Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus
by
Xiao-Jun Li
1,2,†, 
Qin-Peng Zou
3,†,
Xiang Wang
1,
Kwan-Woo Kim
2,
Mao-Fang Lu
1,
Sung-Kwon Ko
4,
Chang-Soo Yook
5,
Youn-Chul Kim
2,* and
Xiang-Qian Liu
1,* 1
School of Pharmacy, Hunan University of Chinese Medicine, Changsha 410208, Hunan, China
2
College of Pharmacy, Wonkwang University, Iksan 570-749, Korea
3
Broad-Ocean Bio-Science and Technique Co., Ltd. of Changsha, Changsha 410205, Hunan, China
4
Department of Oriental Medical Food & Nutrition, Semyung University, Jecheon 27136, Korea
5
School of Pharmacy, KyungHee University, Seoul 130-701, Korea
*
Authors to whom correspondence should be addressed.
†
These authors contribute equally to this work.
Molecules 2018, 23(1), 87; https://doi.org/10.3390/molecules23010087
Submission received: 1 December 2017
/
Revised: 27 December 2017
/
Accepted: 29 December 2017
/
Published: 1 January 2018
(This article belongs to the Section Natural Products Chemistry)
Abstract
:The phytochemical study on the leaves of Acanthopanax gracilistylus (Araliaceae) resulted in the discovery of a new lupane-triterpene compound, acangraciligenin S (1), and a new lupane-triterpene glycoside, acangraciliside S (2), as well as two known ones, 3α,11α-dihydroxy-lup-20(29)-en-23,28-dioic acid (3) and acankoreoside C (4). Their chemical structures were elucidated by mass, 1D- and 2D-nuclear magnetic resonance (NMR) spectroscopy. The chemical structures of the new compounds 1 and 2 were determined to be 1β,3α-dihydroxy-lup-20(29)-en-23, 28-dioic acid and 1β,3α-dihydroxy-lup-20(29)-en-23,28-dioic acid 28-O-[α-l-rhamnopyranosyl-(1→4)-β-d-glucopyranosyl-(1→6)-β-d-glucopyranosyl] ester, respectively. The anti-neuroinflammatory activity of the selective compounds, 1 and 3, were evaluated with lipopolysaccharide (LPS)-induced BV2 microglia. The tested compounds showed moderate inhibitory effect of nitric oxide (NO) production.
1. Introduction
Acanthopanax gracilistylus W. W. Smith belongs to Araliaceae, which is widely distributed in China, and its dried roots and stem barks are listed officially in the Chinese Pharmacopoeia (2015 edition) as Acanthopanax Cortex (named as Wujiapi), which has been used as medicine for the treatment of paralysis, arthritis, rheumatism, lameness, and liver disease [1,2,3]. Previous phytochemical investigations of this plant have led to the identification of triterpenoids [4,5,6,7], diterpenoids [8,9], monoterpenoids [10,11,12], lignans [12], steroids, cerebrosides [13], and volatile components [10,14,15,16,17,18,19], which showed diverse biological activities, such as anti-tumor [20,21], anti-inflammatory [22,23,24], and liver protective effects [3]. As part of our ongoing research to identify bioactive substances from the genus Acanthopanax, we investigated a MeOH extract of the leaves of A. gracilistylus and identified two new lupane triterpenoids (1 and 2), and two known compounds (3 and 4) by using high-speed countercurrent chromatography (HSCCC) in conjunction with a high-performance liquid chromatography (HPLC) isolation system (Figure 1). In this paper, the isolation and structural elucidation of the new isolates, as well as an evaluation of their bio-activities, are reported.
2. Results and Discussion
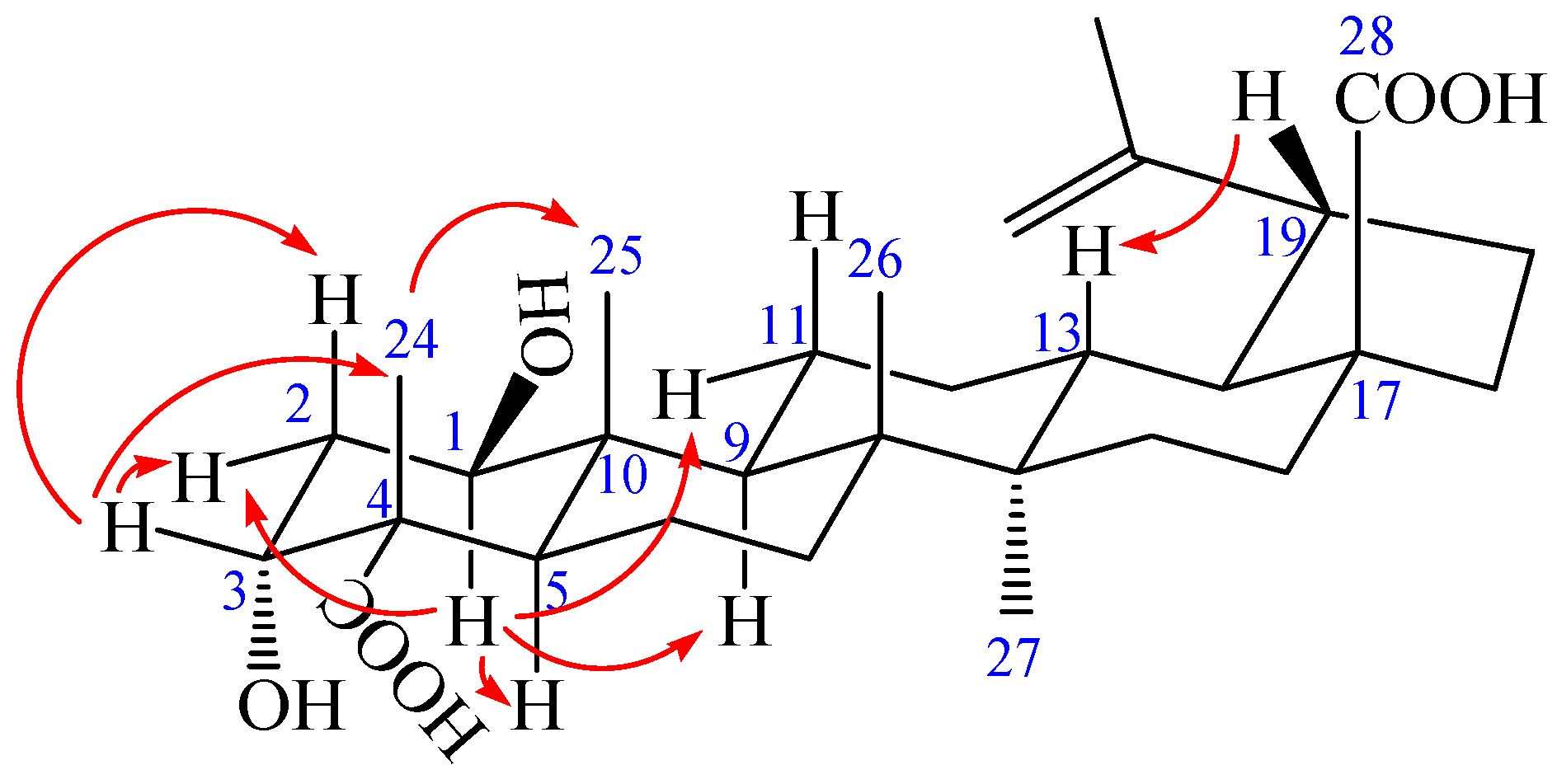
Compound 1 was obtained as a colorless needle, m.p. 282.6 °C, and gave a positive reaction in the Liebermann–Burchard test. Its molecular formula of C30H46O6 was determined on the basis of ESI-MS at m/z = 501.3 [M − H]− (negative) and HR-ESI-MS at m/z = 501.3221 [M − H]− (calcd. for C30H45O6: 501.3222), indicating eight degrees of unsaturation. The 1H-NMR spectrum of 1 (in acetone-d6) showed the following signals: five tertiary methyl groups at δ 0.97, 0.99, 1.07, 1.17 and 1.71 (each 3H, s); two olefinic protons at δ 4.58 (1H, m) and 4.72 (1H, d, J = 1.68 Hz); two hydroxy-substituted methines at δ 3.82 (1H, t, J = 2.40 Hz) and 3.84 (1H, dd, J = 8.16, 3.96 Hz). The 13C-NMR and distortionless enhancement by polarization transfer (DEPT) spectra revealed 30 carbon signals. The skeleton of 1 was recognized to be lupane triterpenoid by 1H- and 13C-NMR analysis (see Table 1), with the typical olefinic carbons at δ 151.89 (C-20) and 110.26 (C-29), five quaternary methyl carbons at δ 13.24, 15.40, 17.31, 17.92, and 19.72, two oxymethine carbons at δ 73.99 and 75.46, and two carboxyl signals at δ 177.84 and 178.16, respectively. The assignment of the α-hydroxyl group at C-3 was performed by comparing its spectral data with literature values [25,26,27,28]. The chemical shifts of C-3 (δ 73.99) and C-4 (δ 52.27) further confirmed the axial configuration of the 3-hydroxyl group by comparing with the corresponding data of the 3β-hydroxy-lup-20(29)-en-23,28-dioic acid [δ values for 84.4 (C-3) and 43.0 (C-4)] [29]. Additionally, the carbon chemical shifts of C-23 (δ 178.16) and C-28 (δ 177.84) were also similar to those of 3β-hydroxy-lup-20(29)-en-23,28-dioic acid [δ values for 178.7 (C-23) and 177.3 (C-28)] suggesting that the two carboxyl groups were at