
Application of the Asymmetric Pictet–Spengler Reaction in the Total Synthesis of Natural Products and Relevant Biologically Active Compounds

Abstract
:
{kind=link}
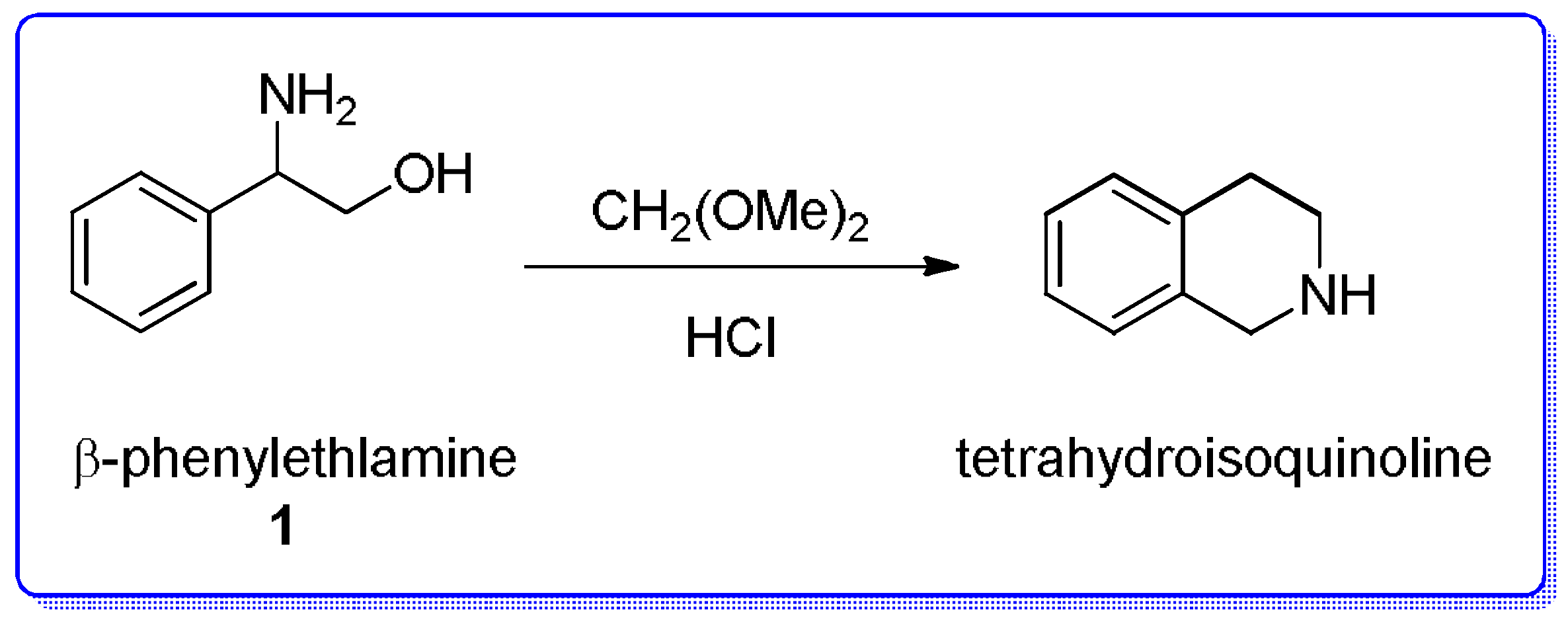{kind=link}
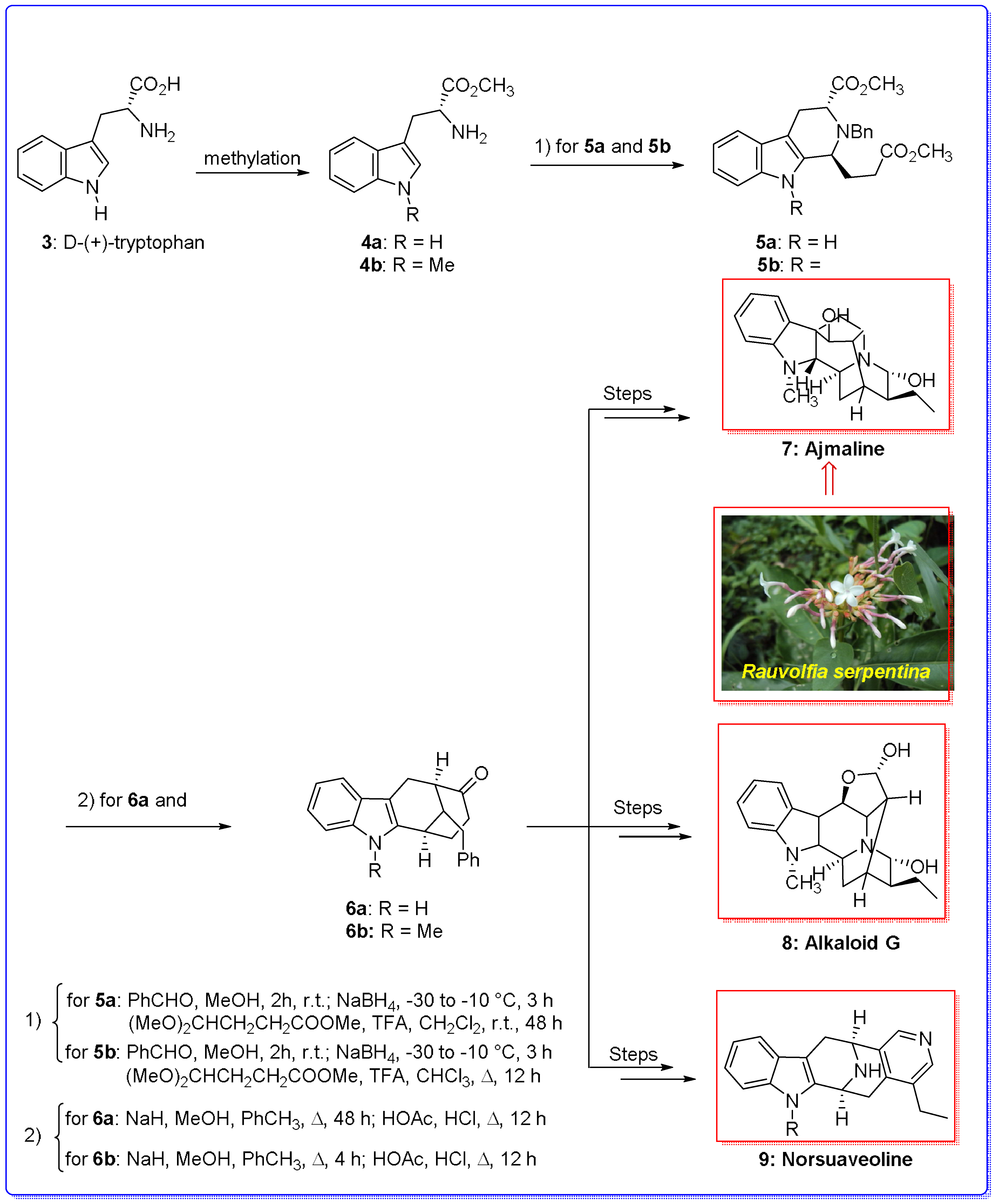{kind=link}
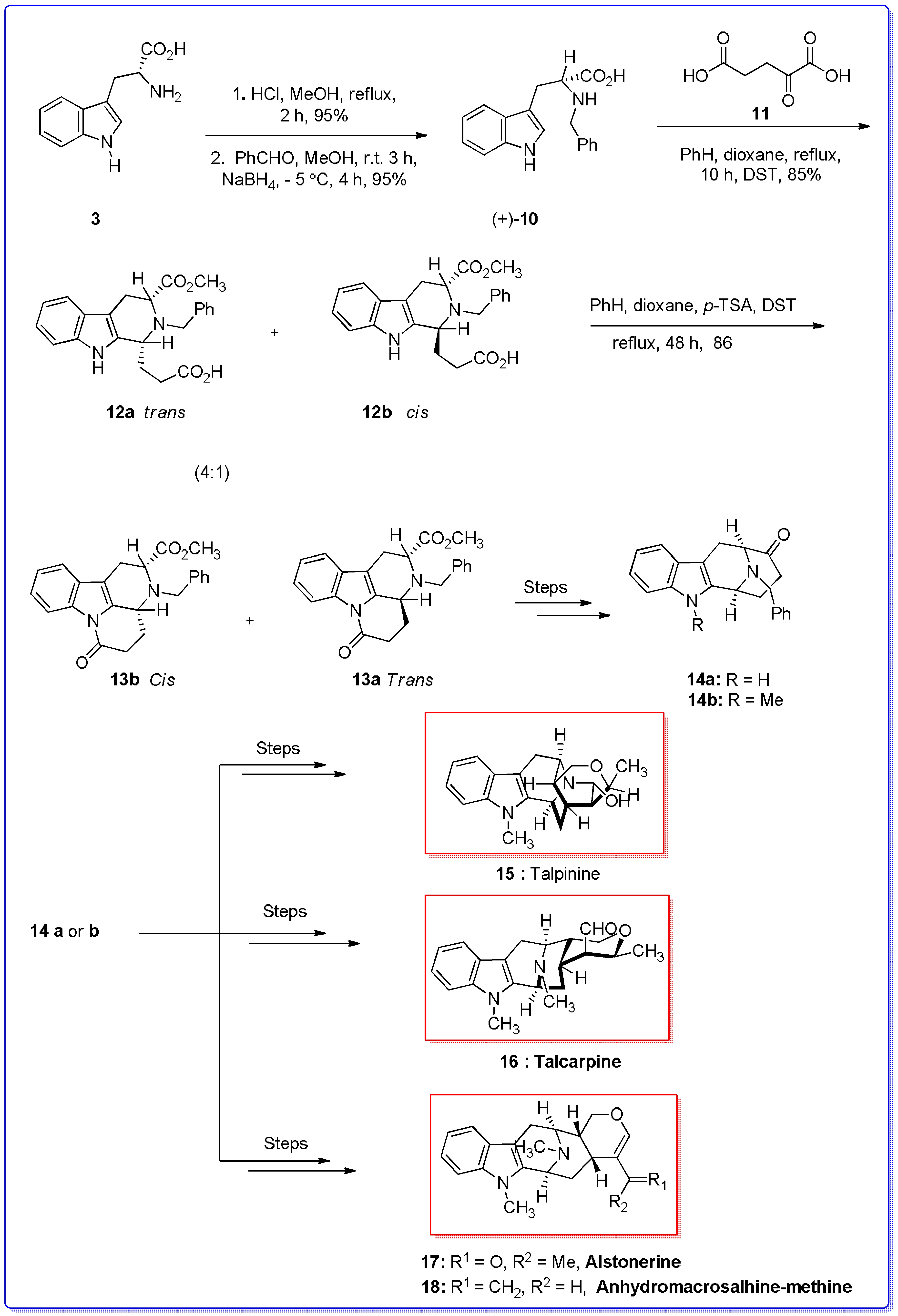{kind=link}
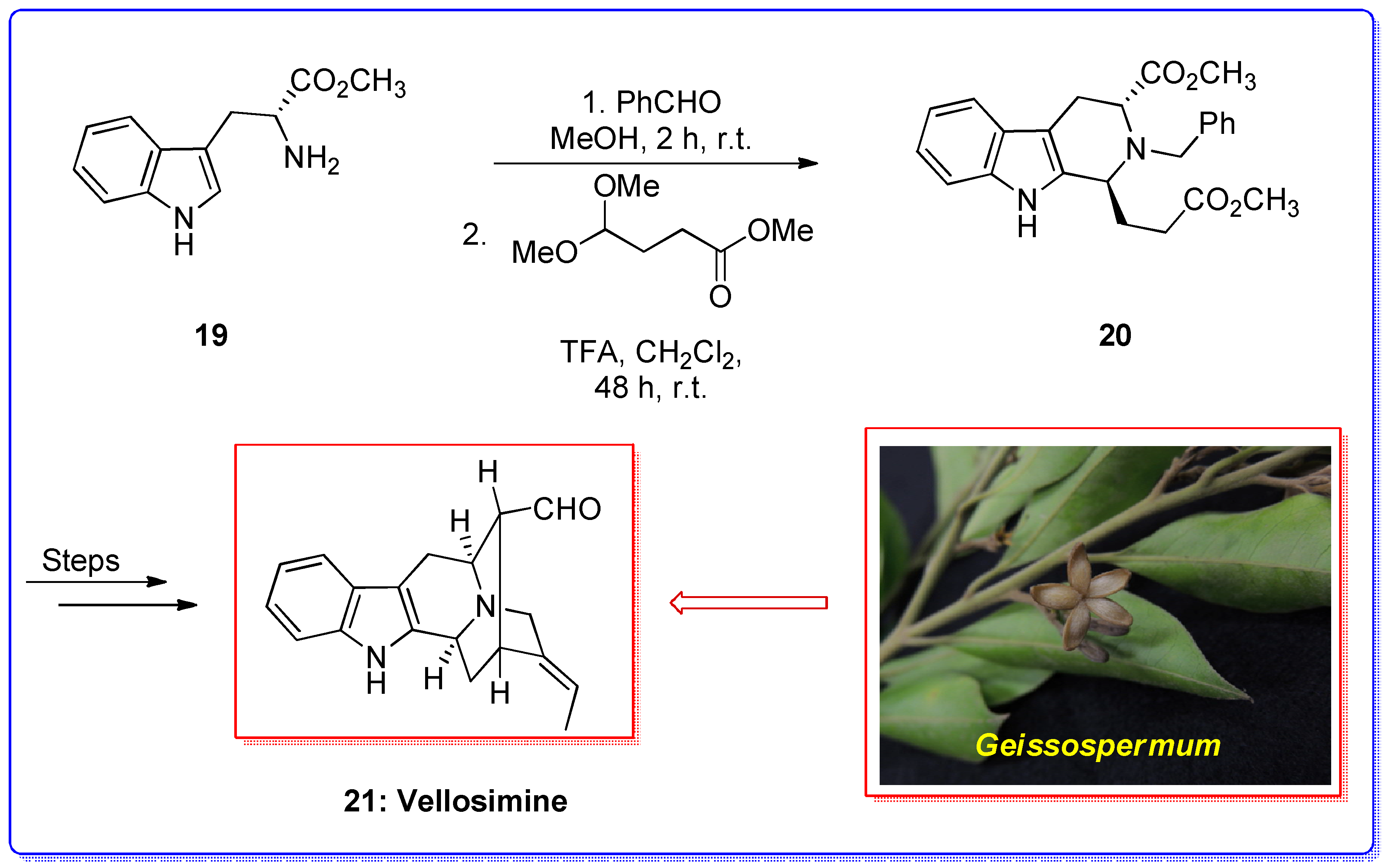{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Pictet–Spengler Reaction in the Total Synthesis of Natural Products
2.1. Indole Scaffold
2.2. Phenyl (Tetrahydroisoquinoline) Scaffold
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seigler, D.S. Plant Secondary Metabolism; Springer: New York, NY, USA, 2001; p. 628. [Google Scholar]
- Rao, R.N.; Maiti, B.; Chanda, K. Application of Pictet-Spengler reaction to indole based alkaloids containing tetrahydro-β-carboline scaffold in combinatorial chemistry. ACS Comb. Sci. 2017, 19, 199–228. [Google Scholar] [CrossRef] [PubMed]
- Manske, R.H.F. The Alkaloids, Chemistry and Physiology; Academic Press: New York, NY, USA, 1981; Volume 19, p. 227. [Google Scholar]
- Pictet, A.; Spengler, T. Pictet-Spengler reaction. Ber. Dtsch. Chem. Ges. 1911, 44, 2030–2036. [Google Scholar] [CrossRef]
- Pictet, A.; Court, G. Uber einige neue pflanzenalkaloide. Ber. Dtsch. Chem. Ges. 1907, 40, 3771–3783. [Google Scholar] [CrossRef]
- Pictet, A. Uber die bildungsweise der alkaloide in der pflanze. Arch. Pharma. 1906, 244, 389–396. [Google Scholar] [CrossRef]
- Hahn, G.; Ludewig, H. Synthese von Tetrahydro-harman-Derivaten unter physiologischen Bedingungen. Chem. Ber. 1934, 67, 2031–2035. [Google Scholar] [CrossRef]
- Tatsui, G.J. Preparation of 1-methyl-1,2,3,4-tetrahydro-β-carboline. J. Pharm. Soc. Jpn. 1928, 48, 92. [Google Scholar]
- Smith, G.N. Strictosidine: A key intermediate in the Biogenesis of indole alkaloids. Chem. Commun. 1968, 912–914. [Google Scholar] [CrossRef]
- Battersby, A.R.; Burnett, A.R.; Parsons, P.G. Preparation of secologanin: Its conversion into ipecoside and its role in indole alkaloid biosynthesis. Chem. Commun. 1968, 1280–1281. [Google Scholar] [CrossRef]
- Klausen, R.S.; Jacobsen, E.N. Weak Brønsted acid−thiourea co-catalysis: Enantioselective, Catalytic Protio-Pictet−Spengler reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.D.; Hollinshead, S.P.; McLay, N.R. Exceptional stereochemical control in the Pictet-Spengler reaction. Tetrahedron Lett. 1987, 28, 5177–5180. [Google Scholar] [CrossRef]
- Lee, M.; Domínguez-Gil, T.; Hesek, D.; Mahasenan, K.V.; Lastochkin, E.; Hermoso, J.A.; Mobashery, S. Turnover of Bacterial Cell wall by SltB3, a Multidomain Lytic Transglycosylase of Pseudomonas aeruginosa. ACS Chem. Biol. 2016, 11, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, R.; Nakagawa, M.; Nishida, A. An efficient synthetic approach to optically active β-carboline derivatives via Pictet–Spengler reaction promoted by trimethylchlorosilane. Tetrahedron Asymmetry 2003, 14, 177–180. [Google Scholar] [CrossRef]
- Kawate, T.; Yamanaka, M.; Nakagawa, M. Chiral auxiliary approach to the asymmetric Pictet-Spengler reaction of Tryptamines. Heterocycles 1999, 2, 1033–1039. [Google Scholar]
- Herlé, B.; Wanner, M.J.; van Maarseveen, J.H.; Hiemstra, H. Total synthesis of (+)-Yohimbine via an enantioselective organocatalytic Pictet–Spengler reaction. J. Org. Chem. 2011, 76, 8907–8912. [Google Scholar] [CrossRef] [PubMed]
- Sewgobind, N.V.; Wanner, M.J.; Ingemann, S.; de Gelder, R.; vanMaarseveen, J.H.; Hiemstra, H. Enantioselective Binol-Phosphoric acid catalyzed Pictet−Spengler reactions of n-Benzyltryptamine. J. Org. Chem. 2008, 73, 6405–6408. [Google Scholar] [CrossRef] [PubMed]
- Seayad, J.; Seayad, A.M.; List, B. Catalytic Asymmetric Pictet−Spengler reaction. J. Am. Chem. Soc. 2006, 128, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Dalpozzo, R. The Chiral pool in the Pictet–Spengler reaction for the synthesis of β-Carbolines. Molecules 2016, 21, 699. [Google Scholar] [CrossRef] [PubMed]
- Nic, M.; Jirat, J.; Kosata, B.; Jenkins, A. IUPAC Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Manske, R.H.F. The Alkaloids: Chemistry and physiology; Academic Press: New York, NY, USA, 1965; Volume VIII, p. 673. [Google Scholar]
- Kittakoop, P.; Mahidol, C.; Ruchirawat, S. Alkaloids as Important scaffolds in therapeutic Drugs for the Treatments of Cancer, Tuberculosis, and Smoking Cessation. Curr. Top. Med. Chem. 2014, 14, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Frustaci, A.; Del Bufalo, A.; Fini, M.; Cesario, A. Multitarget Drugs of Plants Origin Acting on Alzheimer’s Disease. Curr. Med. Chem. 2013, 20, 1686–1993. [Google Scholar] [CrossRef] [PubMed]
- Sinatra, R.S.; Jahr, J.S.; Watkins-Pitchford, M. The Essence of Analgesia and Analgesics; Sinatra, R.S., Jahr, J.S., Watkins-Pitchford, M., Eds.; Cambridge University Press: Cambridge, UK, 2010; p. 82. [Google Scholar]
- Cushnie, T.P.; Cushnie, B.; Lamb, A.J. Alkaloids: An overview of their Antibacterial, Antibiotic-enhancing and Antivirulence Activities. Int. J. Antimicrob. Agents 2014, 44, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Sun, H.; Zhang, A.H.; Xu, H.Y.; Yan, G.L.; Han, Y.; Wang, X.J. Natural Alkaloids: Basic Aspects, Biological roles, and future Perspectives. Chin. J. Nat. Med. 2014, 12, 401–406. [Google Scholar] [CrossRef]
