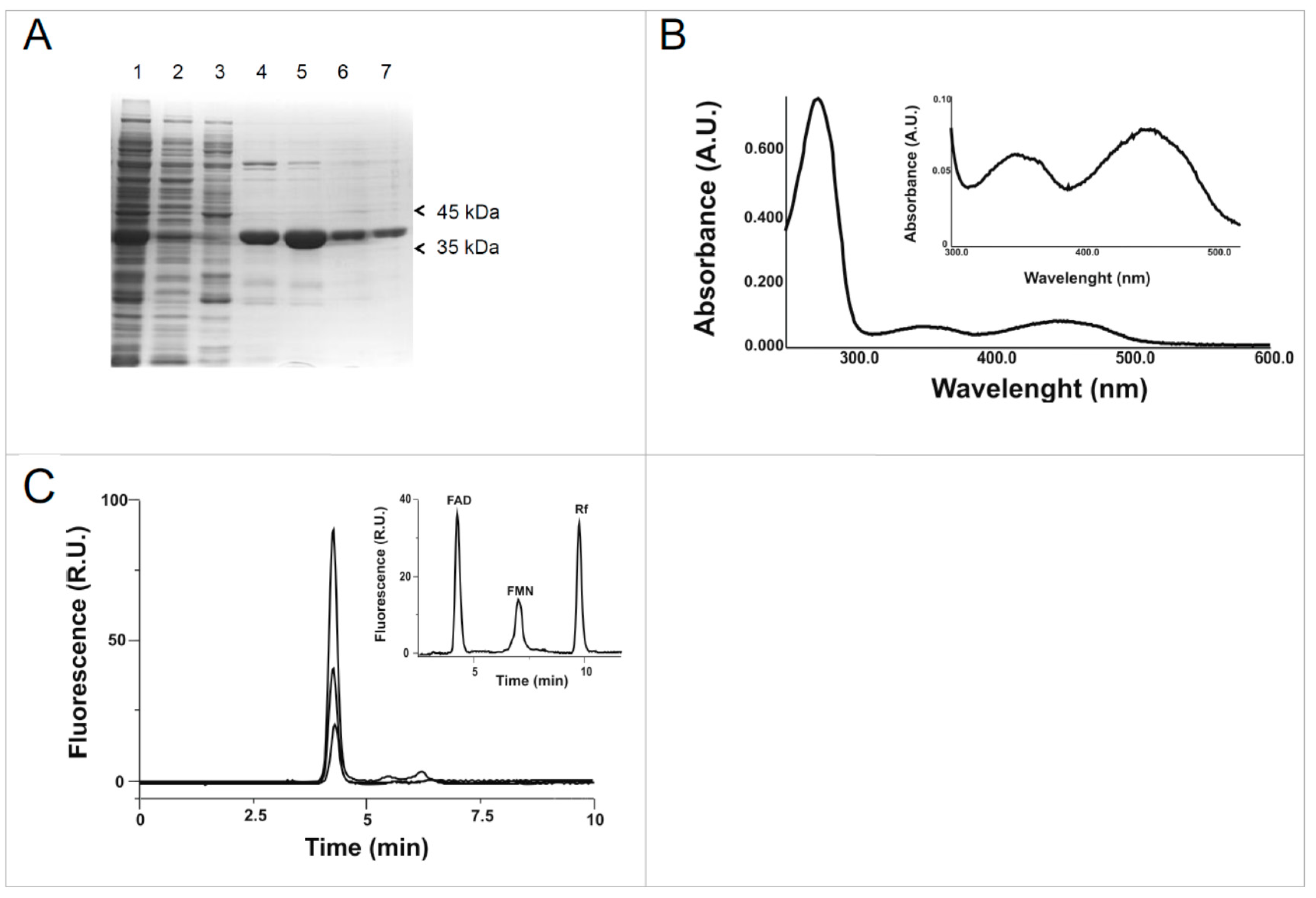
2.1. Cloning, Expression and Purification of the hFADS6 Isoform
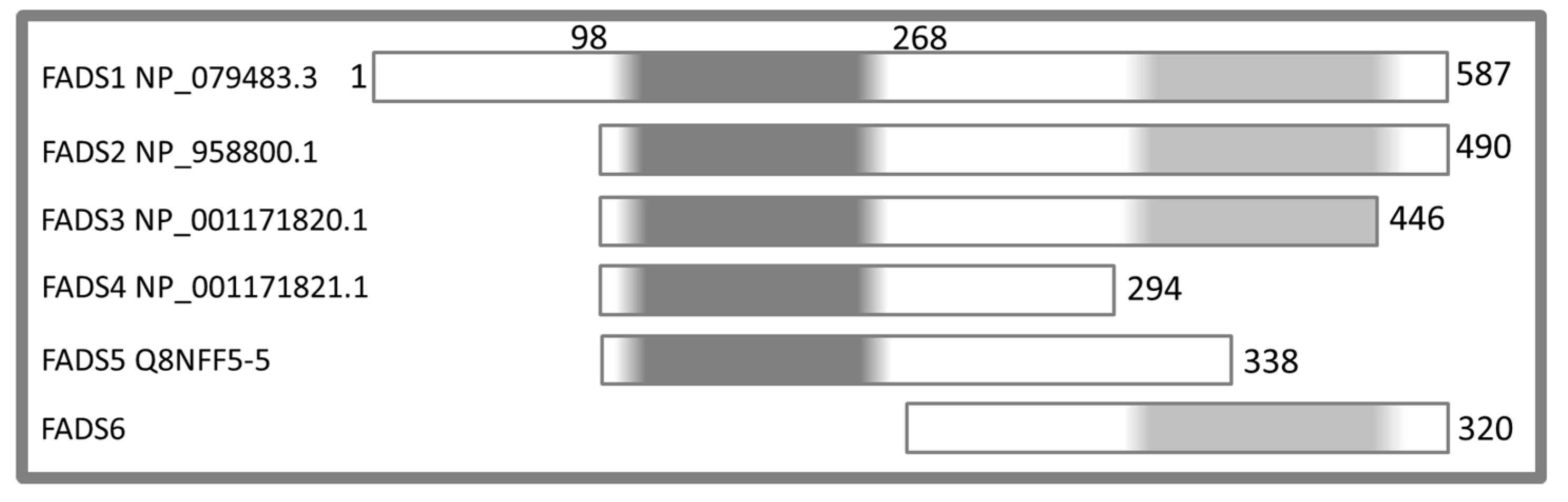
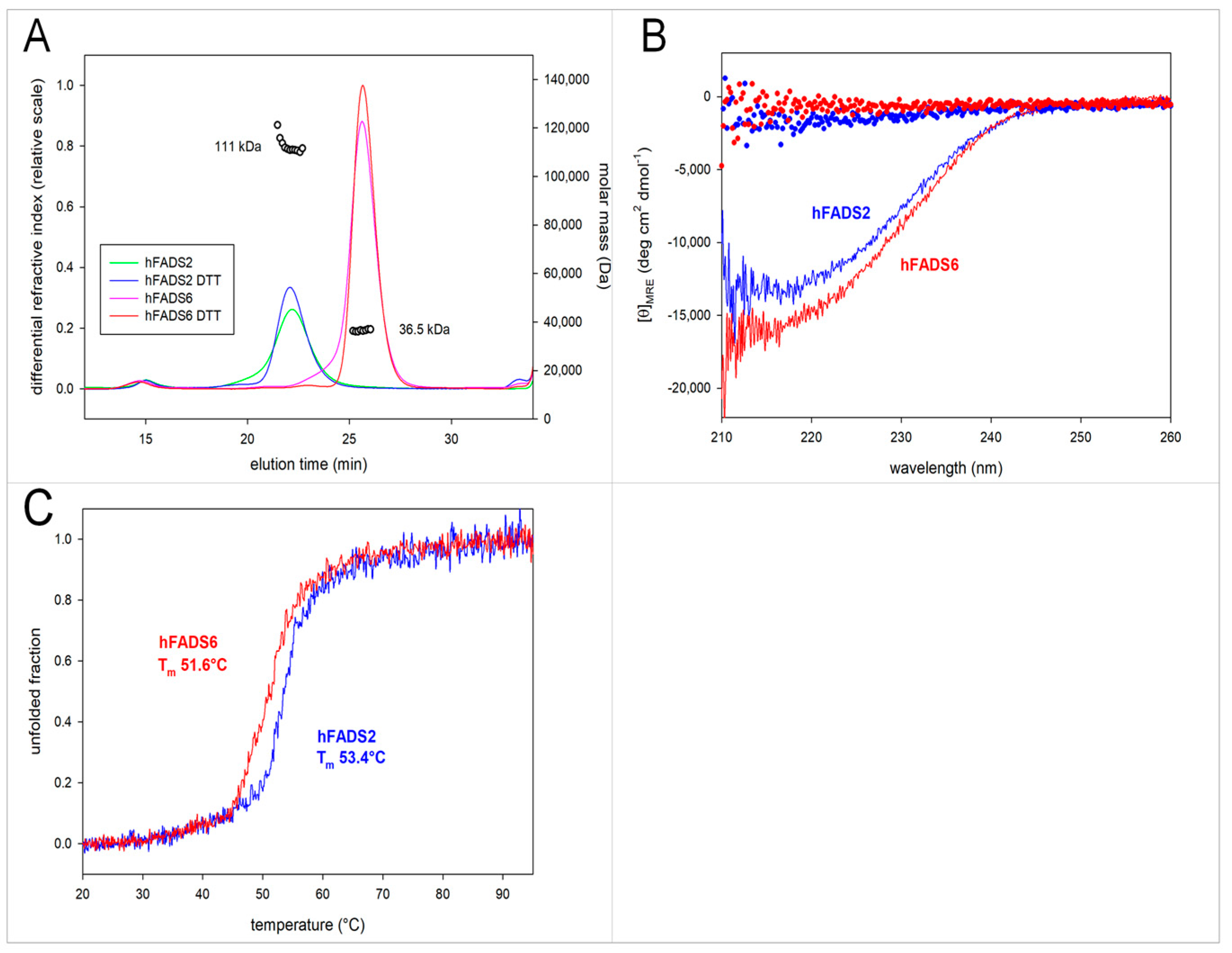
2.2. Molecular Characterization of hFADS6
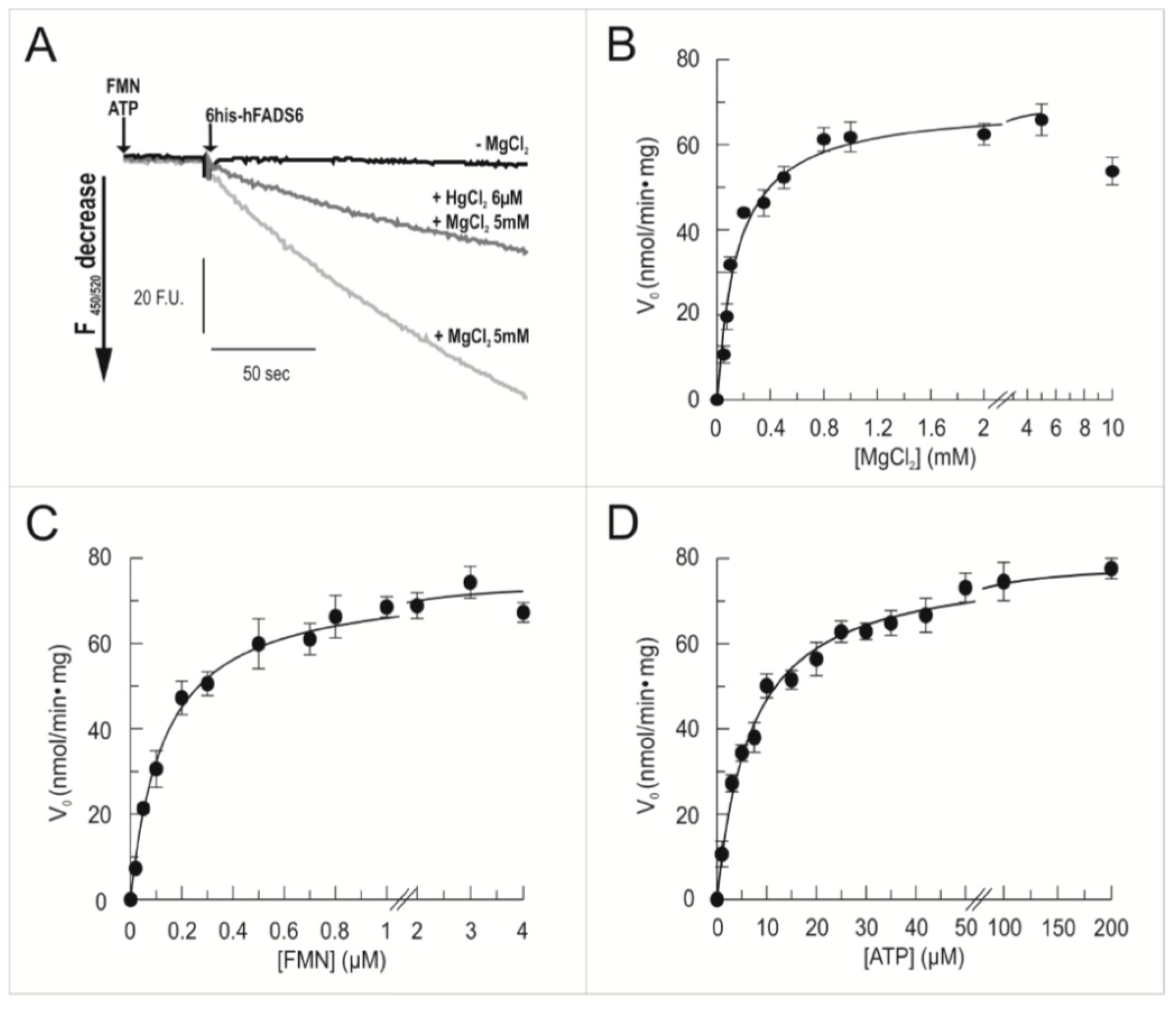
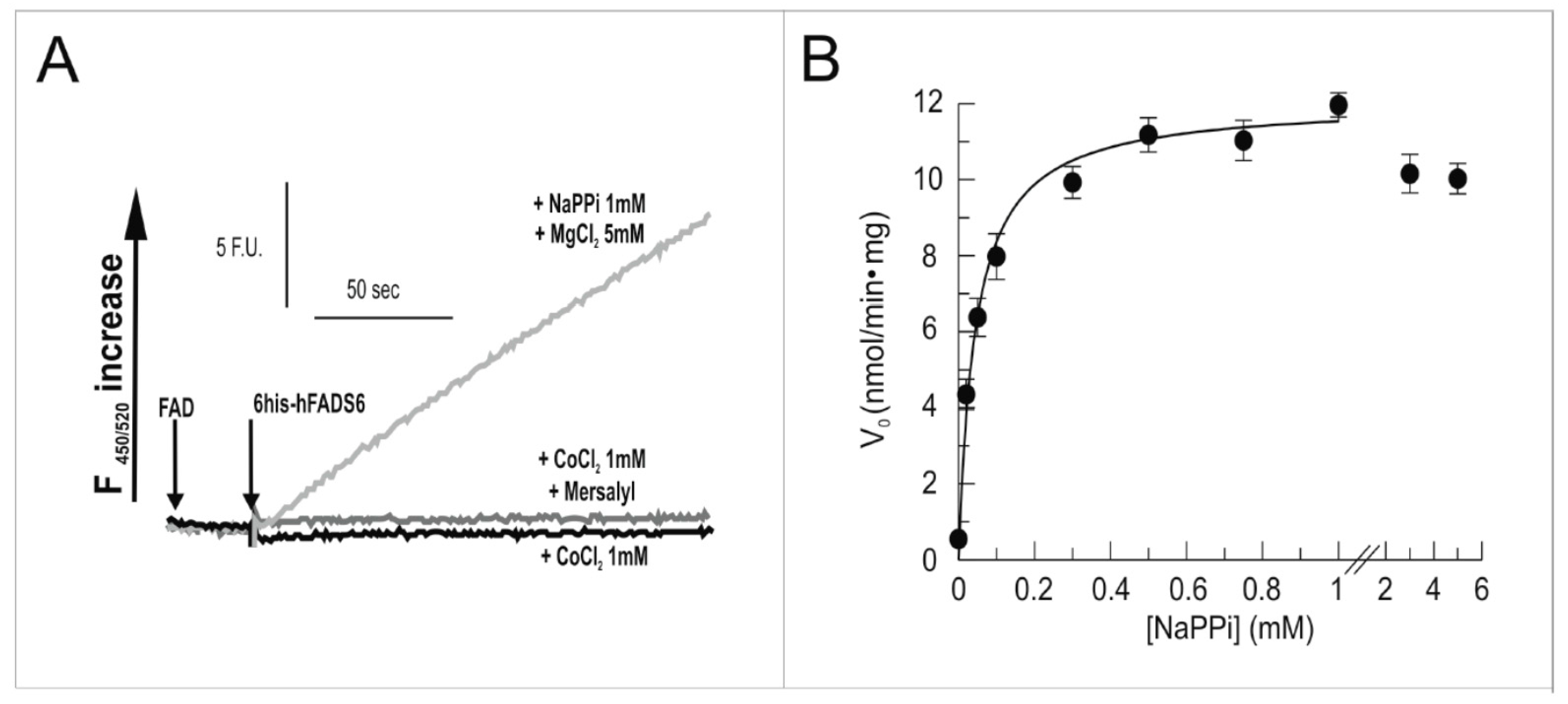
2.3. Functional Characterization of hFADS6 Recombinant Protein
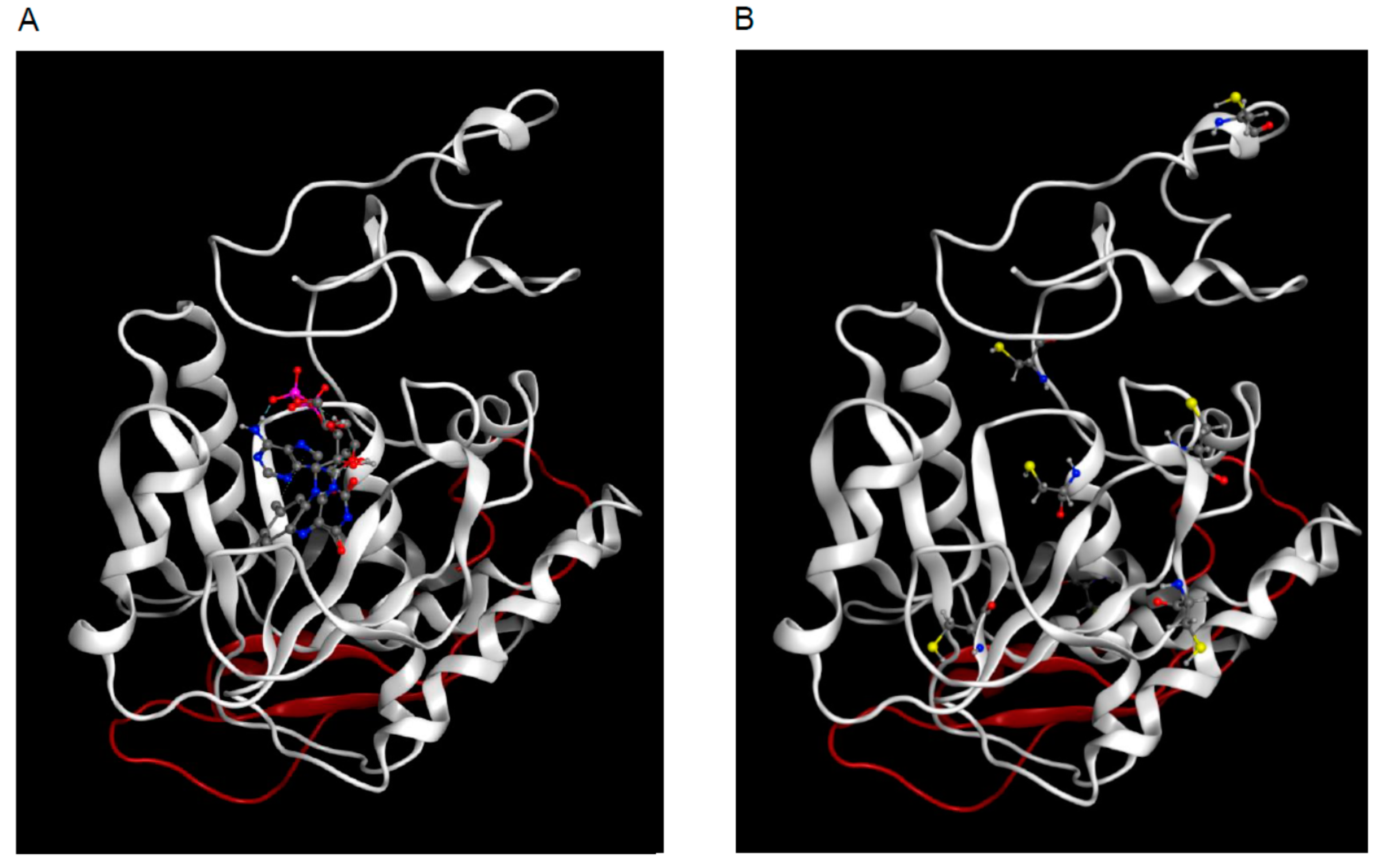
2.4. 3D Model of hFADS6
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cloning of cDNA Coding for hFADS6
4.3. Expression of Recombinant hFADS6 Protein in E. coli
4.4. Purification of Recombinant 6-His-hFADS6
4.5. Protein Concentration and FAD/Protein Monomer Ratio Measurements
4.6. FAD Content as Assayed by HPLC Analysis
4.7. Circular Dichroism Measurements
4.8. Molecular Parameters
4.9. Measurements of FAD Synthesis and FAD Pyrophosphorolysis Rate
4.10. Electrophoretic Analysis
4.11. Immunoblotting
4.12. 3D Modeling of hFADS6
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barile, M.; Giancaspero, T.A.; Brizio, C.; Panebianco, C.; Indiveri, C.; Galluccio, M.; Vergani, L.; Eberini, I.; Gianazza, E. Biosynthesis of flavin cofactors in man: Implications in health and disease. Curr. Pharm. Des. 2013, 19, 2649–2675. [Google Scholar] [CrossRef] [PubMed]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol. 2007, 11, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Gudipati, V.; Macheroux, P. The human flavoproteome. Arch. Biochem. Biophys. 