C-23 and C-28 [29]. In heteronuclear multiple bond connectivity (HMBC) spectrum, the H-1 proton signal at δ 3.84 (1H, dd, J = 8.16, 3.96 Hz) correlated with carbons C-2 (δ 37.36), C-9 (δ 52.98), C-10 (δ 44.33), and C-25 (δ 13.24); protons H-2 at δ 1.82 (1H, m) and H-25 at δ 0.97 (3H, s) correlated with carbon C-1 (δ 75.46); the H-3 proton signal at δ 3.82 (1H, t, J = 2.40 Hz) correlated with carbons C-5 (δ 45.25) and C-23 (δ 178.16); protons H-2 at δ 1.82 (1H, m) and H-24 at δ 1.17 (3H, s) correlated with carbon C-3 (δ 73.99); protons H-5 at δ 1.95 (1H, m) and H-24 at δ 1.17 (3H, s) correlated with carbon C-23 (δ 178.16); protons H-16 at δ 1.48 (1H, m), H-18 at δ 1.64 (1H, m), and H-22 at δ 1.92 (1H, m) correlated with carbon C-28 (δ 177.84); the correlations from H-29 at δ 4.58 (1H, m, Ha) and δ 4.72 (1H, d, J = 1.68 Hz, Hb) to C-19 (δ 48.24), C-20 (δ 151.89), and C-30 (δ 19.72); the correlations from H-30 at δ 1.71 (3H, s) to C-19 (δ 48.24), C-20 (δ 151.89), and C-29 (δ 110.26). These pieces of evidence confirmed that the double bond was at C-20/C-29 and the other hydroxyl group was at C-1 (Figure 2 and Table S1). The nuclear Overhauser enhancement spectroscopy (NOESY) spectrum of 1 showed the correlations between the proton of H-1 and H-2eq, H-1 and H-5, H-1, and H-9, H-1 and H-11eq, suggesting that H-1 was axial in orientation, which, in turn, suggested that the hydroxyl group at C-1 was β-positioned. Similarly, cross-peaks between H-24 and H-25, as well as H-3, H-3 and H-2eq as well as H-2ax, which indicated that the methyl group (H-24) was axial and H-3 was equatorial, which, in turn, suggested that the hydroxyl group at C-3 and carboxyl group at C-4 were α-positioned. Furthermore, cross-peaks between H-19 and H-13 confirmed that the isopropenyl group at C-19 was α-positioned (Figure 3). Based on the above spectral data, the structure of 1 was determined as 1β,3α-dihydroxy-lup-20(29)-en-23,28-dioic acid, a new compound named acangraciligenin S.
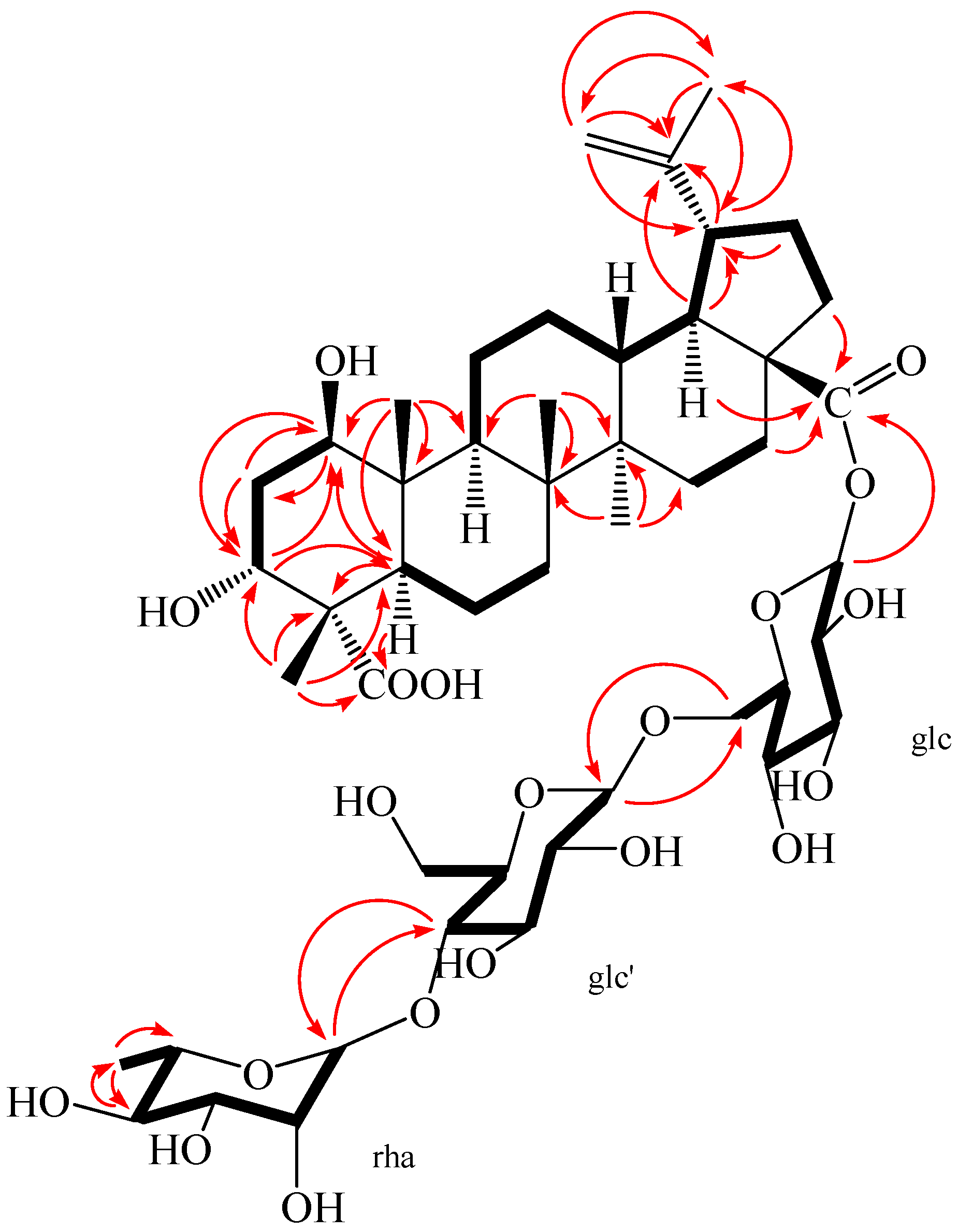
Compound 2 was obtained as a white amorphous powder, m.p. 230.5 °C, and gave positive responses in Liebermann–Burchard and Molish tests. Its molecular formula of C48H76O20 was determined on the basis of ESI-MS at m/z = 972.5 [M]+ and HR-ESI-MS at m/z = 971.4865 [M − H]− (negative) (calcd. for C48H75O20: 971.4857), indicating eleven degrees of unsaturation. The 1H-NMR spectrum of 2 (in methanol-d4) showed the following signals: five tertiary methyl groups at δ 0.95, 0.98, 1.03, 1.09 and 1.70 (each 3H, s); one secondary methyl group at δ 1.25 (3H, d, J = 4.96 Hz), assigned to H-6″ of the rhamnose; and two olefinic protons at δ 4.58 (1H, brs) and 4.72 (1H, brs); two hydroxy-substituted methines at δ 3.67 (1H, m) and 3.80 (1H, m). In addition, three anomeric protons were at δ 4.37 (1H, d, J = 6.28 Hz), 4.84 (1H, overlapped), and 5.45 (1H, d, J = 6.56 Hz), suggesting the appearance of three sugar units. The 13C-NMR and DEPT spectra revealed 48 carbon signals, of which, 30 signals were assigned to a triterpenoid sapogenol moiety and 18 signals belong to three monosaccharide moieties. The aglycone of 2 was recognized to be lupane triterpene type by 1H- and 13C-NMR analysis (see Table 1), with the typical olefinic carbons at δ 151.77 (C-20) and 110.41 (C-29), five quaternary methyl carbons at δ 13.17, 15.10, 17.14, 18.08, and 19.49, two oxymethine carbons at δ 74.14 and 76.33, and two carboxyl signals at δ 176.40 and 182.60, respectively. Compared with the 1H- and 13C-NMR data of the aglycone of 2 and compound 1, the high similarity indicated that the aglycone of 2 was the same as that of compound 1. Assignments of the β-hydroxyl group at C-1 (δ 76.33), α-hydroxyl group at C-3 (δ 74.14), and two carboxyl groups at C-23 (δ 182.60) and C-28 (δ 176.40) were performed by comparing its spectral data with compound 1 [δ values for 75.46 (C-1), 73.99 (C-3), 178.16 (C-23) and 177.84 (C-28)] and literature values [28]. In HMBC spectrum, the H-1 proton signal at δ 3.80 (1H, m) correlated with carbons C-2 (δ 36.79), C-3 (δ 74.14), C-9 (δ 53.06), C-10 (δ 44.47), and C-25 (δ 13.17); protons H-2 at δ 1.72 (1H, m), H-3 at δ 3.67 (1H, m), H-5 at δ 1.87 (1H, m), and H-25 at δ 0.95 (3H, s) correlated with carbon C-1 (δ 76.33); the H-3 proton signal at δ 3.67 (1H, m) correlated with carbons C-1 (δ 76.33) and C-5 (δ 46.11); protons H-2 at δ 1.80 (1H, m) and H-24 at δ 1.09 (3H, s) correlated with carbon C-3 (δ 74.14); protons H-5 at δ 1.87 (1H, m) and H-24 at δ 1.09 (3H, s) correlated with carbon C-23 (δ 182.60); protons H-16 at δ 1.44 (1H, m), H-18 at δ 1.65 (1H, m), and H-22 at δ 1.94 (1H, m) correlated with carbon C-28 (δ 176.40); the correlations from H-29 at δ 4.58 (1H, brs, Ha) and δ 4.72 (1H, brs, Hb) to C-19 (δ 48.36), C-20 (δ 151.77), and C-30 (δ 19.49); the correlations from H-30 at δ 1.70 (3H, s) to C-19 (δ 48.36), C-20 (δ 151.77), and C-29 (δ 110.41). These pieces of evidences further confirmed that the double bond was at C-20/C-29 and the two hydroxyl groups were at C-1 and C-3, respectively (Figure 4 and Table S2).