- Robbers, J.E.; Speedie, M.K.; Tyler, V.E. Chapter 9: Alkaloids. In Pharmacognosy and Pharmacobiotechnology; Lippincott. Williams & Wilkins: Philadelphia, NY, USA, 1996; pp. 143–185. [Google Scholar]
- Rhoades, D.F. Evolution of plant chemical defense against Herbivores. In Their Interaction with Secondary Plant Metabolites; Academic Press: New York, NY, USA, 1979; p. 41. [Google Scholar]
- Ho, B.T.; McIsaac, W.M.; Walker, K.A.; Estevez, V. Inhibitors of Monoamine Oxidase. Influence of Methyl substitution on the inhibitory activity of 8-Carbolines. J. Pharm. Sci. 1968, 57, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K. Consice Textbook of Forensic Medicine & Toxicology; Elsevier: New Delhi, India, 2008. [Google Scholar]
- Roquebert, J.; Demichel, P. Inhibition of the alpha 1 and alpha 2-adrenoceptor-mediated pressor response in Pithed Rats by Raubasine, Tetrahydroalstonine and Akuammigine. Eur. J. Pharm. 1984, 106, 203–205. [Google Scholar] [CrossRef]
- Müller, J.M.; Schlittler, E.; Bein, H.J. Reserpin, the sedative principle from Rauwolfia serpentina B. Experientia 1952, 8, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Neuss, N.; Gorman, M.; Hargrove, W.; Cone, N.J.; Biemann, K.; Buchi, G.; Manning, R.E. The structures of the Oncolytic alkaloids Vinblastine (VLB) and Vincristine (VCR). J. Am. Chem. Soc. 1964, 86, 1440–1442. [Google Scholar] [CrossRef]
- Van der Heijden, R.; Jacobs, D.; Snoeijer, W.; Hallard, D.; Verpoorte, R. The Catharanthus alkaloids: Pharmacognosy and biotechnology. Curr. Med. Chem. 2004, 11, 607–628. [Google Scholar] [CrossRef] [PubMed]
- Southin, T.W.; Buckingham, J. Dictionary of Alkaloids; Chapman&Hall: London, UK, 1979; p. 3. [Google Scholar]
- Nicolaou, K.C.; Montagnon, T.; Snyder, S.A. Tandem reactions, Cascade sequences, and Biomimetic strategies in total synthesis. Chem. Commun. 2003, 5, 551–564. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Edmonds, D.J.; Bulger, P.C. Cascade reactions in total synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo, R.M.; Christmann, M. Organocatalytic synthesis of drugs and bioactive natural products. Eur. J. Org. Chem. 2007, 38, 2575–2600. [Google Scholar] [CrossRef]
- Tietze, L.F.; Beifuss, U. Sequential transformations in organic chemistry: A synthetic strategy with a future. Angew. Chem. Int. Ed. 1993, 32, 131–163. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic Cascade reactions as a new tool in total synthesis. Nature Chem. 2010, 2, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Poisson, T. Macmillan’s imidazolidinones: Powerful chiral organocatalysts. Synlett 2008, 1, 147–148. [Google Scholar] [CrossRef]
- Souza, D.M.D.; Mueller, T.J.J. Multi-component syntheses of heterocycles by transition-metal catalysis. Chem. Soc. Rev. 2007, 36, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hill, M.; Wang, M.; Panjikar, S.; Stockigt, J. Structural basis and enzymatic mechanism of the biosynthesis of C9- from C10-monoterpenoid indole alkaloids. Angew. Chem. Int. Ed. 2009, 48, 5211–5213. [Google Scholar] [CrossRef] [PubMed]
- Cordell, G.A. The Alkaloids: Chemistry and Biology; Academic Press: San Diego, CA, USA, 1998; Volume 51, p. 199. [Google Scholar]
- Heravi, M.M.; Khaghaninejad, S.; Nazari, N. Chapter five-Bischler–Napieralski reaction in the syntheses of Isoquinolines. Adv. Heterocycl. Chem. 2015, 112, 183–226. [Google Scholar]
- Sadjadi, S.; Heravi, M.M.; Nazari, N. Isocyanide-based multicomponent reactions in the synthesis of heterocycles. RSC Adv. 2016, 6, 53203–53272. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E.; Nazari, N. Negishi coupling: An easy progress for C-C bond construction in total synthesis. Mol. Divers. 2014, 18, 441–472. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Hashemi, E.; Azimian, F. Recent developments of the Stille reaction as a revolutionized method in total synthesis. Tetrahedron 2014, 70, 7–21. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E.; Ghobadi, N. Development of recent total syntheses based on the Heck reaction. Curr. Org. Chem. 2013, 17, 2192–2224. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E. Recent applications of the Suzuki reaction in total synthesis. Tetrahedron 2012, 68, 9145–9178. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Oxazolidinones as chiral auxiliaries in asymmetric Aldol reactions applied to total synthesis. Tetrahedron Asymmetry 2013, 24, 1149–1188. [Google Scholar] [CrossRef]
- Heravi, M.M.; Vavsari, V.F. Recent applications of Intramolecular Diels–Alder reaction in total synthesis of natural products. RSC Adv. 2015, 5, 50890–50912. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hamidi, H.; Zadsirjan, V. Recent applications of Click reaction in the syntheses of 1,2,3-Triazoles. Curr. Org. Chem. 2014, 11, 647–675. [Google Scholar] [CrossRef]
- Heravi, M.M.; Lashaki, T.B.; Fattahi, B.; Zadsirjan, V. Application of asymmetric Sharpless Aminohydroxylation in total synthesis of natural products and some synthetic complex bio-active molecules. RSC Adv. 2018, 8, 6634–6659. [Google Scholar] [CrossRef]
- Koshvandi, A.T.K.; Heravi, M.M. Current applications of Suzuki-Miyaura coupling reaction in the total synthesis of natural products: An update: Applications of Suzuki-Miyaura coupling reaction in natural products. Appl. Organomet. Chem. 2018. [Google Scholar] [CrossRef]