2013, 535, 150–162. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.B. A trail of research on cofactors: An odyssey with friends. J. Nutr. 2000, 130 (Suppl. 2S), 323S–330S. [Google Scholar] [PubMed]
- Heikal, A.A. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark. Med. 2010, 4, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, S.; Yuan, H.; Wang, Y.F.; Orville, A.M.; Weber, I.T.; Gadda, G. Structural and kinetic studies on the Ser101Ala variant of choline oxidase: Catalysis by compromise. Arch. Biochem. Biophys. 2010, 501, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, N.; Corydon, T.J.; Gregersen, N.; Olsen, R.K. Cellular consequences of oxidative stress in riboflavin responsive multiple acyl-CoA dehydrogenation deficiency patient fibroblasts. Hum. Mol. Genet. 2014, 23, 4285–4301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Frerman, F.E.; Kim, J.J. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. USA 2006, 103, 16212–16217. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Tan, Y.; Wang, L.; Wang, Z.; Wang, B.; Wen, X.; Yang, G.; Xi, Z.; Shen, Y. Structural insight into human variegate porphyria disease. FASEB J. 2011, 25, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Inui, K. Novel riboflavin transporter family RFVT/SLC52: Identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol. Asp. Med. 2013, 34, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O’Brien, P.J. Mitochondrial function and toxicity: Role of B vitamins on the one-carbon transfer pathways. Chem. Biol. Interact. 2006, 163, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Gherasim, C.G.; Zaman, U.; Raza, A.; Banerjee, R. Impeded electron transfer from a pathogenic FMN domain mutant of methionine synthase reductase and its responsiveness to flavin supplementation. Biochemistry 2008, 47, 12515–12522. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, K.R.; Scrutton, N.S. Protein interactions in the human methionine synthase-methionine synthase reductase complex and implications for the mechanism of enzyme reactivation. Biochemistry 2007, 46, 6696–6709. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, S.; Pedrolli, D.B.; Stolz, J.; Vogl, C.; Lehmann, M.; Mack, M. RibM from Streptomyces davawensis is a riboflavin/roseoflavin transporter and may be useful for the optimization of riboflavin production strains. BMC Biotechnol. 2011, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Angulo, V.A. Overlapping riboflavin supply pathways in bacteria. Crit. Rev. Microbiol. 2017, 43, 196–209. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M. Recent advances in transport of water-soluble vitamins in organs of the digestive system: A focus on the colon and the pancreas. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G601–G610. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.J. Brown-Vialetto-Van Laere and Fazio Londe syndromes: Defects of riboflavin transport with biochemical similarities to multiple acyl-CoA dehydrogenation defects (MADD). J. Inherit. Metab. Dis. 2012, 35, 941–942. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Makowski, C.; Yao, Y.; Graf, E.; Hempel, M.; Wieland, T.; Tauer, U.; Ahting, U.; Mayr, J.A.; Freisinger, P.; et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012, 35, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Abeling, N.G.; Ijlst, L.; Knoester, H.; van der Pol, W.L.; Stroomer, A.E.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2011, 34, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Brizio, C.; Galluccio, M.; Wait, R.; Torchetti, E.M.; Bafunno, V.; Accardi, R.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem. Biophys. Res. Commun. 2006, 344, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Galluccio, M.; Brizio, C.; Torchetti, E.M.; Ferranti, P.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli, purification and characterization of isoform 2 of human FAD synthetase. Protein Expr. Purif. 2007, 52, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Sebastian, M.; Arilla-Luna, S.; Baquedano, S.; Herguedas, B.; Velazquez-Campoy, A.; Martinez-Julvez, M.; Medina, M. The trimer interface in the quaternary structure of the bifunctional prokaryotic FAD synthetase from Corynebacterium ammoniagenes. Sci. Rep. 2017, 7, 404. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Sebastian, M.; Arilla-Luna, S.; Baquedano, S.; Pallares, M.C.; Lostao, A.; Herguedas, B.; Velazquez-Campoy, A.; Martinez-Julvez, M.; Medina, M. Quaternary organization in a bifunctional prokaryotic FAD synthetase: Involvement of an arginine at its adenylyltransferase module on the riboflavin kinase activity. Biochim. Biophys. Acta 2015, 1854, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Torchetti, E.M.; Bonomi, F.; Galluccio, M.; Gianazza, E.; Giancaspero, T.A.; Iametti, S.; Indiveri, C.; Barile, M. Human FAD synthase (isoform 2): A component of the machinery that delivers FAD to apo-flavoproteins. FEBS J. 2011, 278, 4434–4449. [Google Scholar] [CrossRef] [PubMed]
- Miccolis, A.; Galluccio, M.; Nitride, C.; Giancaspero, T.A.; Ferranti, P.; Iametti, S.; Indiveri, C.; Bonomi, F.; Barile, M. Significance of redox-active cysteines in human FAD synthase isoform 2. Biochim. Biophys. Acta 2014, 1844, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Torchetti, E.M.; Brizio, C.; Colella, M.