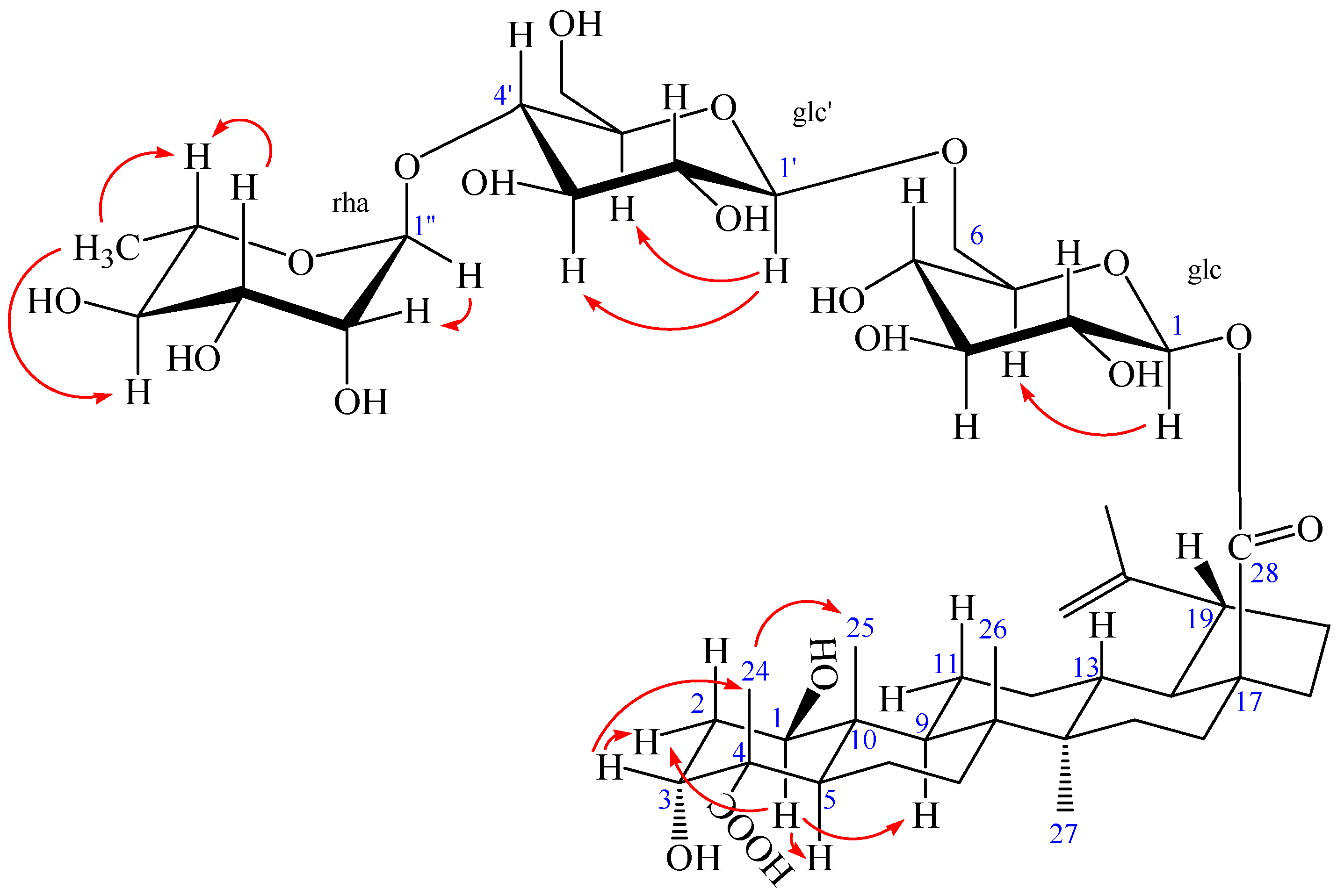
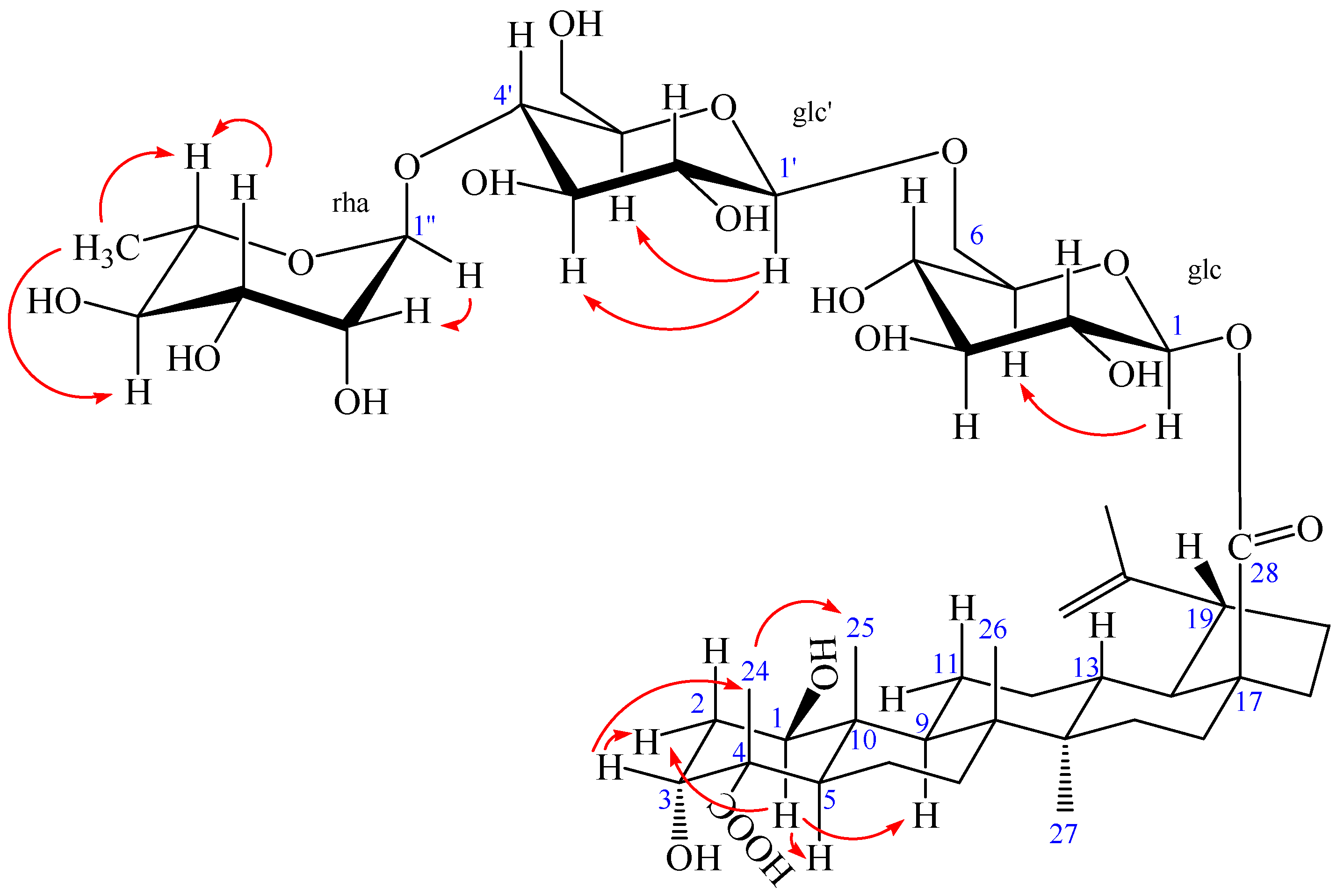
The NOESY spectrum of 2 showed the correlations between the proton of H-1 and H-2eq, H-1 and H-5, H-1, and H-9, suggesting that H-1 had an axial orientation, which, in turn, suggested that the hydroxyl group at C-1 was β-positioned. Similarly, cross-peaks between H-24 and H-25, as well as H-3, H-3 and H-2eq, indicated that the methyl group (H-24) was axial and H-3 was equatorial, which, in turn, suggested that the hydroxyl group at C-3 and carboxyl group at C-4 were α-positioned (Figure 5). Alkaline hydrolysis of 2 with 5% KOH in MeOH gave a sapogenol (2a), a colorless needle, mp 282.5 °C, together with a mixture of sugars. The sugar mixture was identified to be composed of D-glucose and L-rhamnose by thin-layer chromatography (TLC) with their authentic sample. The 1H and 13C NMR data of 2a (see Experimental Section) were the same as those of 1, which further confirmed that this aglycone (2a) was 1β,3α-dihydroxy-lup-20(29)-en-23,28-dioic acid. Moreover, the HMBC correlations between the inner glc H-1 (δ 5.45) and C-28 of the aglycone (δ 176.40), between outer glc H-1′ (δ 4.37) and inner glc C-6 (δ 69.55), between rha H-1″ (δ 4.84) and glc C-4′ (δ 79.51) were observed (Figure 4). In addition, the NOESY correlations between the inner glc H-1 and glc H-5, between outer glc H-1′ and glc H-3′ as well as glc H-5′, between rha H-1″ and rha H-2″, rha H-5″ and rha H-3″, as well as rha H-6″, rha H-4″ and rha H-6″, which further confirmed the inner glucose, outer glucose, and rhamnose were β-D, β-D, and α-L positioned, respectively (Figure 5). These results suggest the sequence of sugar linkages of 2. The carbon signals of the sugar moiety were superimposable on those of characteristic triterpene glycosides isolated from Acanthopanax species [4,5,6,7,25,26,27,28]. Consequently, the structure of 2 was determined as 1β,3α-dihydroxy-lup-20(29)-en-23,28-dioic acid 28-O-[α-l-rhamnopyranosyl-(1→4)-β-d-glucopyranosyl-(1→6)-β-d-glucopyranosyl] ester, a new compound named acangraciliside S.
Compounds 3 and 4 were identified as 3α,11α-dihydroxy-lup-20(29)-en-23,28-dioic acid [30] and acankoreoside C [31], respectively, by comparing their NMR and mass spectral data with the literature values. (The 1H- and 13C-NMR data of 3 and 4 see Supplementary Materials: Table S3)
Moreover, the cytotoxicity and inhibition of production of nitric oxide (NO) of selected isolates (1 and 3) from A. gracilistylus were investigated on LPS-stimulated BV2 microglias. Compounds 2 and 4 were excluded from biological evaluation because of sample shortage. As shown in Table 2, the production of NO was down-regulated moderately when their concentration was 80 μM with the inhibition as 38.6% and 40.4%, respectively.
3. Experimental Section
3.1. General Experimental Procedures
Melting points (uncorrected) were measured using a Boetius micromelting point apparatus (Beijing Jingjing Science and Technology Co., Ltd., Beijing, China). 