- Heravi, M.M.; Rohani, S.; Zadsirjan, V.; Zahedi, N. Fischer Indole synthesis applied to the total synthesis of natural products. RSC Adv. 2017, 7, 52852–52887. [Google Scholar] [CrossRef]
- Koshvandi, A.T.K.; Heravi, M.M. Applications of Danishefsky’s Dienes in asymmetric Oxo-Diels-Alder reactions. Tetrahedron Asymmetry 2017, 28, 1506–1556. [Google Scholar] [CrossRef]
- Talaei, B.; Heravi, M.M.; Oskooie, H.A.; Rezvanian, A. An approach to the diastereoselective synthesis of cyclohexane-1,3-dicarboxamide derivatives via a Pseudo five-componentreaction based on diketene. Synlett 2018, 29, 225–229. [Google Scholar]
- Heravi, M.M.; Moradi, R.; Malmir, M. Recent advances in the application of the Heck reaction in the synthesis of heterocyclic compounds: An update. Curr. Org. Chem. 2017, 22, 165–198. [Google Scholar] [CrossRef]
- Seigler, D.S. Plant Secondary Metabolism. New Phytol. 2000, 147, 483–485. [Google Scholar]
- Knunyants, I.L. Chemical Encyclopedia; Wiley: New York, NY, USA, 1988; Volume 1, pp. 1–623. [Google Scholar]
- Siddiqui, S.; Siddiqui, R.H. Chemical examination of the roots of Rauwolfia serpentina Benth. J. Indian Chem. Soc. 1931, 8, 667–680. [Google Scholar]
- Koskinen, A.; Lounasmaa, M. The Sarpagine-Ajmaline group of indole alkaloids. In Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G.W., Eds.; Springer-Verlag: New York, NY, USA, 1983; Volume 43, p. 275. [Google Scholar]
- Brugada, J.; Brugada, P. What to do in patients with no structural heart Disease and sudden arrhythmic Death? Am. J. Cardiol. 1996, 78, 69–75. [Google Scholar] [CrossRef]
- Slowinski, S.; Rajch, D.; Zabowka, M. Myocardial infarction in a man with. Wolff-Parkinson-White syndrome. Przegl. Lek. 1996, 53, 196–198. [Google Scholar] [PubMed]
- Mest, H.J.; Winkler, J.; Foerster, W. The antiarrhythmic effect of Prostaglandin E2 on Catecholamine-induced Arrhythmias. Acta Biol. Med. Ger. 1977, 36, 1193–1196. [Google Scholar] [PubMed]
- Bi, Y.; Hamaker, L.K.; Cook, J.M. The synthesis of Macroline related alkaloids. In Studies in Natural Products Chemistry, Bioactive Natural Products; Part A; Rahman, A., Basha, F.Z., Eds.; Elsevier Science: Amsterdam, The Netherlands, 1993; Volume 13, p. 383. [Google Scholar]
- Endreb, S.; Takayama, H.; Suda, S.; Kitajima, M.; Aimi, N.; Sakai, S.-I.; Sto¨ckigt, J. Alkaloids from Rauwolfia serpentina cell cultures treated with Ajmaline. Phytochemistry 1993, 32, 725–730. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Yu, P.; Peterson, A.; Weber, R.; Soerens, D.; Grubisha, D.; Bennett, D.; Cook, J.M. General approach for the synthesis of Ajmaline/Sarpagine Indole alkaloids: Enantiospecific total synthesis of (+)-Ajmaline, Alkaloid G, and Norsuaveoline via the asymmetric Pictet-Spengler reaction. J. Am. Chem. Soc. 1999, 121, 6998–7010. [Google Scholar] [CrossRef]
- Hamaker, L.K.; Cook, J.M. The synthesis of Macroline related alkaloids. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S.W., Ed.; Elsevier Science: New York, NY, USA, 1995; Volume 9, p. 23. [Google Scholar]
- Wong, W.-H.; Lim, P.-B.; Chuah, C.-H. Oxindole alkaloids from alstonia macrophylla. Phytochemistry 1996, 41, 313–315. [Google Scholar] [CrossRef]
- Cook, J.M.; LeQuesne, P.W. Macralstonine from alstonia muelleriana. Phytochemistry 1971, 10, 437–439. [Google Scholar] [CrossRef]
- Chatterjee, A.; Parks, L.M. The Structure of Verbenalin. J. Am. Chem. Soc. 1949, 71, 2249–2250. [Google Scholar] [CrossRef]
- Wright, C.W.; Allen, D.; Cai, Y.; Phillipson, J.D.; Said, I.M.; Kirby, G.C.; Warhurst, D.C. In vitro antiamoebic and antiplasmodial activities of alkaloids isolated from Alstonia Angustifolia Roots. Phytother. Res. 1992, 6, 121–124. [Google Scholar] [CrossRef]
- Leclercq, J.; de Pauw-Gillet, M.-C.; Bassleer, R.; Angenot, L. Screening of Cytotoxic activities of Strychnos alkaloids (methods and results). J. Ethnopharmacol. 1986, 15, 305–316. [Google Scholar] [CrossRef]
- Tan, G.T.; Pezzuto, J.M.; Kinghorn, A.D.; Hughes, L.S.H. Evaluation of natural products as inhibitors of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase. J. Nat. Prod. 1991, 54, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.T.; Miller, J.F.; Kinghorn, A.D.; Hughes, S.H.; Pezzuto, J.M. HIV-1 and HIV-2 reverse transcriptases: A comparative study of sensitivity to inhibition by selected natural products. Biochem. Biophys. Res. Commun. 1992, 185, 370–378. [Google Scholar] [CrossRef]
- Nordman, C.E.; Kumra, S.K. The structure of Villalstonine. J. Am. Chem. Soc. 1965, 87, 2059–2060. [Google Scholar] [CrossRef]
- Waldner, E.E.; Hesse, M.; Taylor, W.I.; Schmid, H. Über die. Konstitution des Macralstonidins.124. Mitteilung über Alkaloide. Helv. Chim. Acta 1967, 50, 1926–1939. [Google Scholar] [CrossRef] [PubMed]
- Talapatra, S.K.; Adityachaudhury, N. Macralstonine, an alkaloid of the trunk bark of Alstonia macrophylla. Wall. Sci. Culture 1958, 24, 243–270. [Google Scholar]
- Soerens, D.; Sandrin, J.; Ungemach, F.; Mokry, P.; Wu, G.S.; Yamanaka, E.; Hutchins, L.; DiPierro, M.; Cook, J.M. Study of the Pictet–Spengler reaction in aprotic media: Synthesis of the β-Galactosidase inhibitor, Pyridindolol. J. Org. Chem. 1979, 44, 535–545. [Google Scholar] [CrossRef]
- Shimizu, M.; Ishikawa, M.; Komoda, Y.; Nakajima, T.; Yamaguchi, K.; Sakai, S. Asymmetric synthesis and absolute configuration of (−)-Trypargine. Chem. Pharm. Bull. 1984, 32, 1313–1325. [Google Scholar] [CrossRef]