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Roberti, M.; Barile, M. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion 2010, 10, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Galluccio, M.; Miccolis, A.; Leone, P.; Eberini, I.; Iametti, S.; Indiveri, C.; Barile, M. Human FAD synthase is a bi-functional enzyme with a FAD hydrolase activity in the molybdopterin binding domain. Biochem. Biophys. Res. Commun. 2015, 465, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Miccolis, A.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Barile, M. Bacterial over-expression and purification of the 3′phosphoadenosine 5′phosphosulfate (PAPS) reductase domain of human FAD synthase: Functional characterization and homology modeling. Int. J. Mol. Sci. 2012, 13, 16880–16898. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Konarikova, E.; Giancaspero, T.A.; Mosegaard, S.; Boczonadi, V.; Matakovic, L.; Veauville-Merllie, A.; Terrile, C.; Schwarzmayr, T.; Haack, T.B.; et al. Riboflavin-Responsive and -Non-responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2016, 98, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Rhead, W.; Roettger, V.; Marshall, T.; Amendt, B. Multiple acyl-coenzyme A dehydrogenation disorder responsive to riboflavin: Substrate oxidation, flavin metabolism, and flavoenzyme activities in fibroblasts. Pediatr. Res. 1993, 33, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Huerta, C.; Borek, D.; Machius, M.; Grishin, N.V.; Zhang, H. Structure and mechanism of a eukaryotic FMN adenylyltransferase. J. Mol. Biol. 2009, 389, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Locato, V.; Barile, M. A regulatory role of NAD redox status on flavin cofactor homeostasis in S. cerevisiae mitochondria. Oxid. Med. Cell. Longev. 2013, 2013, 612784. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Barile, M.; Brizio, C.; Valenti, D.; De Virgilio, C.; Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 2000, 267, 4888–4900. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Yang, J.T. A new approach to the calculation of secondary structures of globular proteins by optical rotatory dispersion and circular dichroism. Biochem. Biophys. Res. Commun. 1971, 44, 1285–1291. [Google Scholar] [CrossRef]
- Barile, M.; Brizio, C.; De Virgilio, C.; Delfine, S.; Quagliariello, E.; Passarella, S. Flavin adenine dinucleotide and flavin mononucleotide metabolism in rat liver—The occurrence of FAD pyrophosphatase and FMN phosphohydrolase in isolated mitochondria. Eur. J. Biochem. 1997, 249, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Brizio, C.; Brandsch, R.; Bufano, D.; Pochini, L.