1H-NMR (400 MHz), 13C-NMR (100 MHz) and 2D-NMR were recorded at room temperature in CD3COCD3 or CD3OD using Bruker DRX-400 NMR spectrometer (Bruker Biospin, Zurich, Switzerland) and chemical shifts were presented in δ (ppm) values relative to tetramethylsilane (TMS) as internal standard. HR-ESI-MS was performed on an API QSTAR spectrometer (Agilent Technologies, Santa Clara, CA, USA). The high-speed countercurrent chromatography (HSCCC) instrument was performed in the present study using a model an TBE-300A HSCCC (Shanghai Tauto Biotech Co., Ltd., Shanghai, China). Semi-preparative HPLC was performed on an LC-20A liquid chromatograph (Shimadzu Technologies, Kyoto, Japan). Column chromatography was carried out on silica gel (200–300 mesh and 100–200 mesh, Qingdao Marine Chemical Co., Ltd., Qingdao, China), and Sephadex LH-20 (Merck, Darmstadt, Germany). RP-TLC was measured on a precoated RP-18F254s (Merck, Darmstadt, Germany) plates. TLC was conducted on self-made silica gel G (Qingdao Marine Chemical Industry, Qingdao, China) plates and spots were visualized by spraying with 10% H2SO4 in ethanol (v/v) followed by heating at 105 °C. The standard d-glucose and l-rhamnose were purchased from Beijing North Carolina Souren Biotechnology Research Institute (Beijing, China).
3.2. Collection and Identification of Biological Materials
The leaves of A. gracilistylus were collected from its natural habitat in Yuanling, Hunan Province, China, in July 2015 and identified by X.Q. Liu, the corresponding author of this work. A voucher specimen has been deposited in the Herbarium of Hunan University of Chinese Medicine, Hunan, China (no. 201507).
3.3. Extraction and Isolation
The dried, powdered leaves of A. gracilistylus (600 g) were extracted with methanol under reflux three times (3 × 3 L). The solvent was then removed under reduced pressure to yield a residue (120 g), which was suspended in distilled water and successively partitioned with petroleum ether (PE, 60–90), EtOAc, and n-BuOH, respectively. The EtOAc extract (28 g) was subjected to column chromatography (CC) on silica gel eluting with a gradient of CHCl3-MeOH (35:1 to 10:1, v/v) to obtain the discolored fraction (E1). E1 was partitioned by HSCCC using two-phase solvent system composing of n-hexane-ethyl acetate-methanol-water (1:2:1.6:1, v/v/v/v) with the flow rate of mobile phase of 2.0 mL/min, rotation rate of 800 r.p.m, to generate five fractions (F1–F5). Compounds 1 (5.0 mg) and 3 (6.0 mg) were yielded from F2 (25 mg) by using Pre-HPLC with CH3CN-H2O (40:60, v/v) for further purification. Similarly, the n-BuOH extract (25 g) was partitioned by HSCCC using two-phase solvent system composing of ethyl acetate-butanol-methanol-water (3:0.3:0.8:4, v/v/v/v) with the flow rate of mobile phase of 2.0 mL/min, rotation rate of 900 r.p.m, to obtain five fractions (F6–F10). F6 (35 mg) was further chromatographed on Pre-HPLC with CH3CN-H2O (25:75, v/v) to produce two compounds 2 (11.3 mg) and 4 (5.3 mg).
Acangraciligenin S (1) was obtained as a colorless needle: mp. 282.6 °C; 1H- and 13C-NMR data: (Table 1); (−)-HR-ESI-MS m/z 501.3221 [M − H]− (calcd. for C30H45O6: 501.3222).
Acangraciliside S (2) was obtained as a white amorphous powder: mp. 230.5 °C; 1H- and 13C-NMR data: (Table 1); (−)-HR-ESI-MS m/z 971.4865 [M − H]− (calcd. for C48H75O20: 971.4857).
3.4. Alkaline Hydrolysis of 2
Compound 2 (10 mg) was hydrolyzed with 8 mL of 5% KOH in MeOH for 2 h at 80 °C. The reaction mixture was neutralized with 2 M HCl in H2O and extracted with EtOAc. The aqueous layer was filtered, concentrated and chromatographed on TLC plate in which d-glucose and l-rhamnose were detected by comparing with standard samples [4]. The EtOAc layer was evaporated in vacuo and the residue was performed on a preparative HPLC column by using elution with CH3CN-H2O (40:60, v/v) to obtain sapogenol fraction 2a (2 mg).