- Yu, P.; Wang, T.; Li, J.; Cook, J.M. Enantiospecific total synthesis of the Sarpagine related indole alkaloids Talpinine and Talcarpine as well as the improved total synthesis of Alstonerine and Anhydromacrosalhine-methine via the asymmetric Pictet-Spengler reaction. J. Org. Chem. 2000, 65, 3173–3191. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, H.; Onak, T.P.; Hughes, N.A.; Reinecke, M.G. Alkaloids of Geissospermum vellosii. J. Am. Chem. Soc. 1958, 80, 1601–1604. [Google Scholar] [CrossRef]
- Rapoport, H.; Moore, E. Alkaloids of Geissospermum vellosii. Isolation and structure determinations of Vellosimine, Vellosiminol, and Geissolosimine. J. Org. Chem. 1962, 27, 2981–2985. [Google Scholar] [CrossRef]
- Bertho, A.; von Schuckmann, G.; Schönberger, W. Kurchi-Alkaloide, I. Mitteil.: Über einige neue Basen aus Holarrhena anti-dysenterica. Ber. Dtsch. Chem. Ges. 1933, 66, 786–790. [Google Scholar] [CrossRef]
- Ferreira, F.S.; Brito, S.V.; Ribeiro, S.C.; Saraiva, A.A.; Almeida, W.O.; Alves, R.R. Animal-based folk remedies sold in public markets in Crato and Juazeiro do Norte, Ceará, Brazil. BMC Complement. Altern. Med. 2009, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Arthur, H.R.; Johns, S.R.; Lamberton, J.A.; Loo, S.N. Identification of Vellosimine and Peraksine, and demonstration from N.M.R. Data that Peraksine is a mixture of two epimers. Aust. J. Chem. 1968, 21, 1399–1401. [Google Scholar] [CrossRef]
- Deng, R.X.; Yu, L.B.; Zhang, H.B.; Geng, R.L.; Ye, K.L.; Zhang, D.F. Studied on antimalarial agents-α-(alkylaminomethyl)-halogenated-4-fluorinemetha-nol. Acta Pharm. Sin. 1981, 16, 920–924. [Google Scholar]
- Chatterjee, A.; Bandyopadhyay, S. Vellosimine, an alkaloid of Rauwolfia vomitoria. Indian J. Chem. 1979, 18B, 87–88. [Google Scholar]
- Sierra, P.; Novotny, L.; Samek, Z.; Budesinsky, M.; Dolejs, L.; Blaha, K. Alkaloids of Rauvolfia salicifolia Griseb species. Collect. Czech. Chem. Commun. 1982, 47, 2912–2921. [Google Scholar] [CrossRef]
- Lin, M.; Yang, B.Q.; Yu, D.Q. Studies on the quaternary alkaloids of Rauvolfia verticillata. Acta Pharm. Sin. 1986, 21, 114–118. [Google Scholar]
- Ponglux, D.; Wongseripipatana, S.; Subhadhirasakul, S.; Takayama, H.; Yokota, M.; Ogata, K.; Phisalaphong, C.; Aimi, N.; Sakai, S. Studies on the indole alkaloids of Gelsem1um elegans (thailand): Structure elucidation and proposal of biogenetic route. Tetrahedron 1988, 44, 5075–5094. [Google Scholar] [CrossRef]
- Wang, T.; Cook, J.M. General approach for the synthesis of Sarpagine/Ajmaline indole alkaloids. Stereospecific total synthesis of the Sarpagine alkaloid (+)-vellosimine. Org. Lett. 2000, 2, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wang, T.; Yu, F.; Cook, J.M. General approach for the synthesis of Macroline/Sarpagine related indole alkaloids via the asymmetric Pictet-Spengler reaction: The enantiospecific synthesis of the Na-H, azabicyclo[3.3.1]nonone template. Tetrahedron Lett. 1997, 38, 6819–6822. [Google Scholar] [CrossRef]
- Kondo, Y.; Kojima, S.; Sakamoto, T. General and facile synthesis of indoles with Oxygen-bearing substituents at the benzene moiety. J. Org. Chem. 1997, 62, 6507–6511. [Google Scholar] [CrossRef]
- Ma, C.; Liu, X.; Li, X.; Flippen-Anderson, J.; Yu, S.; Cook, J.M. Efficient asymmetric synthesis of biologically important Tryptophan analogues via a Palladium-mediated Heteroannulation reaction. J. Org. Chem. 2001, 66, 4525–4542. [Google Scholar] [CrossRef] [PubMed]
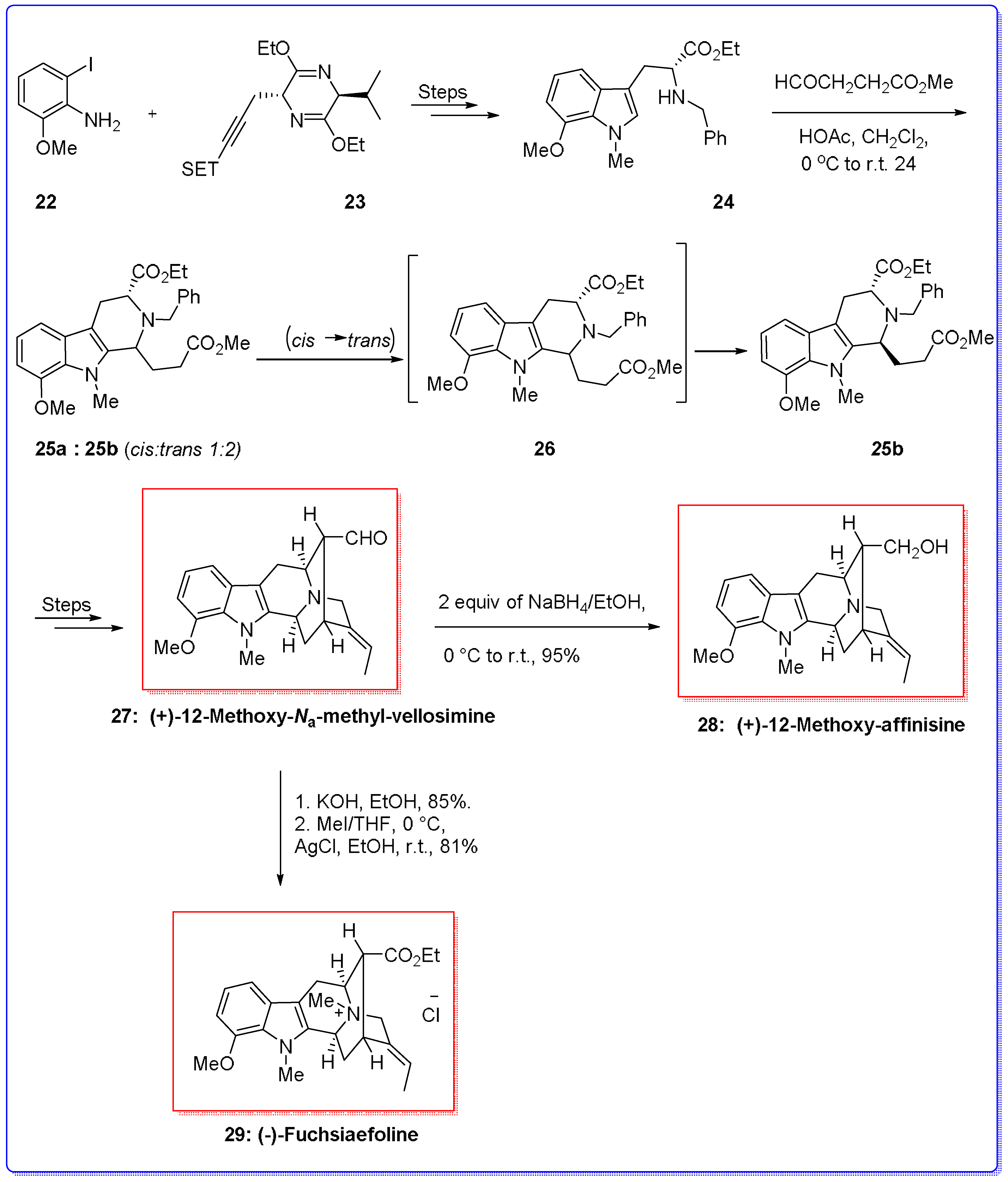
- Zhou, H.; Liao, X.; Cook, J.M. Regiospecific, enantiospecific total synthesis of the 12-alkoxy-substituted indole alkaloids, (+)-12-methoxy-Na-methylvellosimine, (+)-12-Methoxyaffinisine, and (−)-Fuchsiaefoline. Org. Lett. 2004, 6, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.D.; Hamker, L.K.; Li, J.; Yu, P.; Czerwinski, K.M.; Deng, L.; Bennett, D.W.; Cook, J.M.; Watson, W.H.; Krawiec, M. Enantiospecific formation of trans 1,3-disubstituted tetrahydro-β-carbolines by the Pictet−Spengler reaction and conversion of cis diastereomers into their trans counterparts by scission of the C-1/N-2 bond. J. Org. Chem. 1997, 62, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Trudell, M.L.; Hollinshead, S.P.; Cook, J.M. [3,3]-Sigmatropic rearrangements in indoloazabicyclo[3.3.1]nonene systems. Reversal of the stereofacial selectivity in the Claisen vs. The ortho Ester Claisen rearrangement. J. Am. Chem. Soc. 1989, 111, 8263–8265. [Google Scholar] [CrossRef]
- Elderfield, R.C.; Gilman, R.E. Characterization of the Lipids of some Orchids. Phytochemistry 1972, 11, 339–343. [Google Scholar] [CrossRef]
- Keawpradub, N.; Houghton, P.J.; Eno-Amooquaye, E.; Burke, P. Activity of extracts and alkaloids of Thai Alstonia species against human lung cancer cell lines. J. Planta Med. 1997, 63, 97–101. [Google Scholar] [CrossRef]
- Keawpradub, N.; Kirby, G.C.; Steele, J.C.P.; Houghton, P. Antiplasmodial activity of extracts and alkaloids of three Alstonia species from Thailand. J. Planta Med. 1999, 65, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.E.; Cook, J.M.; Le Quesne, P.W. Biomimetic synthesis of the bisindole alkaloids Villalstonine and Alstonisidine. J. Am. Chem. Soc. 1973, 95, 546–552. [Google Scholar] [CrossRef]