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli, functional characterization and refolding of rat dimethylglycine dehydrogenase. Protein Expr. Purif. 2004, 37, 434–442. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leone, P.; Galluccio, M.; Barbiroli, A.; Eberini, I.; Tolomeo, M.; Vrenna, F.; Gianazza, E.; Iametti, S.; Bonomi, F.; Indiveri, C.; et al. Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein? Molecules 2018, 23, 116. https://doi.org/10.3390/molecules23010116
Leone P, Galluccio M, Barbiroli A, Eberini I, Tolomeo M, Vrenna F, Gianazza E, Iametti S, Bonomi F, Indiveri C, et al. Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein? Molecules. 2018; 23(1):116. https://doi.org/10.3390/molecules23010116
Chicago/Turabian StyleLeone, Piero, Michele Galluccio, Alberto Barbiroli, Ivano Eberini, Maria Tolomeo, Flavia Vrenna, Elisabetta Gianazza, Stefania Iametti, Francesco Bonomi, Cesare Indiveri, and et al. 2018. "Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein?" Molecules 23, no. 1: 116. https://doi.org/10.3390/molecules23010116
APA StyleLeone, P., Galluccio, M., Barbiroli, A., Eberini, I., Tolomeo, M., Vrenna, F., Gianazza, E., Iametti, S., Bonomi, F., Indiveri, C., & Barile, M. (2018). Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein? Molecules, 23(1), 116. https://doi.org/10.3390/molecules23010116