Compound 2a: Colorless needles, m.p. 282.5 °C. 1H-NMR (400 MHz, CD3COCD3) δ: 3.84 (1H, dd, J = 8.16, 3.96 Hz, H-1), 3.82 (1H, t, J = 2.40 Hz, H-3), 1.95 (1H, m, H-5), 1.72 (1H, m, H-9), 2.34 (1H, td, J = 10.32, 3.0 Hz, H-13), 1.64 (1H, m, H-18), 3.05 (1H, td, J = 8.76, 4.0 Hz, H-19), 1.17 (3H, s, H-24), 0.97 (3H, s, H-25), 0.99 (3H, s, H-26), 1.08 (3H, s, H-27), 4.59 (1H, m, H-29a), 4.73 (1H, d, J = 1.68 Hz, H-29b), 1.71 (3H, s, H-30); 13C-NMR (100 MHz, CD3COCD3) δ: 75.45 (C-1), 37.35 (C-2), 73.98 (C-3), 52.26 (C-4), 45.23 (C-5), 22.08 (C-6), 35.18 (C-7), 42.75 (C-8), 52.97 (C-9), 44.32 (C-10), 24.66 (C-11), 26.85 (C-12), 39.13 (C-13), 43.71 (C-14), 30.73 (C-15), 33.14 (C-16), 56.96 (C-17), 50.29 (C-18), 48.22 (C-19), 151.88 (C-20), 31.59 (C-21), 37.82 (C-22), 178.14 (C-23), 17.90 (C-24), 13.23 (C-25), 17.29 (C-26), 15.38 (C-27), 177.82 (C-28), 110.25 (C-29), 19.70 (C-30).
3.5. MTT Assay for Cell Viability
BV2 microglias were maintained at 5 × 105 cells/mL in Dulbecco minimum essential medium (DMEM) medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin G (100 U/mL), streptomycin (100 mg/L), and L-glutamine (2 mM) and incubated at 37 °C in a humidified atmosphere containing 5% CO2. Cell viability was determined by adding 100 mg/mL of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) to 1 mL of a cell suspension (1 × 105 cells/mL in 96-well plates) and incubated for 30 min. The formazan formed was dissolved in acidic 2-propanol, and the optical density was measured at 540 nm.
3.6. Nitrite Assay
The concentration of nitric oxide (NO) in the conditioned media was determined by a method based on the Griess reaction [32]. An aliquot of each supernatant (100 mL) was mixed with the same volume of Griess reagent (0.1% (w/v) N-(1-naphathyl)-ethylenediamine and 1% (w/v) sulfanilamide in 5% (v/v) phosphoric acid) for 10 min at room temperature. The absorbance of the final product was measured spectrophotometrically at 540 nm using an enzyme-linked immunosorbent assay (ELISA) plate reader. The nitrite concentration in the samples was determined from a standard curve of sodium nitrite prepared in phenol red-free DMEM.
3.7. Statistical Analysis
All values are expressed as the mean ± S.D. Differences between mean values of normally-distributed data were assessed with one-way analysis of variance (ANOVA) (Newman Keuls t-test). Statistical analysis was performed using GraphPad Prism software, version 3.03 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was accepted at p < 0.05.
4. Conclusions
In summary, in this work, a phytochemical study on the leaves of A. gracilistylus (Araliaceae) by using HSCCC-HPLC combinatorial chromatography method resulted in the discovery of four lupane-triterpenoids, including two new compounds, acangraciligenin S (1) and acangraciliside S (2), as well as two known species, 3α,11α-dihydroxy-lup-20(29)-en-23,28-dioic acid (3) and acankoreoside C (4). Two selected compounds were also evaluated for their inhibitory activity against lipopolysaccharide (LPS)-induced nitric oxide (NO) production in murine microglia BV2 cells. Compounds 1 and 3 showed moderate abilities to inhibit NO production and had no influence on cell viability, as determined by the MTT method.
Supplementary Materials
The following 1H-NMR, 13C-NMR, 2D-NMR, ESI-MS, and HR-ESI-MS spectra are available as supporting data. Supplementary materials are available online.
Acknowledgments
This project was supported financially by the Natural Science Foundation of Hunan Province, China (grant 11JJ2042) and the Key Projects of Changsha City Science and Technology Bureau (Grant k1403122-31).
Author Contributions
Xiang-Qian Liu designed the experiments and revised the paper; Xiao-Jun Li and Qin-Peng Zou performed the experiments, analyzed the data, and wrote the paper; Xiang Wang, Mao-Fang Lu, and Sung-Kwon Ko help to analyze data of experiment; Kwan-Woo Kim and Youn-Chul Kim contributed to bioassay reagents, materials and analyzed the data; and Chang-Soo Yook revised the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia of the People’s Republic of China; Medical Science and Technology Press: Beijing, China, 2015; Volume 1, p. 79. [Google Scholar]
- Ni, N.; Liu, X.Q. Advances in studies on plants of Acanthopanax Miq. in Araliaceae. Chin. Tradit. Herb. Drugs 2006, 37, 1895–1900. [Google Scholar]
- Zhang, B.X.; Li, N.; Zhang, Z.P.; Liu, H.B.; Zhou, R.R.; Zhong, B.Y.; Zou, M.X.; Dai, X.H.; Xiao, M.F.; Liu, X.Q.; et al. Protective effect of Acanthopanax gracilistylus-extracted Acankoreanogenin A on mice with fulminant hepatitis. Int. Immunopharmacol. 2011, 11, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Yook, C.S.; Liu, X.Q.; Chang, S.Y.; Park, S.Y.; Nohara, T. Lupane-triterpene glycosides from the leaves of Acanthopanax gracilistylus. Chem. Pharm. Bull. 2002, 50, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Chang, S.Y.; Park, S.Y.; Nohara, T.; Yook, C.S. A new lupane-triterpene glycoside from the leaves of Acanthopanax gracilistylus. Arch. Pharm. Res. 2002, 25, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Chang, S.Y.; Yook, C.S. Lupane-triterpenoids from the leaves of Acanthopanax gracilistylus. J. Lanzhou Univ. Nat. Sci. 2006, 42, 86–91. [Google Scholar]
- Zou, Q.P.; Liu, X.Q.; Lee, H.K.; Oh, O.J. Lupane-triterpenoids from the methanol extracts of leaves of Acanthopanax gracilistylus W. W. Smith. J. Lanzhou Univ. Nat. Sci. 2011, 47, 120–126. [Google Scholar]
- Tang, X.Y.; Ma, Y.C.; Li, P.J. Separation and identification of the anti-inflammatory diterpene from the root cortices of Acanthopanax gracilistylus W. W. Smith. Chin. J. Chin. Mater. Med. 1995, 20, 231–232. [Google Scholar]