- Esmond, R.W.; Le Quesne, P.W. Biomimetic synthesis of Macroline. J. Am. Chem. Soc. 1980, 102, 7116–7117. [Google Scholar] [CrossRef]
- Burke, D.E.; Cook, G.A.; Cook, J.M.; Haller, K.G.; Lazar, H.A.; Le Quesne, P.W. Further alkaloids of Alstonia muelleriana. Phytochemistry 1973, 12, 1467–1474. [Google Scholar] [CrossRef]
- Garnick, R.L.; Le Quesne, P.W. Biomimetic transformations among monomeric Macroline-related indole alkaloids. J. Am. Chem. Soc. 1978, 100, 4213–4219. [Google Scholar] [CrossRef]
- Hesse, M.; Bodmer, F.; Gemenden, C.W.; Joshi, B.S.; Taylor, W.I.; Schmid, H. Die struktur des Alstonia-alkaloides Villalstonin. Helv. Chim. Acta. 1966, 49, 1173–1179. [Google Scholar] [CrossRef]
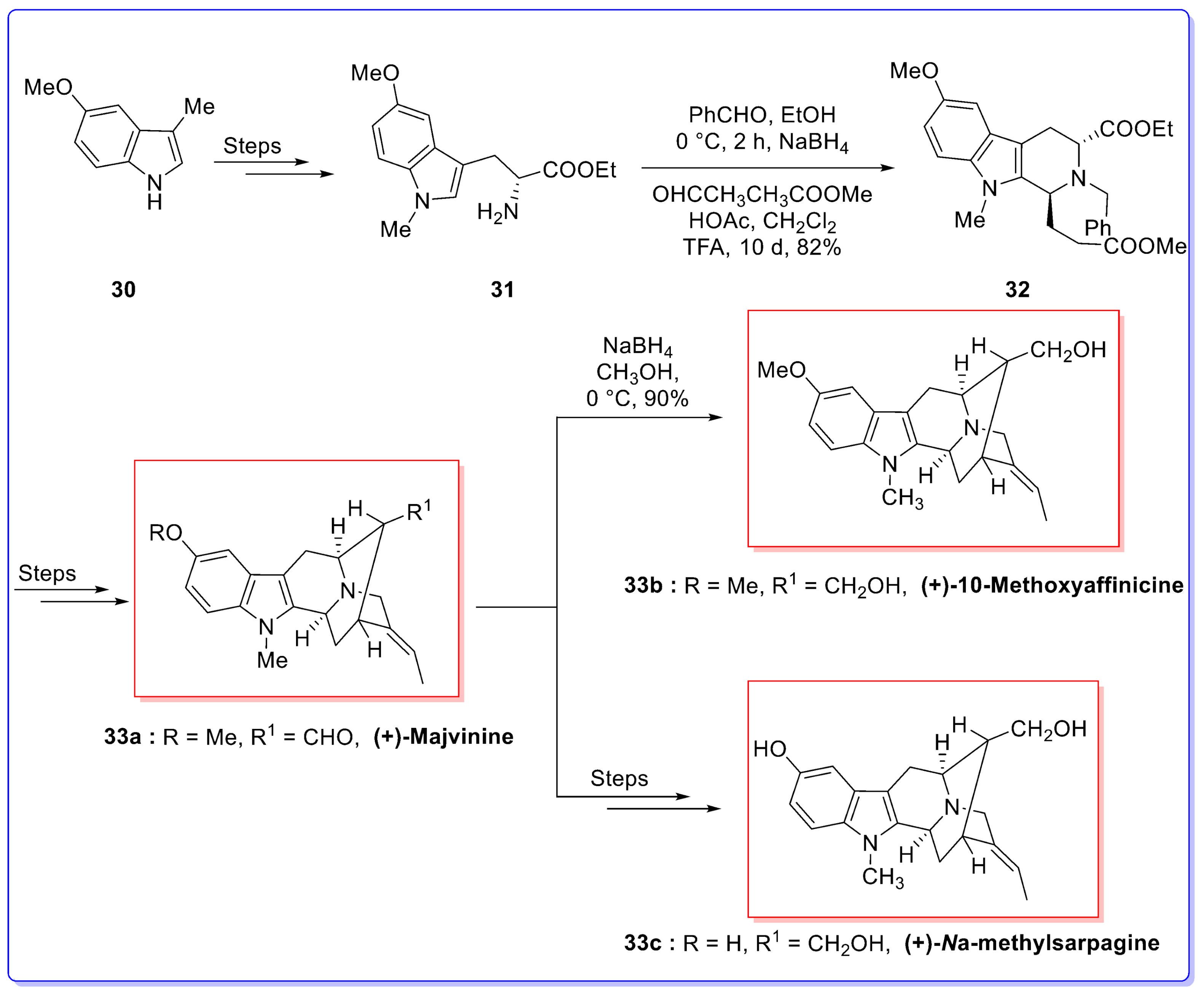
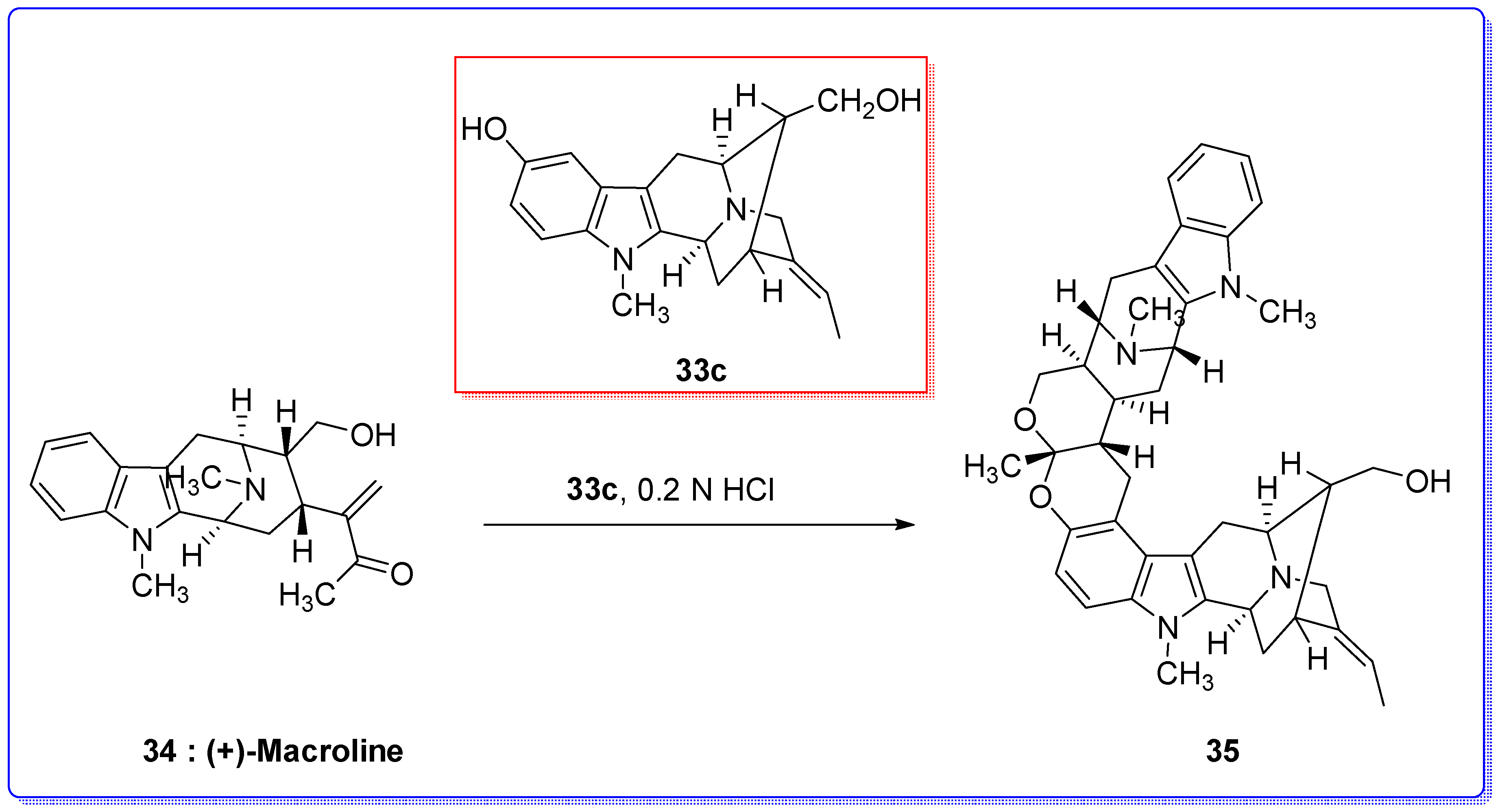
- Zhao, S.; Liao, X.; Cook, J.M. Enantiospecific, stereospecific total synthesis of (+)-Majvinine, (+)-10-methoxyaffinisine, and (+)-Na-methylsarpagine as well as the total synthesis of the Alstonia bisindole macralstonidine. Org. Lett. 2002, 4, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zhao, S.; Cook, J.M. The enantiospecific total synthesis of the Alstonia bisindole alkaloid, Macralstonidine. Presented at the National Organic Symposium, Bozeman, MT, USA, 10–15 June 2001; p. 239. [Google Scholar]
- Heath-Brown, B.; Philpott, P.G. The Fischer indole synthesis. J. Chem. Soc. 1965, 7185–7193. [Google Scholar] [CrossRef]
- Abramovitch, R.A.; Shapiro, D.S. 880. Tryptamines, carbolines, and related compounds. Part II. A convenient synthesis of tryptamines and β-carbolines. J. Chem. Soc. 1956, 4589–4592. [Google Scholar] [CrossRef]
- Angenot, L. Nouveaux alcaloïdes oxindoliques du Strychnos usambarensis GILG. Plant Med. Phytother. 1978, 12, 123–129. [Google Scholar]
- Bassleer, R.; Depauw-Gillet, M.C.; Massart, B.; Marnette, J.-M.; Wiliquet, P.; Caprasse, M.; Angenot, L. Effets de trois alcalóides extraits du Strychnos usambaren- sis sur des cellules cancéreuses en culture. Planta Med. 1982, 45, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.-B.; Kakeya, H.; Osada, H. Spirotryprostatin B, a novel Mammalian cell cycle inhibitor produced by Aspergillus fumigatus. J. Antibiot. 1996, 49, 832–835. [Google Scholar] [CrossRef] [PubMed]
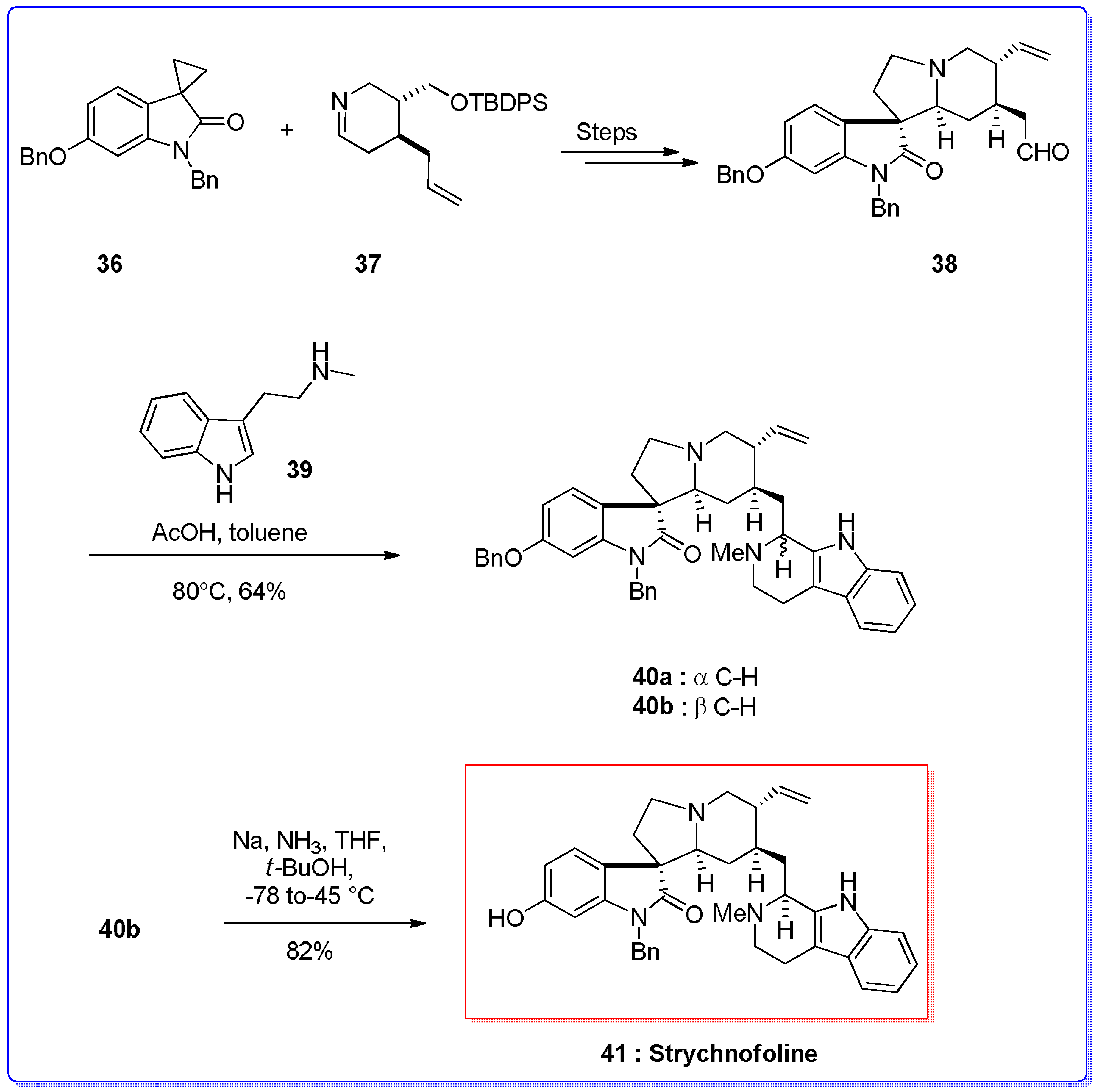
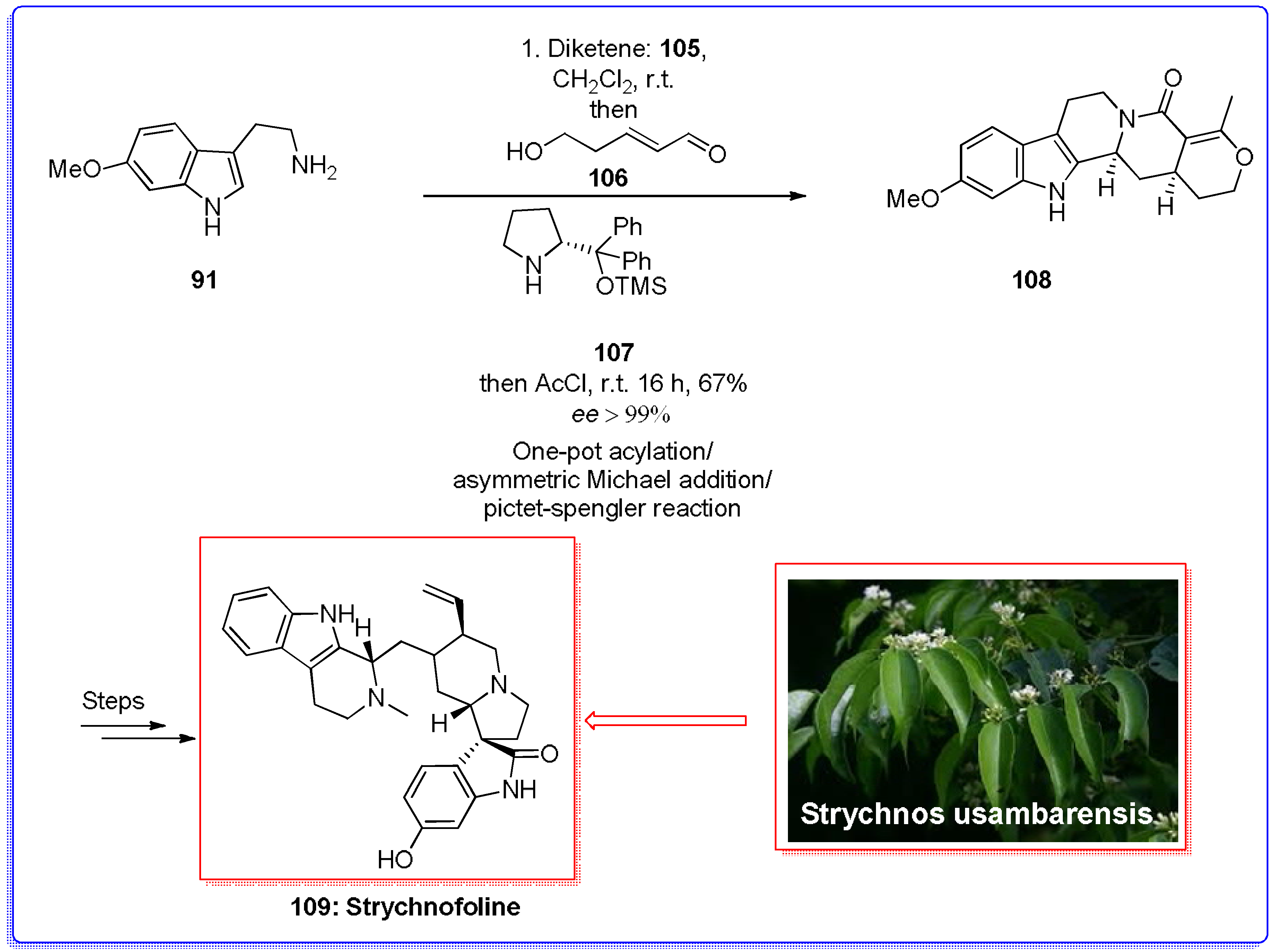
- Lerchner, A.; Carreira, E.M. First total synthesis of (±)-Strychnofoline via a highly selective ring-expansion reaction. J. Am. Chem. Soc. 2002, 124, 14826–14827. [Google Scholar] [CrossRef] [PubMed]
- Burm, B.E.A.; Meijler, M.M.; Korver, J.; Wanner, J.M.; Koomen, G. Synthesis of the brominated marine alkaloids (±)-Arborescidine A, B and C. Tetrahedron 1998, 54, 6135–6146. [Google Scholar] [CrossRef]
- Herdeis, C.; Hubmann, H.P. Synthesis of homochiral R-Baclofen from S-glutamic acid. Tetrahedron Asymmetry 1992, 3, 1213–1221. [Google Scholar] [CrossRef]
- Waldmann, H.; Schmidt, G.; Jansen, M.; Geb, J. Asymmetric steering of the Pictet-Spengler reaction by means of Amino-acid esters as chiral auxiliary groups. Tetrahedron 1994, 50, 11865–11884. [Google Scholar] [CrossRef]
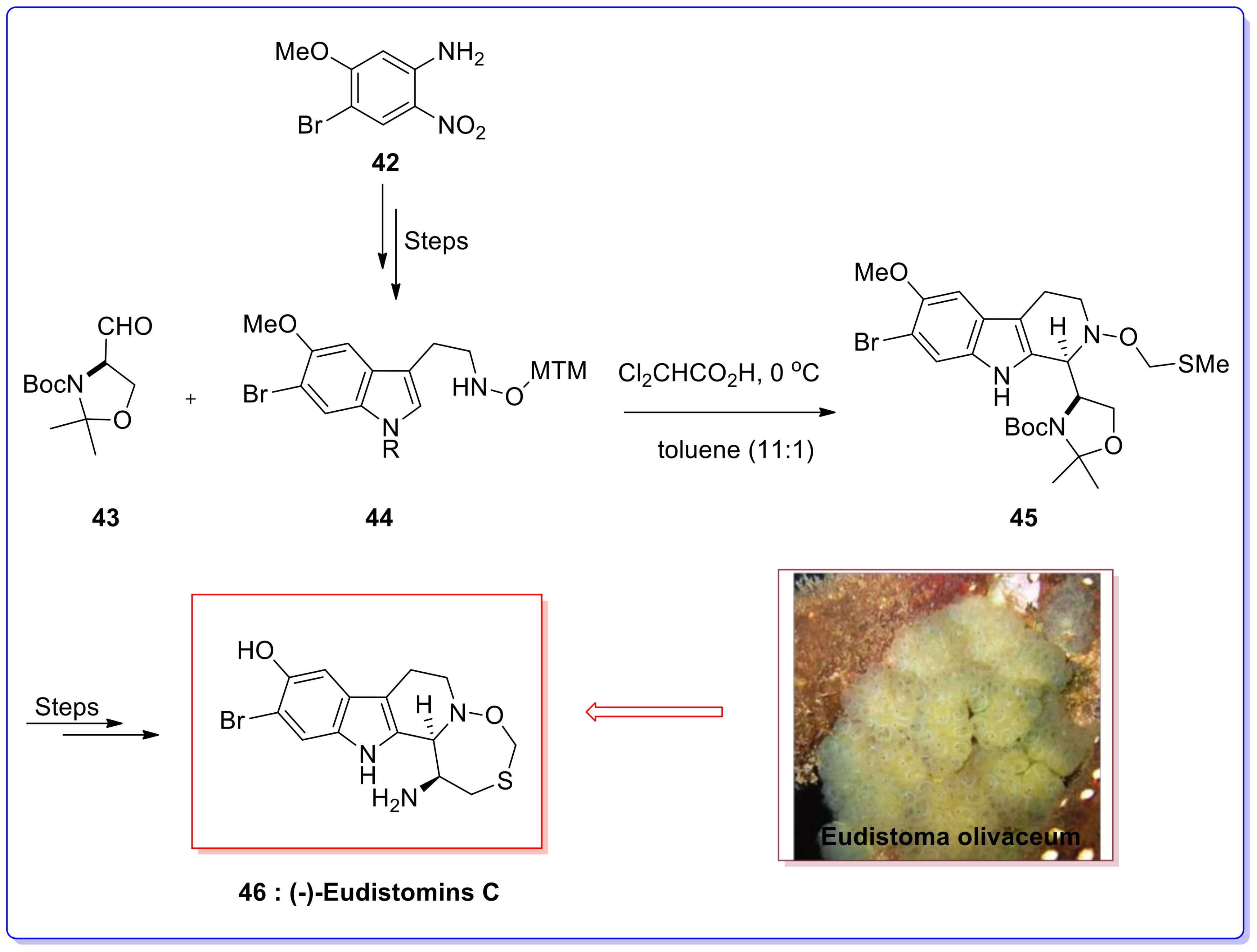
- Rinehart, K.L., Jr.; Kobayashi, J.; Harbour, G.C.; Hughes, R.G., Jr.; Mizsak, S.A.; Scahill, T.A. Eudistomins C, E, K, and l, potent antiviral compounds containing A novel Oxathiazepine ring from the Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1524–1526. [Google Scholar] [CrossRef]
- Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Rinehart, K.L., Jr. Eudistomins A, D, G, H, I, J, M, N, O, P, and Q, Bromo, Hydroxy, Pyrrolyl and iminoazepino. Beta.-carbolines from the antiviral Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1526–1528. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Mascal, M.; Holt, T.G.; Shield, L.S.; Lafargue, F. Eudistomins A–Q,. Beta.-carbolines from the antiviral Caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1987, 109, 3378–3387. [Google Scholar] [CrossRef]
- Lake, R.J.; Blunt, J.W.; Munro, M.H. Eudistomins from the New Zealand Ascidian Ritterella sigillinoides. Aust. J. Chem. 1989, 42, 1201–1206. [Google Scholar] [CrossRef]
- Kauffman, J.M.; Litak, P.T.; Boyko, W.T. Influence of borate complexation on the electrophoretic behavior of carbohydrates in capillary electrophoresis. J. Heterocycl. Chem. 1995, 32, 1541–1547. [Google Scholar] [CrossRef]
- Nakagawa, M.; Liu, J.-J.; Hino, T. Bimetallic solvated metal atom dispersed catalysts. New materials with low-temperature catalytic properties. J. Am. Chem. Soc. 1989, 11, 2721–2722. [Google Scholar] [CrossRef]
- Garner, P.; Park, J.M. 1,1,-dimethylethyl (s)- or (r)-4-formyl-2,2-dimethyl-3 oxazolidinecarboxylate: A useful serinal derivative. Org. Synth. 1992, 70, 18–20. [Google Scholar]
- Dondoni, A.; Perrone, D. Synthesis of 1,1-dimethylethyl (s)-4-formyl-2,2-dimethyl-3-oxazolidinecarboxylate by oxidation of the alcohol. Org. Synth. 1999, 77, 64–67. [Google Scholar]
- Yamashita, T.; Kawai, N.; Tokuyama, H.; Fukuyama, T. Stereocontrolled total synthesis of (−)-Eudistomin C. J. Am. Chem. Soc. 2005, 127, 15038–15039. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D. The Anti-Opium leaf. Pharm. J. 1907, 78, 453–454. [Google Scholar]
- Field, E. Xcviii.—Mitragynine and Mitraversine, two new alkaloids from species of Mitragyne. J. Chem. Soc. Trans. 1921, 119, 887–891. [Google Scholar] [CrossRef]