- Wu, Z.Y.; Zhang, Y.B.; Zhu, K.K.; Luo, C.; Zhang, J.X.; Cheng, C.R.; Feng, R.H.; Yang, W.Z.; Zeng, F.; Wang, Y.; et al. Anti-inflammatory diterpenoids from the root bark of Acanthopanax gracilistylus. J. Nat. Prod. 2014, 77, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Zhang, C.Y.; Yin, W.J.; Liu, Z.X.; Yook, C.S. Analysis of volatile oil components of Acanthopanax gracilistylus. Chin. Tradit. Herb. Drugs 2001, 32, 1074–1075. [Google Scholar]
- Liu, X.Q.; Park, S.Y.; Yook, C.S. Studies on the constituents of the stem barks of Acanthopanax gracilistylus W. W. Smith. Nat. Prod. Sci. 2002, 8, 23–25. [Google Scholar]
- Liu, X.Q.; Yook, C.S.; Chang, S.Y. Chemical constituents of Acanthopanax gracilistylus. Chin. Tradit. Herb. Drugs 2004, 35, 250–252. [Google Scholar]
- Zhang, J.Y.; Pu, S.B.; Qian, S.H.; Liu, D. New cerebrosides from Acanthopanax gracilistylus. Chin. J. Nat. Med. 2011, 9, 105–107. [Google Scholar]
- Liu, X.Q.; Chang, S.Y.; Ro, S.H.; Yook, C.S. Constituents of Acanthopanax gracilistylus W. W. Smith. Nat. Med. 2002, 56, 215–216. [Google Scholar]
- An, S.Y.; Qian, S.H.; Jiang, J.Q.; Wang, K.C. Chemical constituents in leaves of Acanthopanax gracilistylus. Chin. Tradit. Herb. Drugs 2009, 40, 1528–1534. [Google Scholar]
- Xian, L.N.; Qian, S.H.; Li, Z.L. Studies on the chemical constituents from the stems of Acanthopanax gracilistylus. J. Chin. Med. Mater. 2010, 33, 538–542. [Google Scholar]
- Zhang, J.Y.; Pu, S.B.; Qian, S.H.; Liu, D.; Wang, K.C. Studies on the chemical constituents in fruits of Acanthopanax gracilistylus. J. Chin. Med. Mater. 2011, 34, 226–229. [Google Scholar]
- Song, X.H.; Xu, G.J.; Jin, R.L. Studies on chemical constituents of the root bark of Acanthopanax gracilistylus. J. Chin. Pharm. Univ. 1987, 18, 203–204. [Google Scholar]
- Dai, L.; Liu, X.Q.; Xie, X.; Liu, H.Y. Characterization of stereostructure by X-ray and technology of extracting in combination hydrolysis in situ of acankoreanogenin from leaves of Acanthopanax gracilistylus W. W. Smith. J. Cent. South Univ. 2014, 21, 3063–3070. [Google Scholar] [CrossRef]
- Shan, B.E.; Zeki, K.; Sugiura, T.; Yoshida, Y.; Yamashita, U. Chinese medicinal herb, Acanthopanax gracilistylus, extract induces cell cycle arrest of human tumor cells in vitro. Cancer Sci. 2000, 91, 383–389. [Google Scholar] [CrossRef]
- Shan, B.E.; Si, C.Y.; Zhang, J.Z. The Isolation of Anti-tumor Component of Acanthopanax gracilistylus. Teratog. Carcinog. Mutagen. 2004, 4, 203–205. [Google Scholar]
- Li, X.J.; Dai, L.; Li, Z.; Zhang, X.D.; Liu, X.Q.; Zou, Q.P.; Xie, X. Anti-inflammatory activities of lupane-triterpenoids in vitro and their phytochemical fingerprinting from leaves of Acanthopanax gracilistylus. Nat. Prod. Sci. 2015, 21, 104–110. [Google Scholar]
- Zou, Q.P.; Liu, X.Q.; Huang, J.J.; Yook, C.S.; Whang, W.K.; Lee, H.K.; Kwon, O.K. Inhibitory effects of lupane-type triterpenoid saponins from the leaves of Acanthopanax gracilistylus on lipopolysaccharide-induced TNF-α, IL-1β and high-mobility group box 1 release in macrophages. Mol. Med. Rep. 2017, 16, 9149–9156. [Google Scholar] [PubMed]
- Liu, X.Q.; Zou, Q.P.; Huang, J.J.; Yook, C.S.; Whang, W.K.; Lee, H.K.; Kwon, O.K. Inhibitory effects of 3α-hydroxy-lup-20(29)-en-23, 28-dioic acid on lipopolysaccharide-induced TNF-α, IL-1β, and the high mobility group box 1 release in macrophages. Biosci. Biotechnol. Biochem. 2017, 81, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Yook, C.S.; Kim, I.H.; Hahn, D.R.; Nohara, T.; Chang, S.Y. A lupane-triterpene glycoside from leaves of two Acanthopanax. Phytochemistry 1998, 49, 839–843. [Google Scholar] [CrossRef]
- Chang, S.Y.; Yook, C.S.; Nohara, T. Two new lupane-triterpene glycosides from leaves of Acanthopanax koreanum. Chem. Pharm. Bull. 1998, 46, 163–165. [Google Scholar] [CrossRef]
- Nhiem, N.X.; Tung, N.H.; Van Kiem, P.; Van Minh, C.; Ding, Y.; Hyun, J.H.; Kang, H.K.; Kim, Y.H. Lupane triterpene glycosides from leave of Acanthopanax koreanum and their cytotoxic activity. Chem. Pharm. Bull. 2009, 57, 986–989. [Google Scholar] [CrossRef] [PubMed]
- Nhiem, N.X.; Van Kiem, P.; Van Minh, C.; Tai, B.H.; Yen, P.H.; Tung, N.H.; Tung, N.H.; Hyun, J.H.; Kang, H.K.; Kim, Y.H. Lupane-type triterpene glycosides from the leaves of Acanthopanax koreanum and their in vitro cytotoxicity. Planta Med. 2010, 76, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Jahan, N.; Ahmed, W.; Malik, A. A lupene-type triterpene from Mimusops elengi. Phytochemistry 1995, 39, 255–257. [Google Scholar] [CrossRef]
- Lischewski, M.; Ty, P.D.; Schmidt, J.; Preiss, A.; Phiet, H.V.; Adam, G. Natural products from Vietnamese plants. Part 8. 3α,11α-Dihydroxylup-20(29)-ene-23,28-dioic acid from Schefflera octophylla. Phytochemistry 1984, 23, 1695–1697. [Google Scholar] [CrossRef]
- Chang, S.Y.; Yook, C.S.; Nohara, T. Lupane-triterpene glycosides from leaves of Acanthopanax koreanum. Phytochemistry 1999, 50, 1369–1374. [Google Scholar] [CrossRef]
- Titheradge, M.A. The enzymatic measurement of nitrate and nitrite. Methods Mol. Biol. 1998, 100, 83–91. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
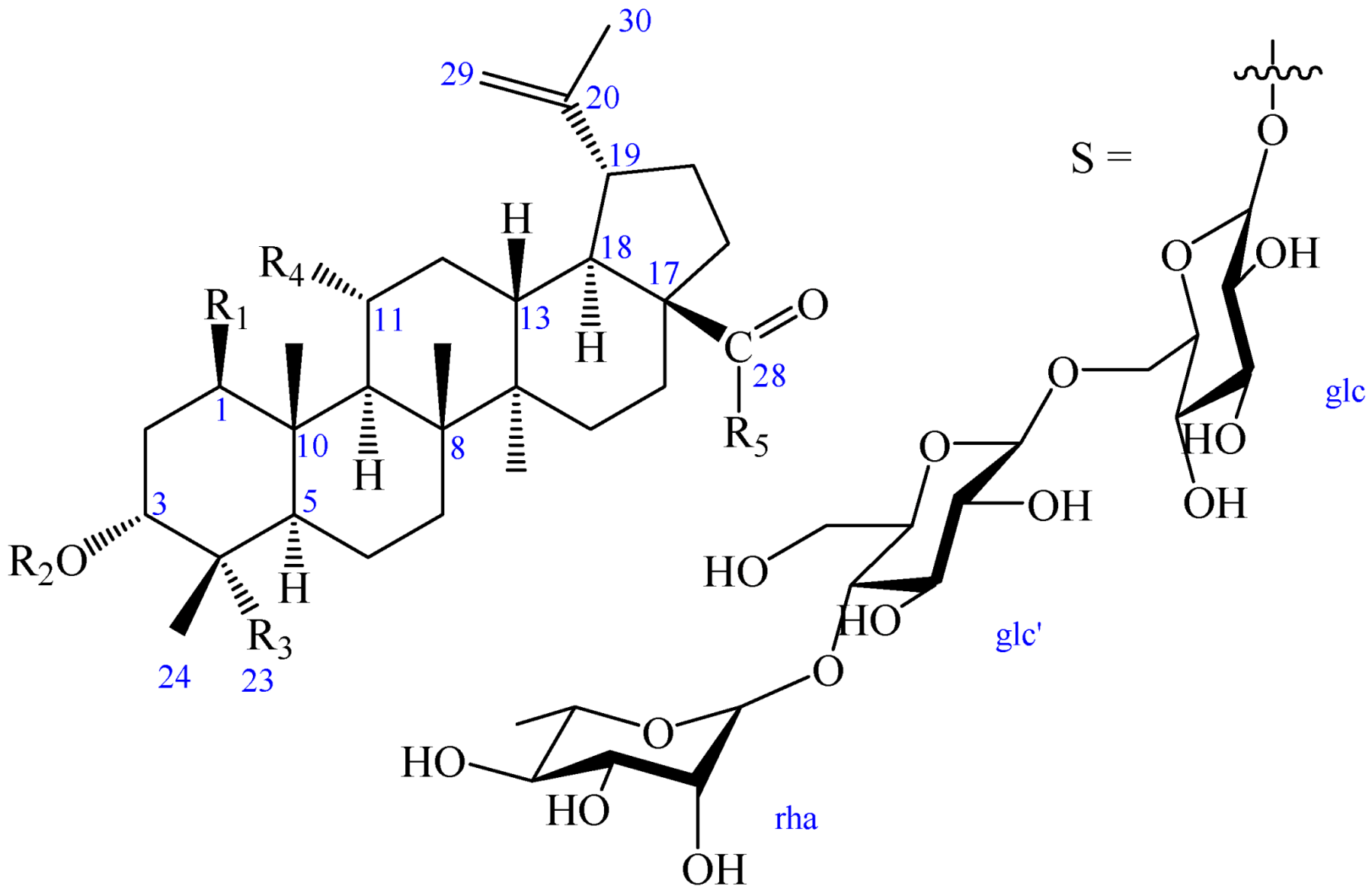
Figure 1.
Chemical structures of compounds 1–4.

| R1 | R2 | R3 | R4 | R5 | |
| 1 | OH | OH | COOH | H | OH |
| 2 | OH | OH | COOH | H | S |
| 2a | OH | OH | COOH | H | OH |
| 3 | H | OH | COOH | OH | OH |
| 4 | H | -β-d-glc | CH3 | OH | S |
Figure 2.
1H-1H COSY (bold) and selected HMBC (arrow) correlations of 1.
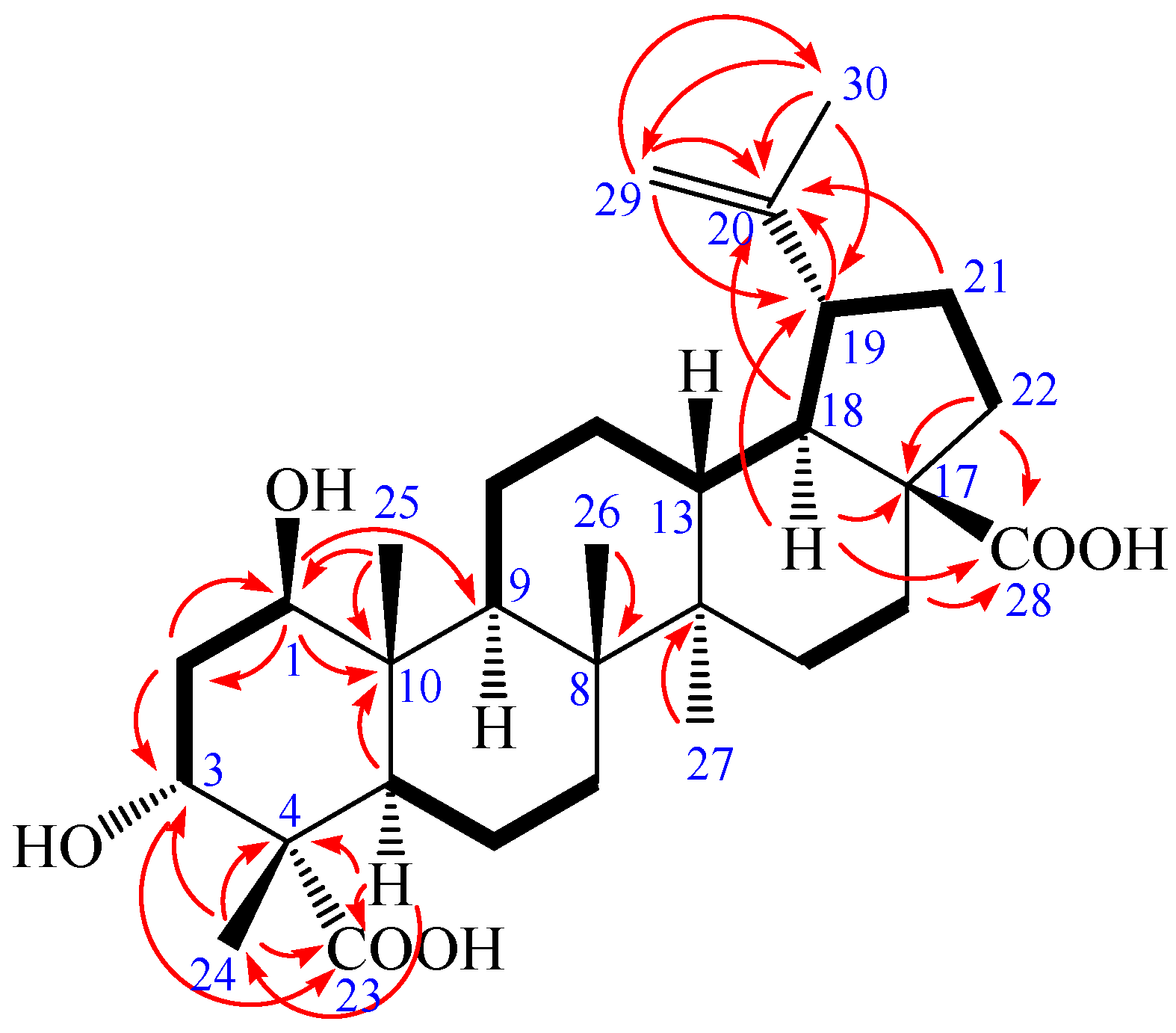
Figure 3.
The key NOESY (arrow) correlations of 1.
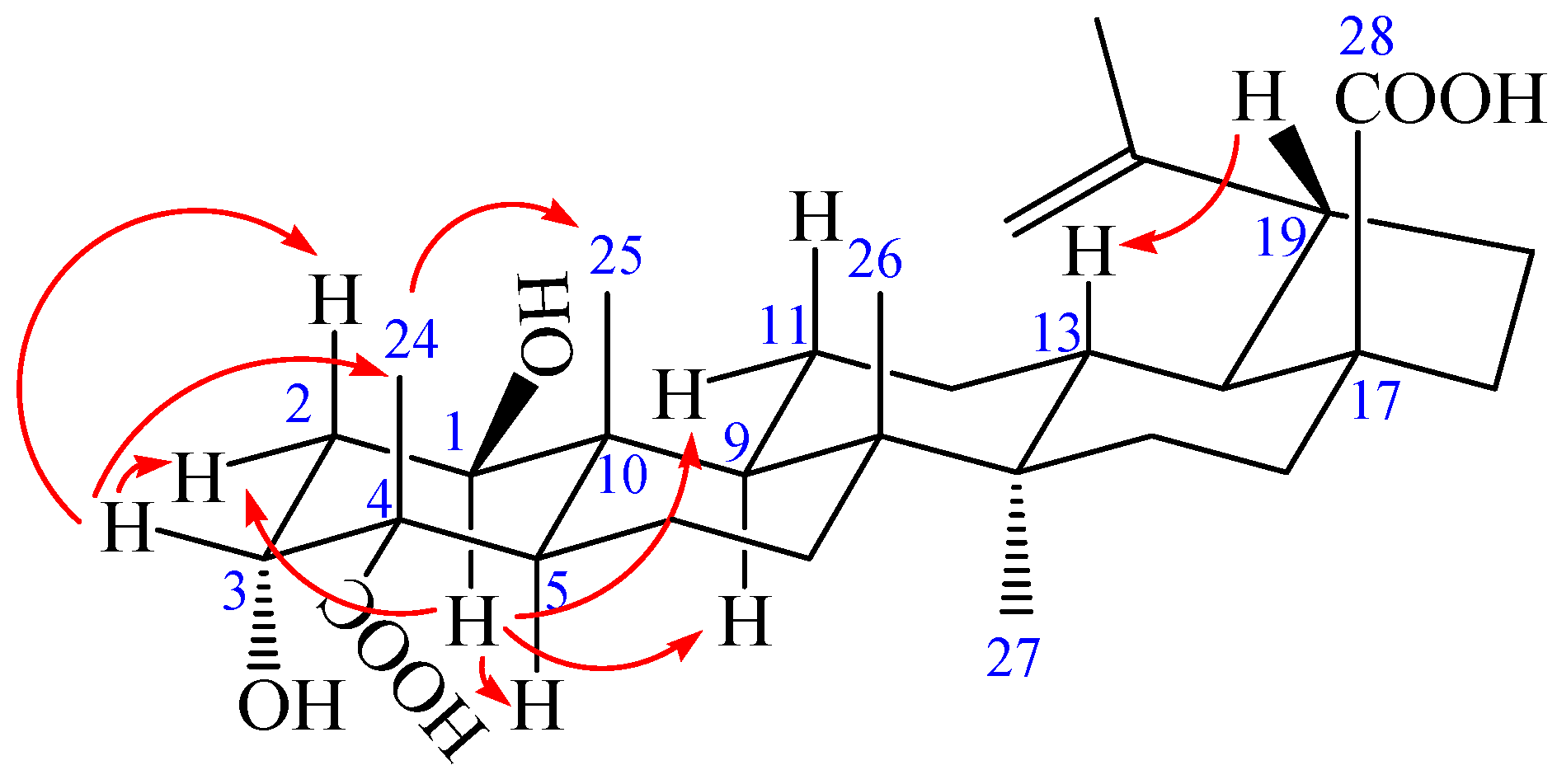
Figure 4.
1H-1H COSY (bold) and selected HMBC (arrow) correlations of 2.