- Zacharias, D.E.; Rosenstein, R.D.; Jeffrey, G.A. The structure of Mitragynine hydroiodide. Acta Cryst. 1965, 18, 1039–1043. [Google Scholar] [CrossRef]
- Ponglux, D.; Wongseripipatana, S.; Takayama, H.; Kikuchi, M.; Kurihara, M.; Kitajima, M.; Aimi, N.; Sakai, S. A new indole alkaloid, 7 α-hydroxy-7H-mitragynine, from Mitragyna speciosa in Thailand. Planta Med. 1994, 60, 580–581. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takayama, H.; Ishikawa, H.; Aimi, N.; Ponglux, D.; Watanabe, K.; Horie, S. Partial agonistic effect of 9-hydroxycorynantheidine on mu-opioid receptor in the Guinea-pig ileum. Life Sci. 2006, 78, 2265–2271. [Google Scholar] [CrossRef] [PubMed]
- Mavar-Manga, H.; Quetin-Leclercq, J.; Labres, G.; Belem-Pinheiro, M.-L.; Da Rocha, A.F.I.; Angenot, L. 9-methoxygeissoschizol, an alkaloid from bark of Strychnos guianensis. Phytochemistry 1996, 43, 1125–1127. [Google Scholar] [CrossRef]
- West, R. New acritarch species from Holocene sediments in central West Greenland. Arch. Int. Pharmacodyn. Ther. 1937, 56, 81–88. [Google Scholar]
- Penelle, J.; Tits, M.; Christen, P.; Molgo, J.; Brandt, V.; Frederich, M.; Angenot, L. Quaternary indole alkaloids from the stem bark of Strychnos guianensis. Phytochemistry 2000, 53, 1057–1066. [Google Scholar] [CrossRef]
- Massiot, G.; Mulamba, T.J. Synthesis of the two enantioners of a tetrahydro-β-carboline from l-(−)-Tryptophan. Chem. Soc. Chem. Commun. 1983, 1147–1149. [Google Scholar] [CrossRef]
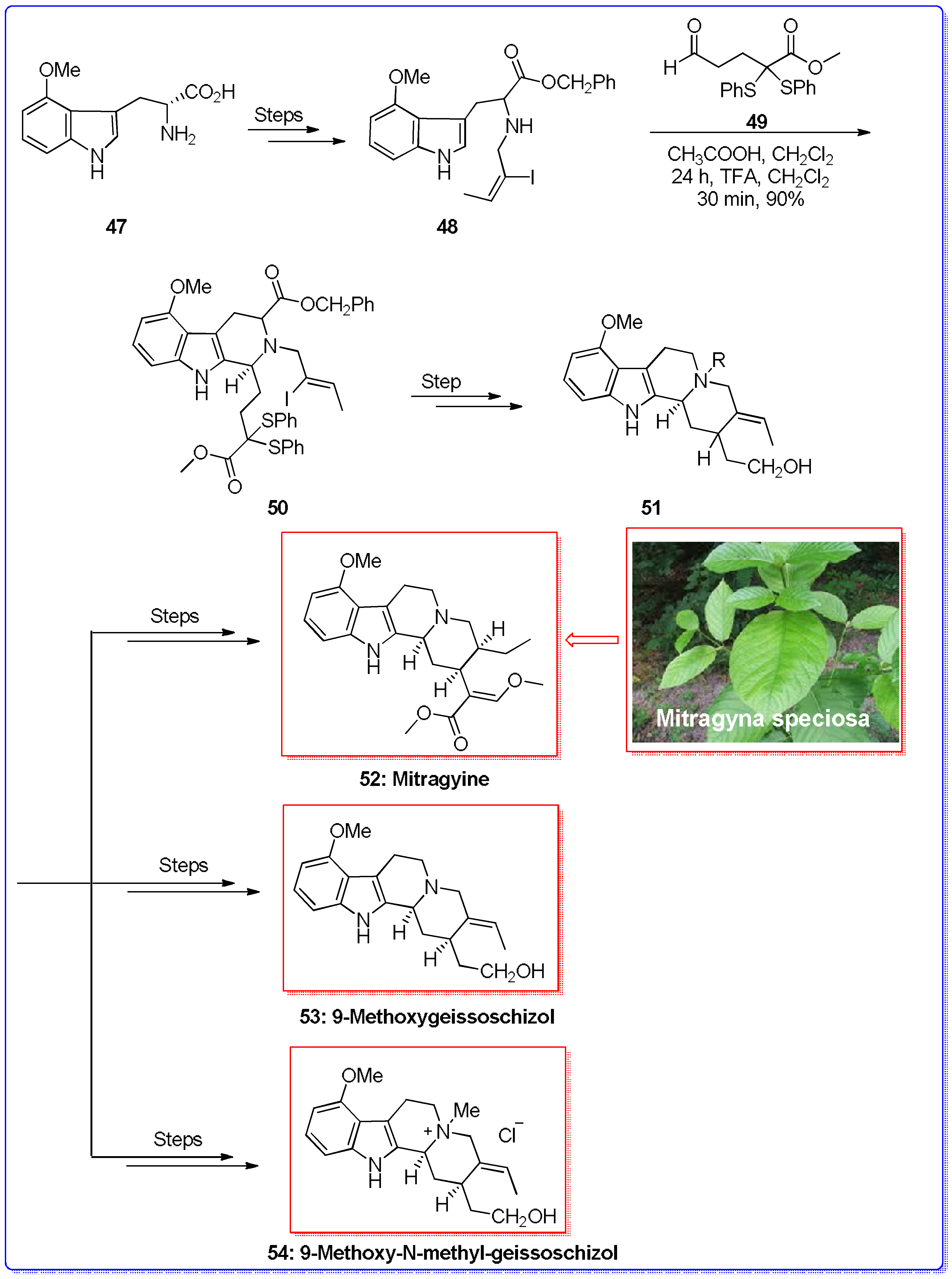
- Yu, S.; Berner, O.M.; Cook, J.M. General Approach for the Synthesis of Indole Alkaloids via the Asymmetric Pictet−Spengler Reaction: First Enantiospecific Total Synthesis of (−)-Corynantheidine as Well as the Enantiospecific Total Synthesis of (−)-Corynantheidol, (−)-Geissoschizol, and (+)-Geissoschizine. J. Am. Chem. Soc. 2000, 122, 7827–7828. [Google Scholar]
- Ma, J.; Yin, W.; Zhou, H.; Cook, J.M. Total synthesis of the opioid agonistic indole alkaloid Mitragynine and the first total syntheses of 9-methoxygeissoschizol and 9-methoxy-Nb-methylgeissoschizol. Org. Lett. 2007, 9, 3491–3494. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.T. In the Chemistry of Heterocyclic Compounds; Taylor, E.C., Ed.; John Wiley and Sons: New York, NY, USA; Chichester, UK; Brisbane, Australian; Toronto, ON, Canada; Singapore, 1983; Volume 25, Part 4; p. 147. [Google Scholar]
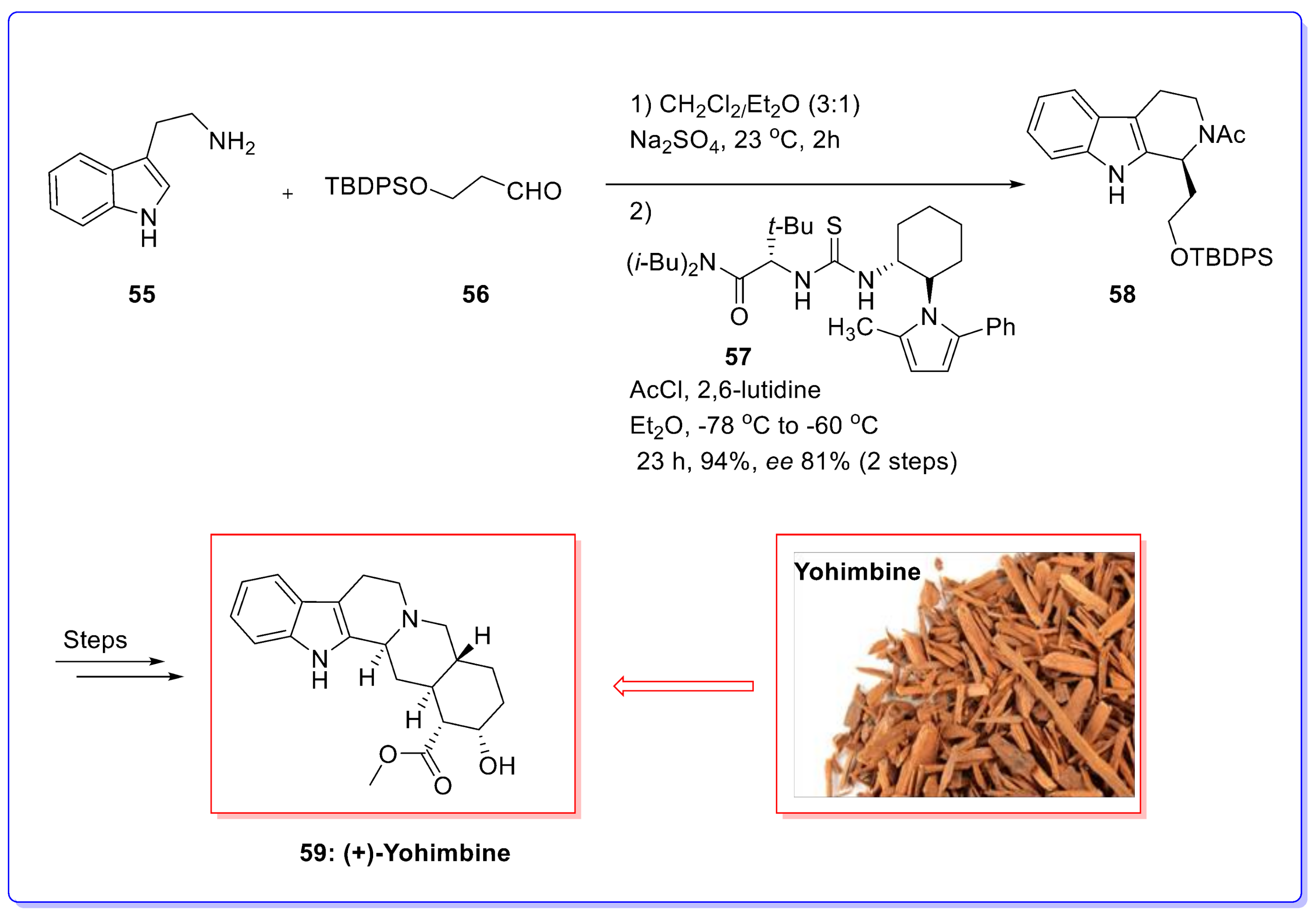
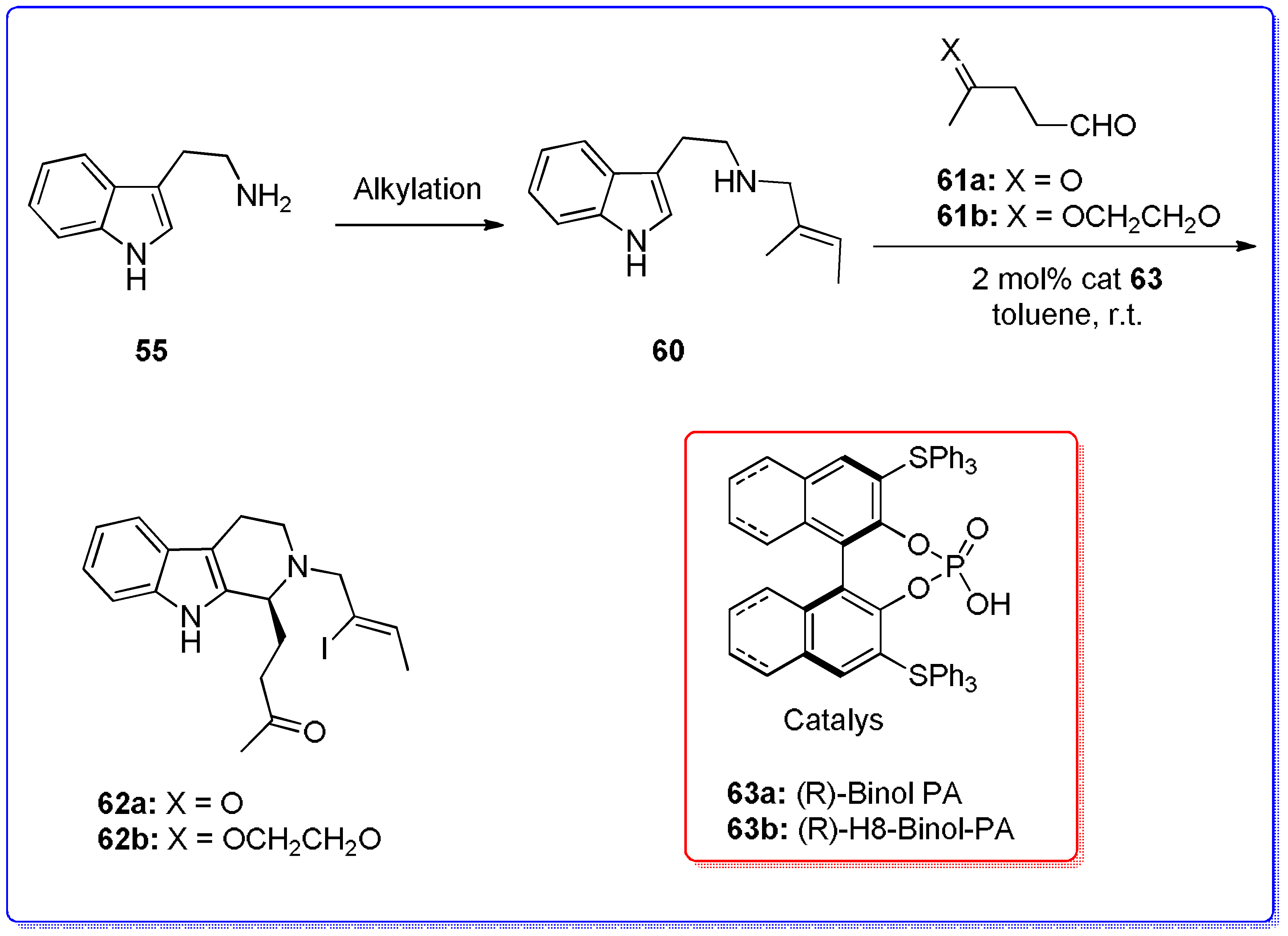
- Goldberg, M.R.; Robertson, D. Yohimbine: A pharmacological probe for study of the alpha 2-adrenoreceptor. Pharmacol. Rev. 1983, 35, 143–180. [Google Scholar] [PubMed]
- Mergott, D.J.; Zuend, S.J.; Jacobsen, E.N. Catalytic asymmetric total synthesis of (+)-Yohimbine. Org. Lett. 2008, 10, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Highly enantioselective catalytic Acyl-Pictet−Spengler reactions. J. Am. Chem. Soc. 2004, 126, 10558–10559. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Safonov, I.G.; Corbett, R.M. Total syntheses of (+)-zampanolide and (+)-dactylolide exploiting A unified strategy. J. Am. Chem. Soc. 2002, 124, 11102–11113. [Google Scholar] [CrossRef] [PubMed]
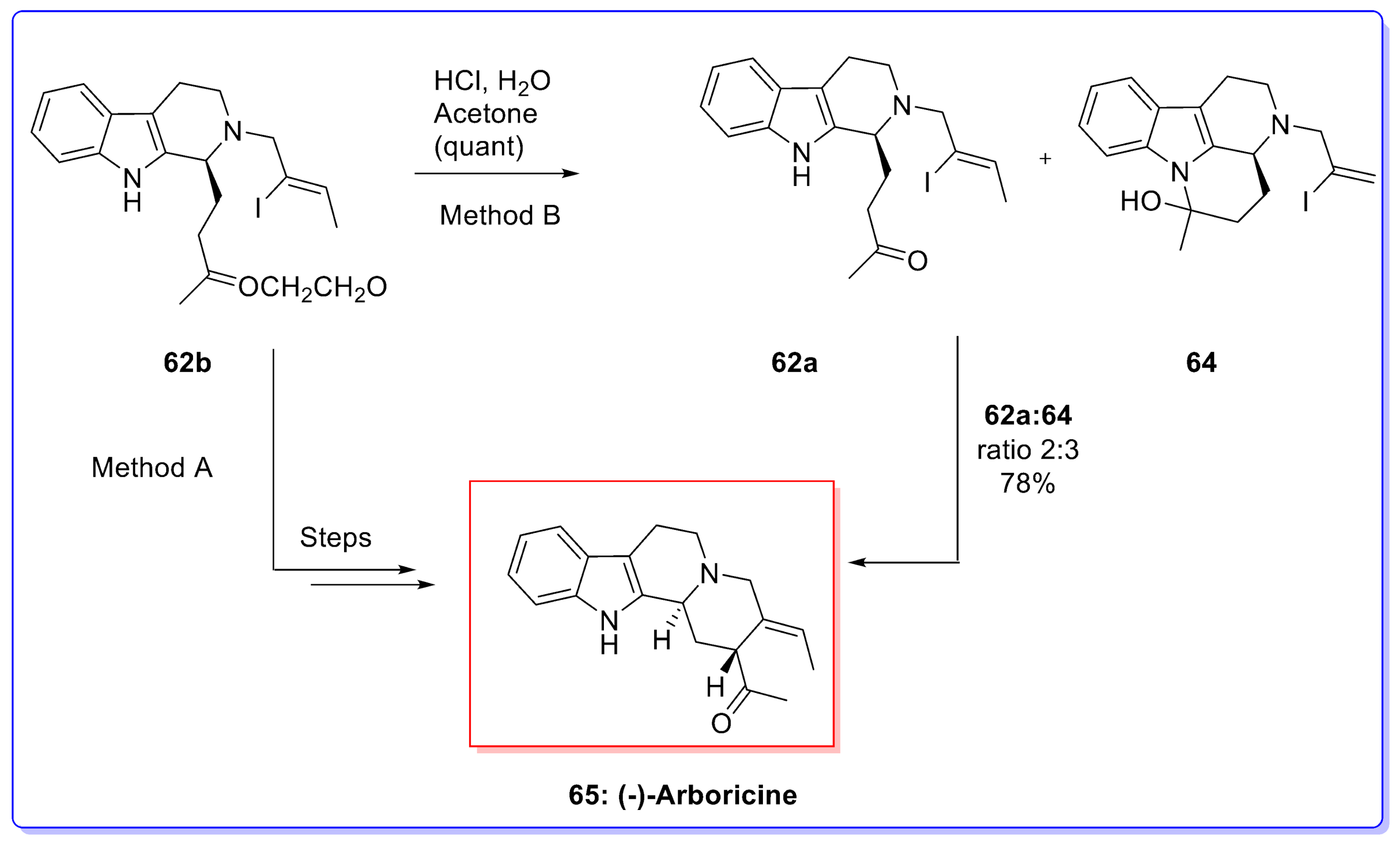
- Lim, K.H.; Komiyama, K.; Kam, T.S. Arboricine and Arboricinine, unusual tetracyclic indole regioisomers from Kopsia. Tetrahedron Lett. 2007, 48, 1143–1145. [Google Scholar] [CrossRef]
- Krafft, M.E.; Cran, J.W. A convenient protocol for the α-iodination of α,β-unsaturated carbonyl compounds with I2 in an aqueous medium. Synlett 2005, 8, 1263–1266. [Google Scholar] [CrossRef]
- Kumpaty, H.J.; van Linn, M.L.; Kabir, M.S.; Forsterling, F.H.; Deschamps, J.R.; Cook, J.M. Study of the cis to trans isomerization of 1-phenyl-2,3-disubstituted tetrahydro-β-carbolines at C(1). Evidence for the carbocation-mediated mechanism. J. Org. Chem. 2009, 74, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Wanner, M.J.; Boots, R.N.A.; Eradus, B.; Gelder, R.; van Maarseveen, J.H.; Hiemstra, H. Total synthesis of Arboricine. Org. Lett. 2009, 11, 2579–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Yang, R.; Cheng, Q.; Ofuji, K. Henrycinols A and B, two novel indole alkaloids isolated from melodinus Henryi Craib. Helv. Chim. Acta. 2003, 86, 415–419. [Google Scholar] [CrossRef]
- Rahman, A.U.; Basha, A. Indole Alkaloids; Hardwood Academic: Reading, UK, 1999. [Google Scholar]
- Iida, H.; Yamazaki, N.; Kibayashi, C. Total synthesis of (+)-Nojirimycin and (+)-1-Deoxynojirimycin. J. Org. Chem. 1987, 52, 3337–3342. [Google Scholar] [CrossRef]
- Araki, K.; Suenaga, K.; Sengoku, T.; Uemura, D. Total synthesis of Attenols A and B. Tetrahedron 2002, 58, 1983–1995. [Google Scholar] [CrossRef]
- Jawdosiuk, M.; Cook, J.M. Pictet-Spengler reactions in aprotic media. J. Org. Chem. 1984, 49, 2699–2701. [Google Scholar] [CrossRef]
- Sandrin, J.; Hollinshead, S.P.; Cook, J.M. Pictet-Spengler reactions in aprotic media. Stereospecificity in the Pictet-Spengler reaction. J. Org. Chem. 1989, 54, 5636–5640. [Google Scholar] [CrossRef]
- Van Linn, M.L.; Cook, J.M. Mechanistic studies on the cis to trans epimerization of trisubstituted 1,2,3,4-tetrahydro-β-carbolines. J. Org. Chem. 2010, 75, 3587–3599. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, N.; Ohno, M. An efficient o-monoalkylation of dimethyl l-tartrate viao-stannylene acetal with alkyl halides in the presence of cesium fluoride. Chem. Lett. 1987, 16, 141–144. [Google Scholar] [CrossRef]