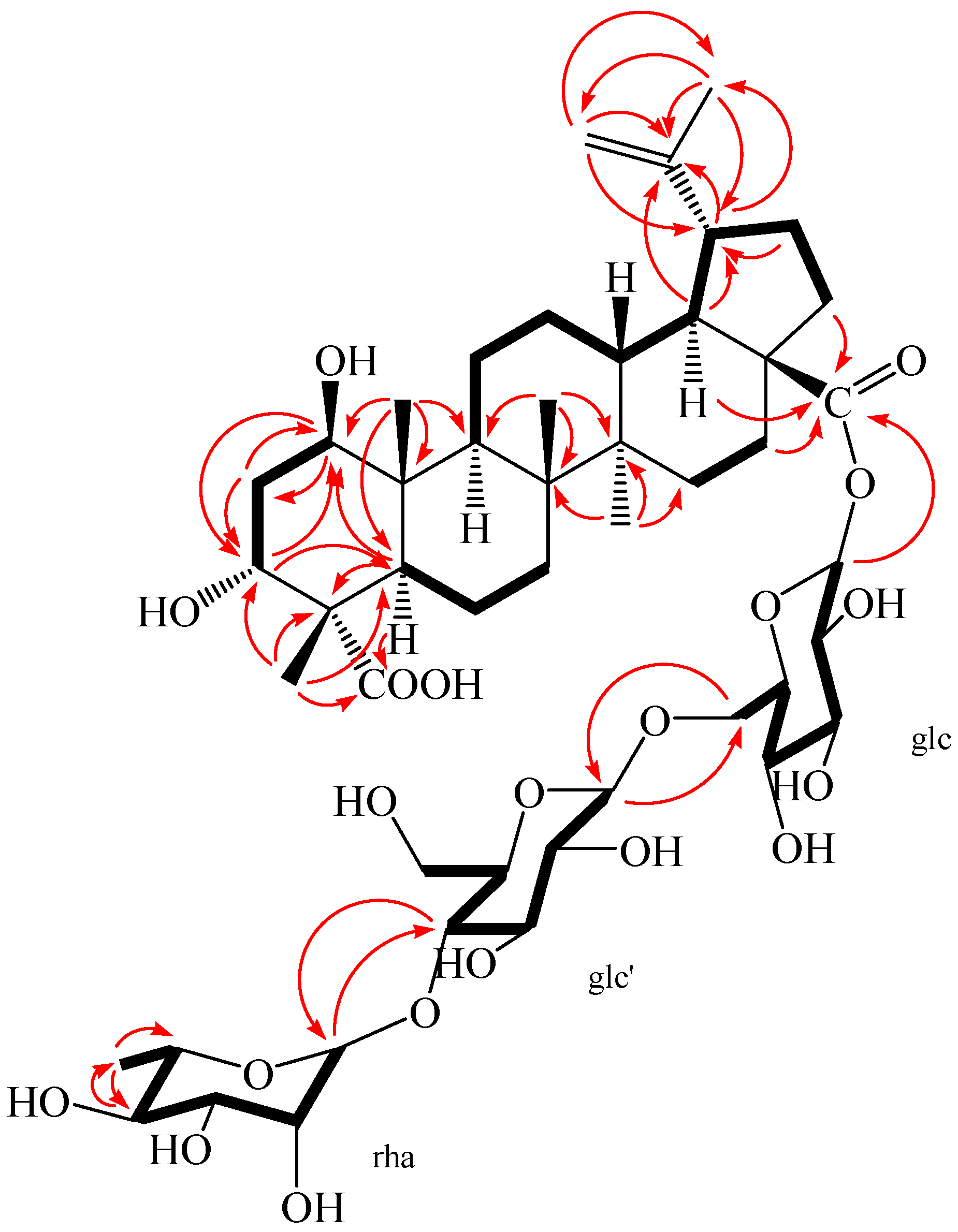
Figure 5.
The key NOESY (arrow) correlations of 2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
NMR spectral data of compounds 1–2.
| Position | 1 δC a,c | δH a,d [mult. (J in Hz)] | 2 δC b,c | δH b,d [mult. (J in Hz)] |
|---|---|---|---|---|
| Aglycone | ||||
| 1 | 75.46 | 3.84 (1H, dd, 8.16, 3.96) | 76.33 | 3.80 (1H, m) |
| 2 | 37.36 | 1.82 (1H, m); 1.87 (1H, m) | 36.79 | 1.72 (1H, m); 1.80 (1H, m) |
| 3 | 73.99 | 3.82 (1H, t, 2.40) | 74.14 | 3.67 (1H, m) |
| 4 | 52.27 | - | 52.24 | - |
| 5 | 45.25 | 1.95 (1H, m) | 46.11 | 1.87 (1H, m) |
| 6 | 22.09 | 1.35 (1H, m); 1.57 (1H, m) | 22.17 | 1.26 (1H, m); 1.58 (1H, m) |
| 7 | 35.19 | 1.31 (1H, m); 1.56 (1H, m) | 35.09 | 1.30 (1H, m); 1.55 (1H, m) |
| 8 | 42.76 | - | 42.96 | - |
| 9 | 52.98 | 1.72 (1H, dd, 10.08, 2.68) | 53.06 | 1.72 (1H, m) |
| 10 | 44.33 | - | 44.47 | - |
| 11 | 24.68 | 1.32 (1H, m); 2.43 (1H, brd, 9.80) | 24.82 | 1.36 (1H, m); 2.28 (1H, m) |
| 12 | 26.87 | 1.12 (1H, dd, 10.52, 3.76); 1.68 (1H, m) | 26.94 | 1.12 (1H, m); 1.68 (1H, m) |
| 13 | 39.14 | 2.34 (1H, td, 10.32, 3.0) | 39.13 | 2.27 (1H, m) |
| 14 | 43.72 | - | 43.82 | - |
| 15 | 30.75 | 1.20 (1H, m); 1.46 (1H, m) | 30.86 | 1.15 (1H, m); 1.54 (1H, m) |
| 16 | 33.15 | 1.48 (1H, m); 2.25 (1H, dt, 9.84, 2.8) | 32.95 | 1.44 (1H, m); 2.33 (1H, m) |
| 17 | 56.98 | - | 57.93 | - |
| 18 | 50.30 | 1.64 (1H, m) | 50.60 | 1.65 (1H, m) |
| 19 | 48.24 | 3.05 (1H, td, 8.76, 4.0) | 48.36 | 3.00 (1H, m) |
| 20 | 151.89 | - | 151.77 | - |
| 21 | 31.61 | 1.34 (1H, m); 1.90 (1H, m) | 31.55 | 1.37 (1H, m); 1.94 (1H, m) |
| 22 | 37.84 | 1.48 (1H, m); 1.92 (1H, m) | 37.68 | 1.48 (1H, m); 1.94 (1H, m) |
| 23 | 178.16 | - | 182.6 | - |
| 24 | 17.92 | 1.17 (3H, s) | 18.08 | 1.09 (3H, s) |
| 25 | 13.24 | 0.97 (3H, s) | 13.17 | 0.95 (3H, s) |
| 26 | 17.31 | 0.99 (3H, s) | 17.14 | 0.98 (3H, s) |
| 27 | 15.40 | 1.07 (3H, s) | 15.10 | 1.03 (3H, s) |
| 28 | 177.84 | - | 176.40 | - |
| 29 | 110.26 | 4.58 (1H, m); 4.72 (1H, d, 1.68) | 110.41 | 4.58 (1H, brs); 4.72 (1H, brs) |
| 30 | 19.72 | 1.71 (3H, s) | 19.49 | 1.70 (3H, s) |
| C-28 O-glc | ||||
| 1 | 95.26 | 5.45 (1H, d, 6.56) | ||
| 2 | 74.00 | 3.33 (1H, m) | ||
| 3 | 78.28 | 3.42 (1H, m) | ||
| 4 | 70.95 | 3.43 (1H, m) | ||
| 5 | 78.06 | 3.54 (1H, m) | ||
| 6 | 69.55 | 3.81 (1H, m); 4.11 (1H, dd, 9.48, 1.36) | ||
| glc′(1→6)glc | ||||
| 1′ | 104.56 | 4.37 (1H, d, 6.28) | ||
| 2′ | 75.32 | 3.23 (1H, m) | ||
| 3′ | 76.71 | 3.45 (1H, m) | ||
| 4′ | 79.51 | 3.53 (1H, m) | ||
| 5′ | 76.89 | 3.30 (1H, m) | ||
| 6′ | 61.90 | 3.65 (1H, m); 3.80 (1H, m) | ||
| rha(1→4)glc′ | ||||
| 1″ | 102.92 | 4.84 (1H, overlapped) | ||
| 2″ | 72.44 | 3.81 (1H, m) | ||
| 3″ | 72.16 | 3.62 (1H, m) | ||
| 4″ | 73.75 | 3.38 (1H, m) | ||
| 5″ | 70.64 | 3.95 (1H, m) | ||
| 6″ | 17.84 | 1.25 (3H, d, 4.96) |
Note: Assignments were performed by HMQC, HMBC, 1H-1H COSY, and NOESY experiments; Glc: d-glucopyranosyl; Rha: l-rhamnopyranosyl; a Measured in CD3COCD3; b Measured in CD3OD; c 100 MHz; d 400 MHz.
Table 2.
Inhibitory effects of compounds 1 and 3 against LPS-Induced NO production in murine microglia BV2 cells a.
Table 2.
Inhibitory effects of compounds 1 and 3 against LPS-Induced NO production in murine microglia BV2 cells a.
| Compounds | Inhibition | Cell Viability |
|---|---|---|
| 1 | 38.6% in 80 μM | 86.44% |
| 3 | 40.4% in 80 μM | 83.90% |
a Data is presented as the mean of three experiments ± SD.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, X.-J.; Zou, Q.-P.; Wang, X.; Kim, K.-W.; Lu, M.-F.; Ko, S.-K.; Yook, C.-S.; Kim, Y.-C.; Liu, X.-Q. Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus. Molecules 2018, 23, 87. https://doi.org/10.3390/molecules23010087
AMA Style
Li X-J, Zou Q-P, Wang X, Kim K-W, Lu M-F, Ko S-K, Yook C-S, Kim Y-C, Liu X-Q. Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus. Molecules. 2018; 23(1):87. https://doi.org/10.3390/molecules23010087
Chicago/Turabian StyleLi, Xiao-Jun, Qin-Peng Zou, Xiang Wang, Kwan-Woo Kim, Mao-Fang Lu, Sung-Kwon Ko, Chang-Soo Yook, Youn-Chul Kim, and Xiang-Qian Liu. 2018. "Lupane Triterpenes from the Leaves of Acanthopanax gracilistylus" Molecules 23, no. 1: 87. https://doi.org/10.3390/molecules23010087