- Nagashima, N.; Ohno, M. Selective monoalkylation of acyclic diols by means of dibutyltin oxide and fluoride salts. Chem. Pharm. Bull. 1991, 39, 1972–1982. [Google Scholar] [CrossRef]
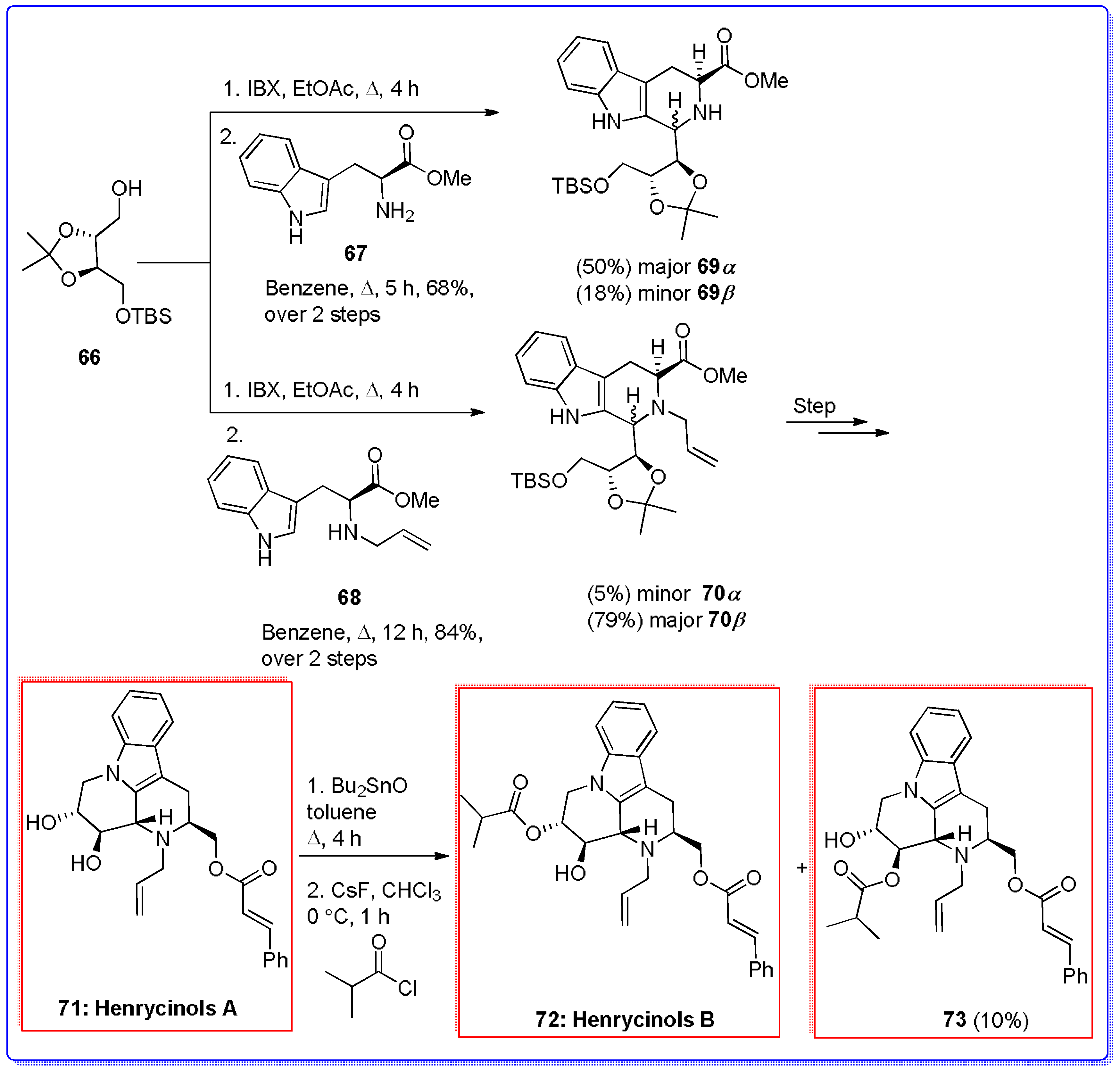
- Prasad, K.R.; Nidhiry, J.E.; Sridharan, M. Total synthesis of the indole alkaloids Henrycinol A and B. Tetrahedron 2014, 70, 4611–4616. [Google Scholar] [CrossRef]
- Saxton, J.E. Recent progress in the chemistry of the monoterpenoid indole alkaloids. Nat. Prod. Rep. 1996, 13, 327–363. [Google Scholar] [CrossRef]
- Bonoczk, P.; Panezel, G.; Nagy, Z. Vinpocetine increases Cerebral blood flow and Oxygenation in stroke patients: A near infrared spectroscopy and transcranial doppler study. Eur. J. Ultrasound 2002, 15, 85–91. [Google Scholar] [CrossRef]
- Sheng, M.H.; Sun, H.B. Research progress toward total synthesis, structural modification and biological evaluation of Vinpocetine. Prog. Pharm. Sci. 2010, 34, 7–17. [Google Scholar]
- Pfaffli, P.; Oppolzer, W.; Wenger, R.; Hauth, H. Stereoselektive synthese von optisch aktivem Vincamin. Helv. Chim. Acta. 1975, 58, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Node, M.; Nagasawa, H.; Fuji, K. Chiral total synthesis of indole alkaloids of the aspidosperma and hunteria types. J. Org. Chem. 1990, 55, 517–521. [Google Scholar] [CrossRef]
- Langlois, Y.; Pouilhes, A.; Genin, D.; Andriamialisoa, R.Z.; Langlois, N. New approaches to the synthesis of eburnane alkaloids. Tetrahedron 1983, 39, 3755–3761. [Google Scholar] [CrossRef]
- Ono, N.; Yoshimura, T.; Saito, T.; Tamura, R.; Tanikaga, R.; Kaji, A. Alkylation and acylation of active methylene compounds using 1,8-diazabicyclo[5.4.0]undec-7-ene as a base. Bull. Chem. Soc. Jpn. 1979, 52, 1716–1719. [Google Scholar] [CrossRef]
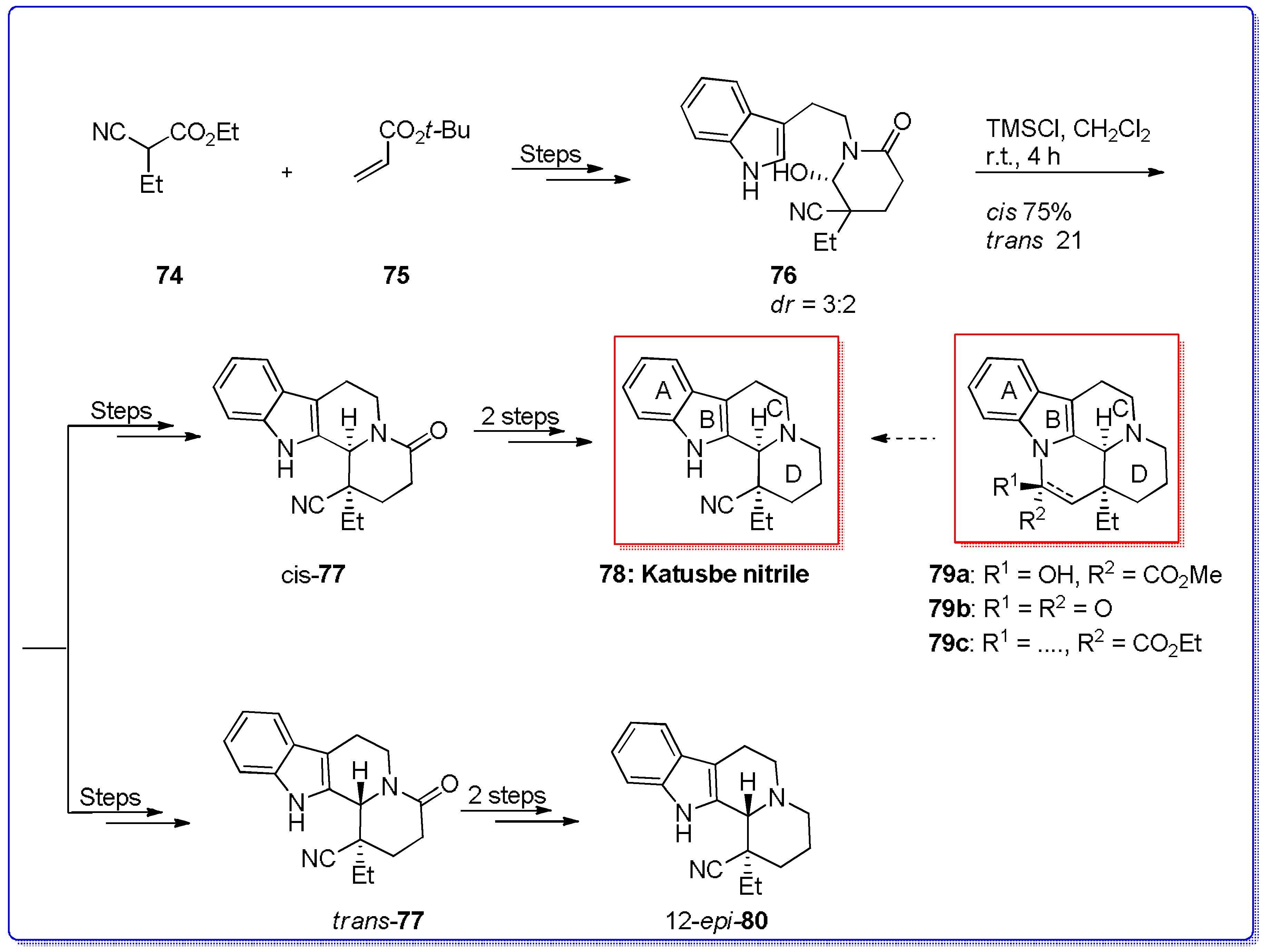
- Chen, X.; Chen, F. An efficient synthesis of Katsube nitrile: A key building block for Eburnamine-Vincamine alkaloids. Synthesis 2014, 46, 1506–1510. [Google Scholar] [CrossRef]
- Bencao, Z. An editorial committee of the administration bureau of traditional Chinese medicine. In Chinese Materia Medica (Zhonghua Benchao); Shanghai Science & Technology Press: Shanghai, China, 1999; Volume 3, p. 56. [Google Scholar]
- Noda, Y.; Mori, A. Antioxidant activities of Uyaku (Lindera Strychnifolia) leaf extract: A natural extract used in traditional medicine. J. Clin. Biochem. Nutr. 2007, 41, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.H.; Wu, T.; Zhang, Z.J.; Chou, G.X.; Wang, Z.T. Evaluation of antioxidant activity of radix Linderae and other two Chinese drugs using tlc-bioautography. Acta Pharm. Sin. 2006, 41, 956–962. [Google Scholar]
- Wang, N.Y.; Minatoguchi, S.; Arai, M.; Uno, Y.; Hashimoto, K.; Chen, X.H.; Fukuda, K.; Akao, S.; Takemura, G.; Fujiwara, H. Lindera strychnifolia is protective against post-ischemic myocardial dysfunction through scavenging hydroxyl radicals and opening the Mitochondrial KATP channels in isolated rat hearts. Am. J. Chin. Med. 2004, 32, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.I.; Meyers, M.J.; Anderson, D.R.; Hegde, S.; Mahoney, M.W.; Vernier, W.F.; Buchler, I.P.; Wu, K.K.; Yang, S.; Hartmann, S.J.; et al. Novel tetrahydro-β-carboline-1-carboxylic acids as inhibitors of Mitogen activated protein Kinase-activated protein Kinase 2 (mk-2). Bioorg. Med. Chem. Lett. 2007, 17, 4657–4663. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.D.; Londregan, A.T.; Ragains, J.R.; Hamann, M.T. Synthesis and biological evaluation of manzamine analogues. Org. Lett. 2006, 8, 3407–3409. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Drees, S.; Geiss, B.; Schmidt, P. Vanadium(V)-catalyzed oxidation of (3R)-Linalool- the selective formation of furanoid Linalool oxides and their conversion into Isocyclocapitelline derivatives. Synlett 2003, 2, 223–226. [Google Scholar] [CrossRef]
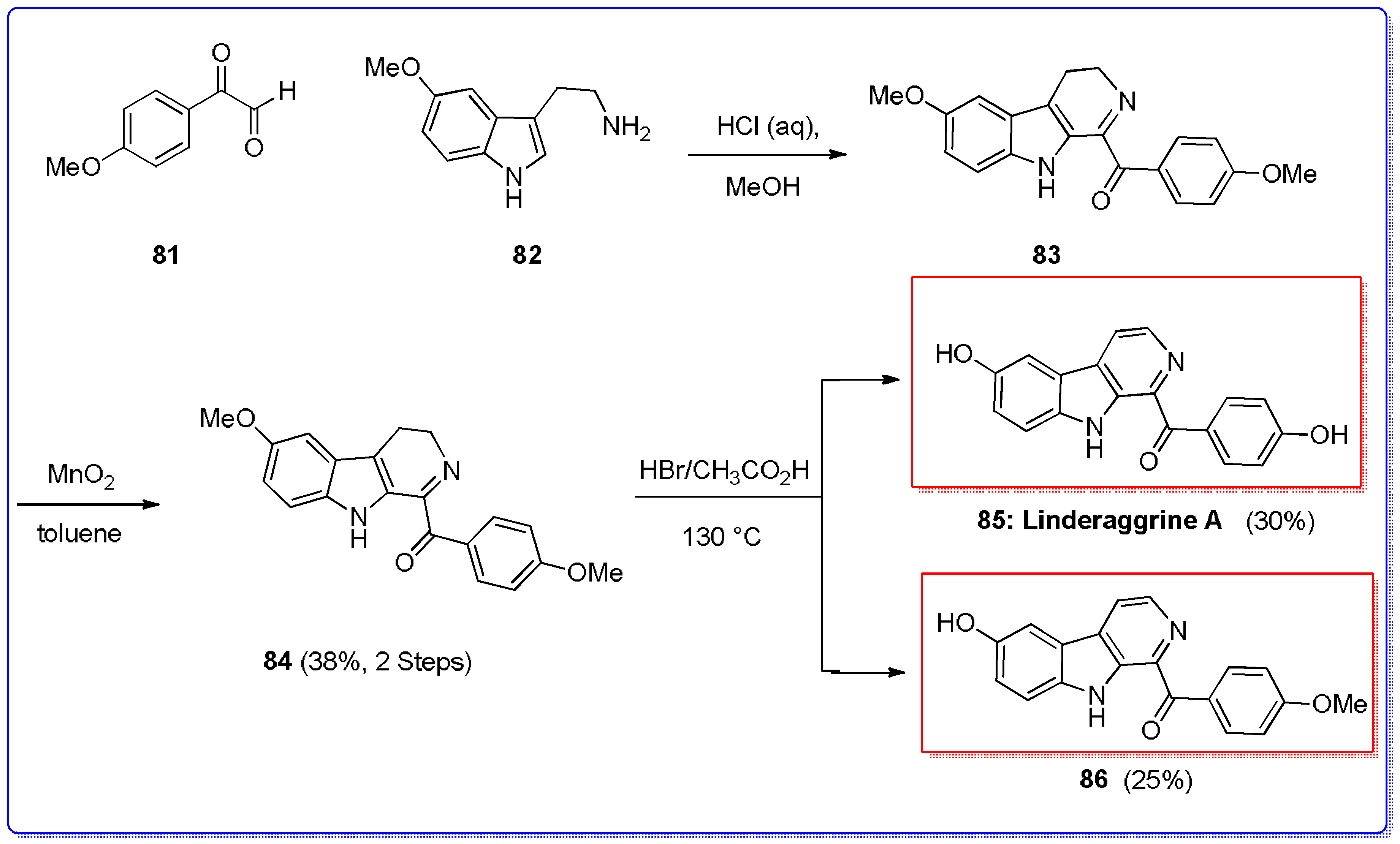
- Yang, M.L.; Kuo, P.C.; Damu, A.G.; Chang, R.J.; Chiou, W.F.; Wu, T.S. A versatile route to the synthesis of 1-substituted β-carbolines by a single step Pictet-Spengler cyclization. Tetrahedron 2006, 62, 10900–10906. [Google Scholar] [CrossRef]
- Kuo, P.C.; Li, Y.-C.; Hwang, T.-L.; Ma, G.-H.; Yang, M.-L.; Lee, E.-J.; Wu, T.-S. Synthesis and structural characterization of an anti-inflammatory principle purified from Lindera aggregata. Tetrahedron Lett. 2014, 55, 108–110. [Google Scholar] [CrossRef]
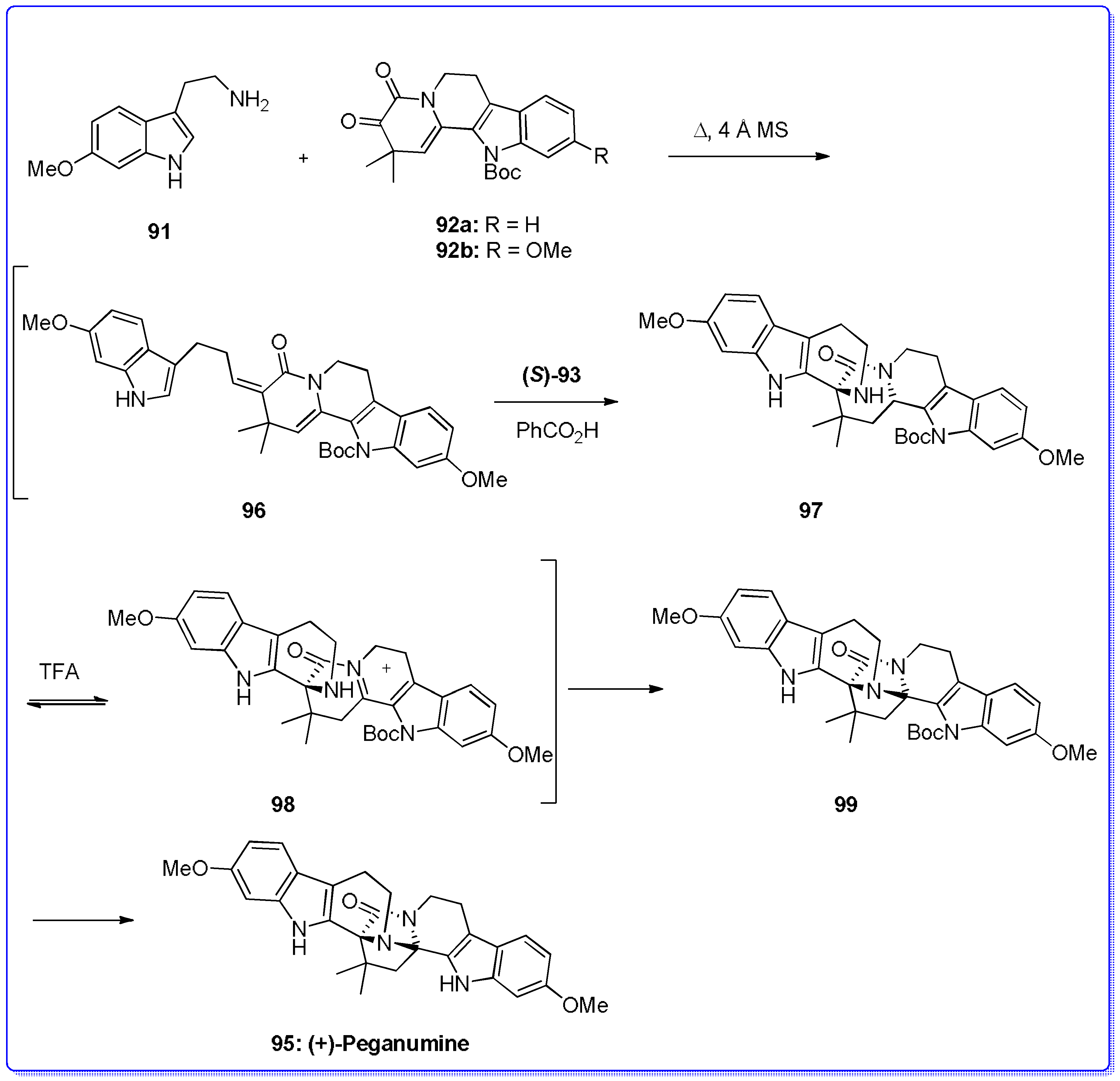
- Wang, K.-B.; Di, Y.-T.; Bao, Y.; Yuan, C.-M.; Chen, G.; Li, D.-H.; Bai, J.; He, H.-P.; Hao, X.-J.; Pei, Y.-H.; et al. Peganumine A, a β-carboline dimer with a new octacyclic scaffold from Peganum harmala. Org. Lett. 2014, 16, 4028–4031. [Google Scholar] [CrossRef] [PubMed]
- Farina, V.; Kapadia, S.; Krishnan, B.; Wang, C.; Liebeskind, L.S. On the nature of the “Copper effect” in the Stille cross-coupling. J. Org. Chem. 1994, 59, 5905–5911. [Google Scholar] [CrossRef]
- Banfi, L.; Riva, R. The Passerini reaction. Org. React. 2005, 65, 1–140. [Google Scholar]
- Wittenberg, R.; Srogl, J.; Egi, M.; Liebeskind, L.S. Ketone synthesis under neutral conditions. Cu(I) diphenylphosphinate-mediated, Palladium-catalyzed coupling of thiol esters and organostannanes. Org. Lett. 2003, 5, 3033–3035. [Google Scholar] [CrossRef] [PubMed]
- Schönherr, H.; Leighton, J.L. Direct and highly enantioselective iso-Pictet–Spengler reactions with α-ketoamides: Access to underexplored indole core structures. Org. Lett. 2012, 14, 2610–2613. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Sun, D.X.; Seidel, D. Conjugate-base-stabilized brønsted acids: Catalytic enantioselective Pictet–Spengler reactions with unmodified Tryptamine. Org. Lett. 2014, 16, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Piemontesi, C.; Wang, Q.; Zhu, J. Enantioselective synthesis of (+)-Peganumine A. J. Am. Chem. Soc. 2016, 138, 11148–11151. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Chong, K.; Lim, K.; Lim, S.; Low, Y.; Kam, T. Arborisidine and Arbornamine, two monoterpenoid indole alkaloids with new polycyclic Carbon–Nitrogen skeletons derived from a common pericine precursor. Org. Lett. 2016, 18, 1618–1621. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, M.; Koyama, T.; Wu, Y.; Kogure, N.; Zhang, R.; Takayama, H. Kopsiyunnanines J1 and J2, new Strychnos-type homo-monoterpenoid indole alkaloids from Kopsia arborea. Nat. Prod. Commun. 2015, 10, 49–51. [Google Scholar] [PubMed]
- Kitajima, M.; Murakami, Y.; Takahashi, N.; Wu, Y.; Kogure, N.; Zhang, R.; Takayama, H. Asymmetric total synthesis of novel pentacyclic indole alkaloid, Kopsiyunnanine e, isolated from Kopsia arborea. Org. Lett. 2014, 16, 5000–5003. [Google Scholar] [CrossRef] [PubMed]
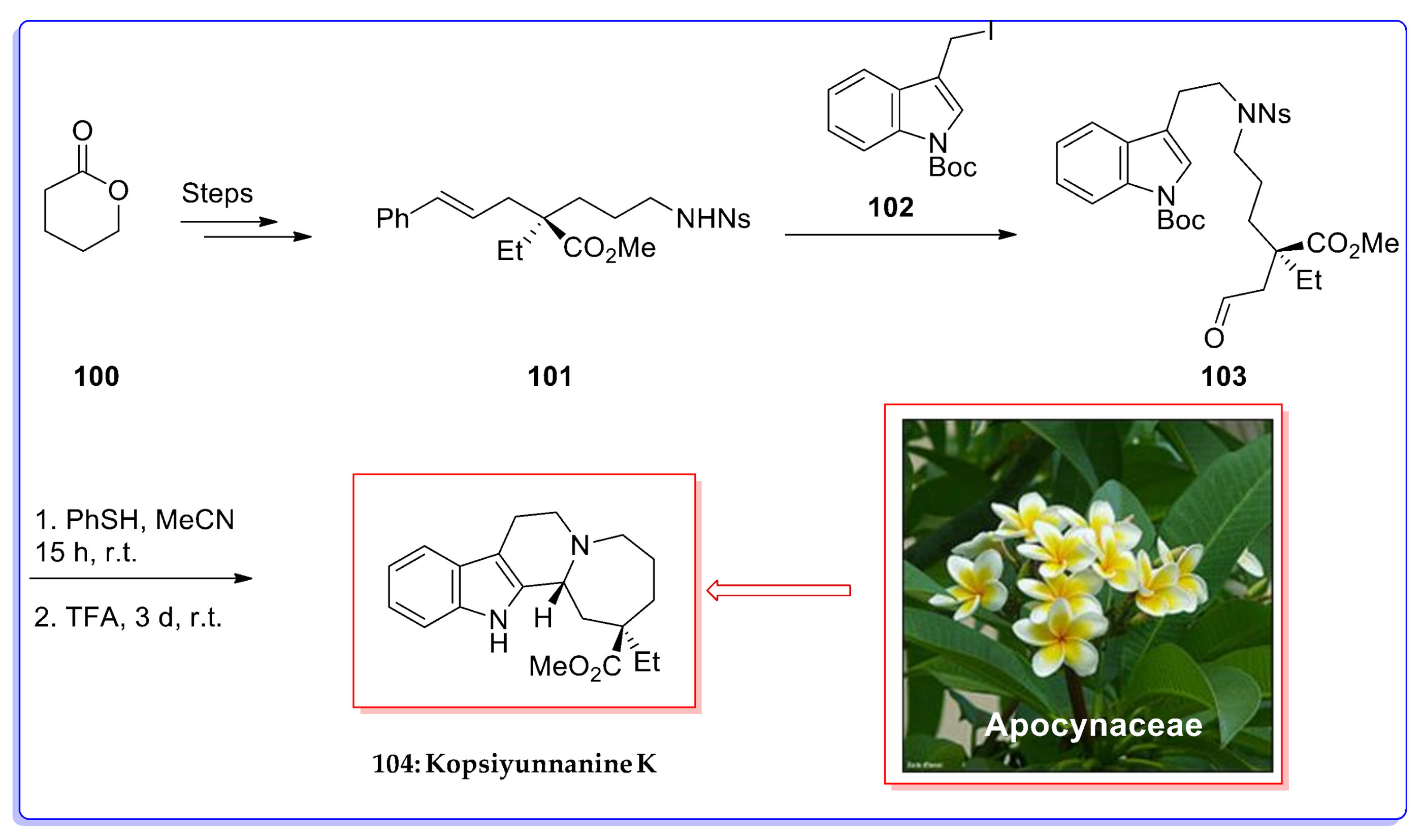
- Tokuda, R.; Okamoto, Y.; Koyama, T.; Kogure, N.; Kitajima, M.; Takayama, H. Asymmetric total synthesis of Kopsiyunnanine K, A monoterpenoid indole alkaloid with a rearranged skeleton. Org. Lett. 2016, 18, 3490–3493. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Danishefsky, S.J. Gelsemine: A thought-provoking target for total synthesis. Angew. Chem. Int. Ed. 2003, 42, 36–51. [Google Scholar] [CrossRef]
- Marti, C.; Carreira, E.M. Construction of spiro[pyrrolidine-3,3′-oxindoles]- recent applications to the synthesis of oxindole alkaloids. Eur. J. Org. Chem. 2003, 2209–2219. [Google Scholar] [CrossRef]
- Borges, J.; Manresa, M.T.; Martin Ramon, J.L.; Pascual, C.; Rumbero, A. Two new oxindole alkaloids isolated from Hamelia patens Jacq. Tetrahedron Lett. 1979, 20, 3197–3200. [Google Scholar] [CrossRef]
- Tsuda, M.; Kasai, Y.; Komatsu, K.; Sone, T.; Tanaka, M.; Mikamiand, Y.; Kobayashi, J. Citrinadin A, a novel pentacyclic alkaloid from Marine-derived Fungus Penicillium Citrinum. Org. Lett. 2004, 6, 3087–3089. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, F.L. Ueber einige Bestandtheile von Gelsemium sempervirens. Ber. Dtsch. Chem. Ges. 1876, 9, 1182–1186. [Google Scholar] [CrossRef]
- Bond, R.F.; Boeyens, J.C.A.; Holzapfel, C.W.; Steyn, P.S. Cyclopiamines A and B, novel oxindole metabolites of Penicillium cyclopium Westling. J. Chem. Soc. Perkin Trans. 1979, 1, 1751–1761. [Google Scholar] [CrossRef]
- Yu, Q.; Guo, P.; Jian, J.; Chen, Y.; Xu, J. Nine-step total synthesis of (−)-Strychnofoline. Chem. Commun. 2018, 54, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Schmeller, T.; Wink, M. Utilization of alkaloids in modern medicine. In Alkaloids: Biochemistry, Ecology, and Medicinal Applications; Roberts, M.F., Wink, M., Eds.; Plenum: New York, NY, USA, 1998; pp. 435–459. [Google Scholar]
- Kam, T.-S.; Choo, Y.-M.; Komiyama, K. Unusual spirocyclic Macroline alkaloids, nitrogenous derivatives, and a cytotoxic bisindole from Alstonia. Tetrahedron 2004, 60, 3957–3966. [Google Scholar] [CrossRef]
- Naranjo, J.; Pinar, M.; Hesse, M.; Schmid, H. Über die Indolalkaloide von Pleíocarpa talbotii Wernham. Helv. Chim. Acta. 1972, 55, 752–771. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Terrazas, C.; Acuña, U.N.; Ninh, T.N.; Chai, H.; de Blanco, E.J.C.; Soejarto, D.D.; Satoskar, A.R.; Kinghorn, A.D. Bioactive indole alkaloids isolated from Alstonia angustifolia. Phytochem. Lett. 2014, 10, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Kiang, A.; Loh, S.; Demanczyk, M.; Gemenden, C.; Papariello, G.; Taylor, W. The structures of peraksine (RP-5) and RP-7 constituents of the leaves and stems of Rauwolfia perakensis. Tetrahedron 1966, 22, 3293–3300. [Google Scholar] [CrossRef]
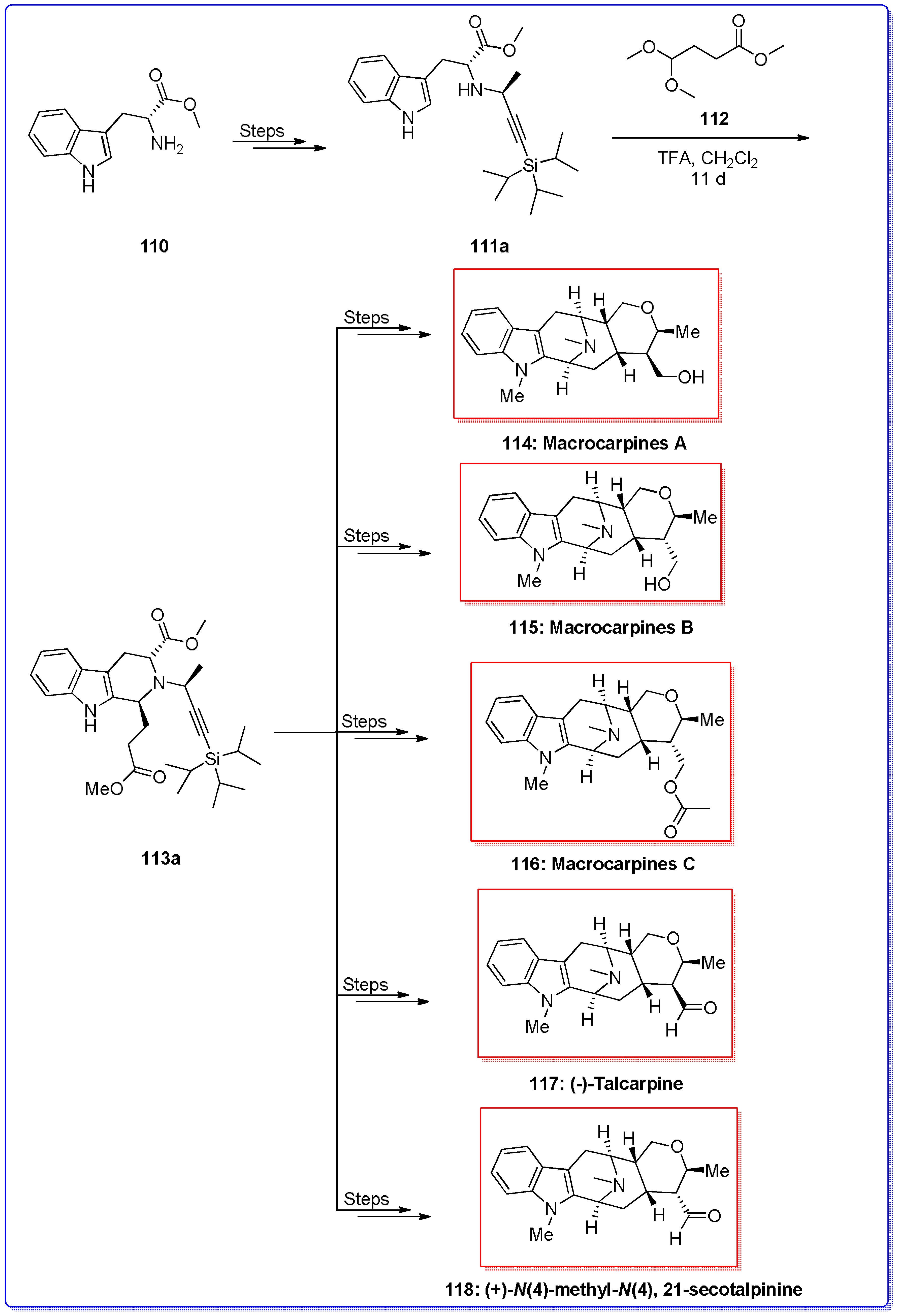
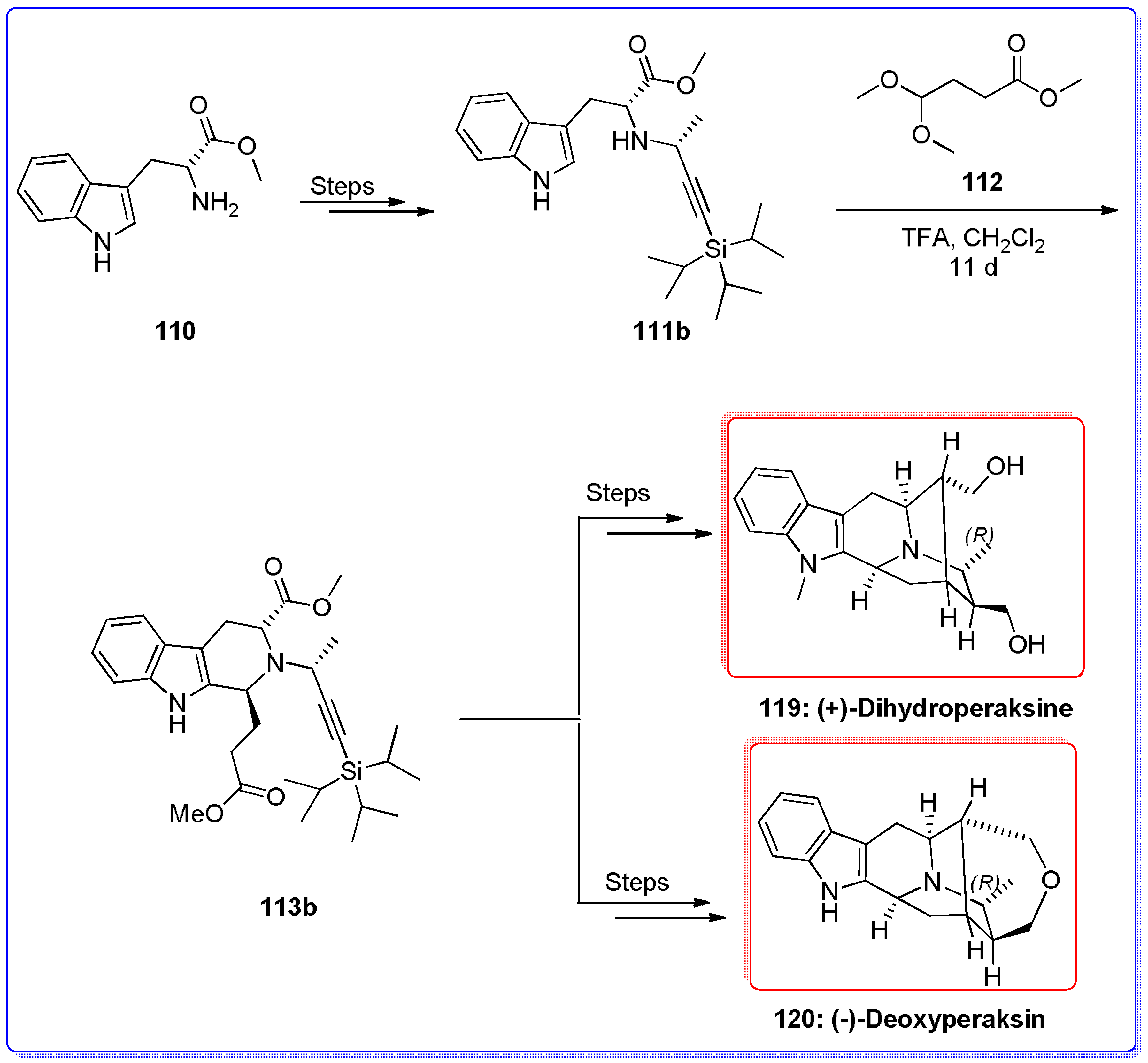
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Cook, J.M. Total synthesis of Sarpagine-related bioactive indole alkaloids. Chem. A Eur. J. 2017. [Google Scholar] [CrossRef] [PubMed]
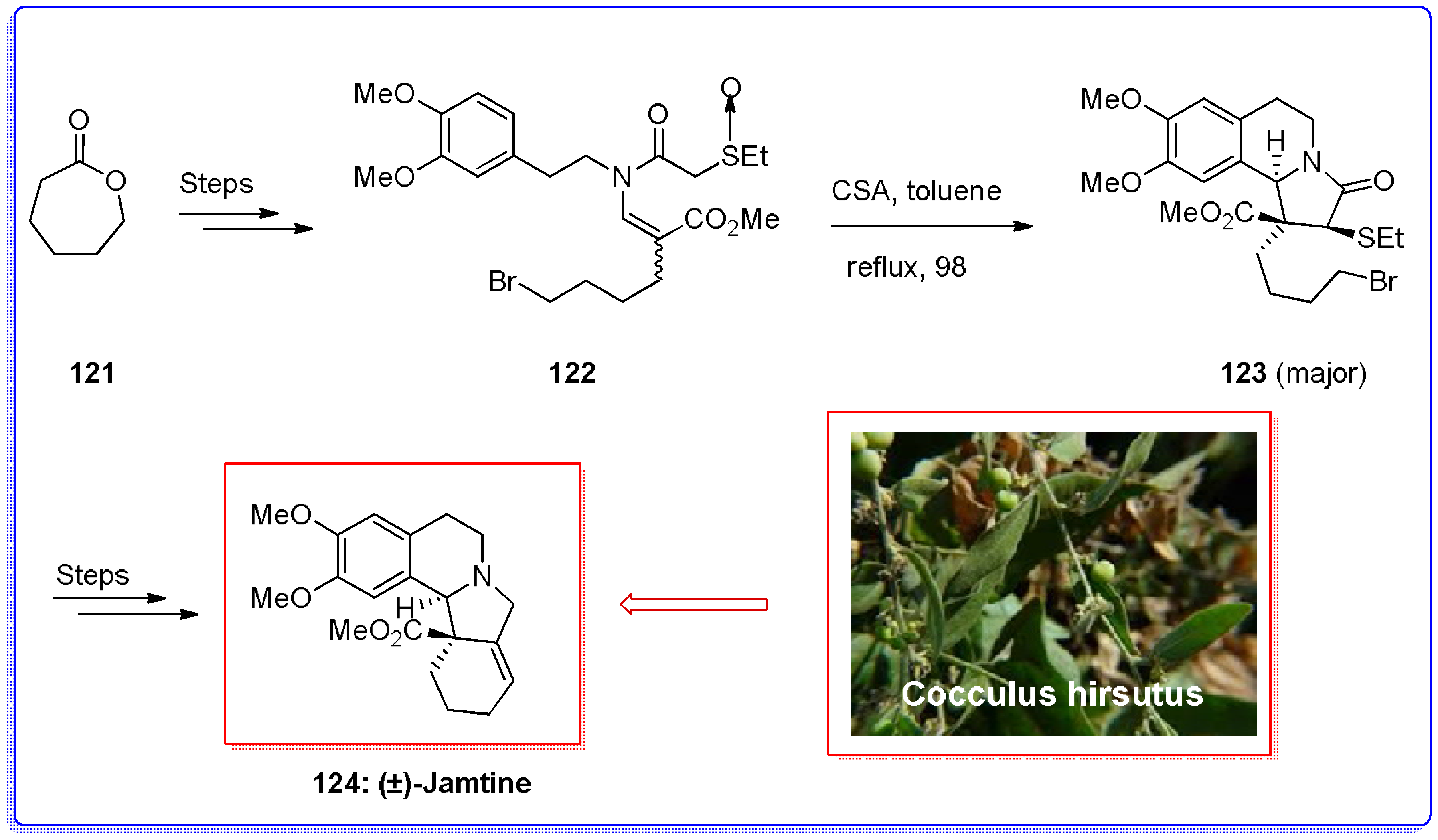
- Ahmad, V.U.; Iqbal, S. Jamtinine, an alkaloid from. Cocculus Hirsutus. Phytochemistry 1993, 3, 735–736. [Google Scholar] [CrossRef]
- Chopra, R.N.; Chopra, I.C.; Handa, K.L.; Kapoor, L.D. Indigenous Drugs of India; U. N. Dhar and Sons Pvt. Ltd.: Calcutta, India, 1958; p. 501. [Google Scholar]
- Ahmad, V.U.; Rahman, A.; Rasheed, T.; Rehman, H. Jamtine-N-oxide a new isoquinoline alkaloid from Cocculus hirsutus. Heterocycles 1987, 26, 1251–1255. [Google Scholar]
- Padwa, A.; Danca, M.D. Total synthesis of (±)-jamtine using a thionium/N-acyliminium ion Cascade. Org. Lett. 2002, 4, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E. Nazarov and related. Cationic Cyclizations. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1991; Volume 5, p. 751. [Google Scholar]
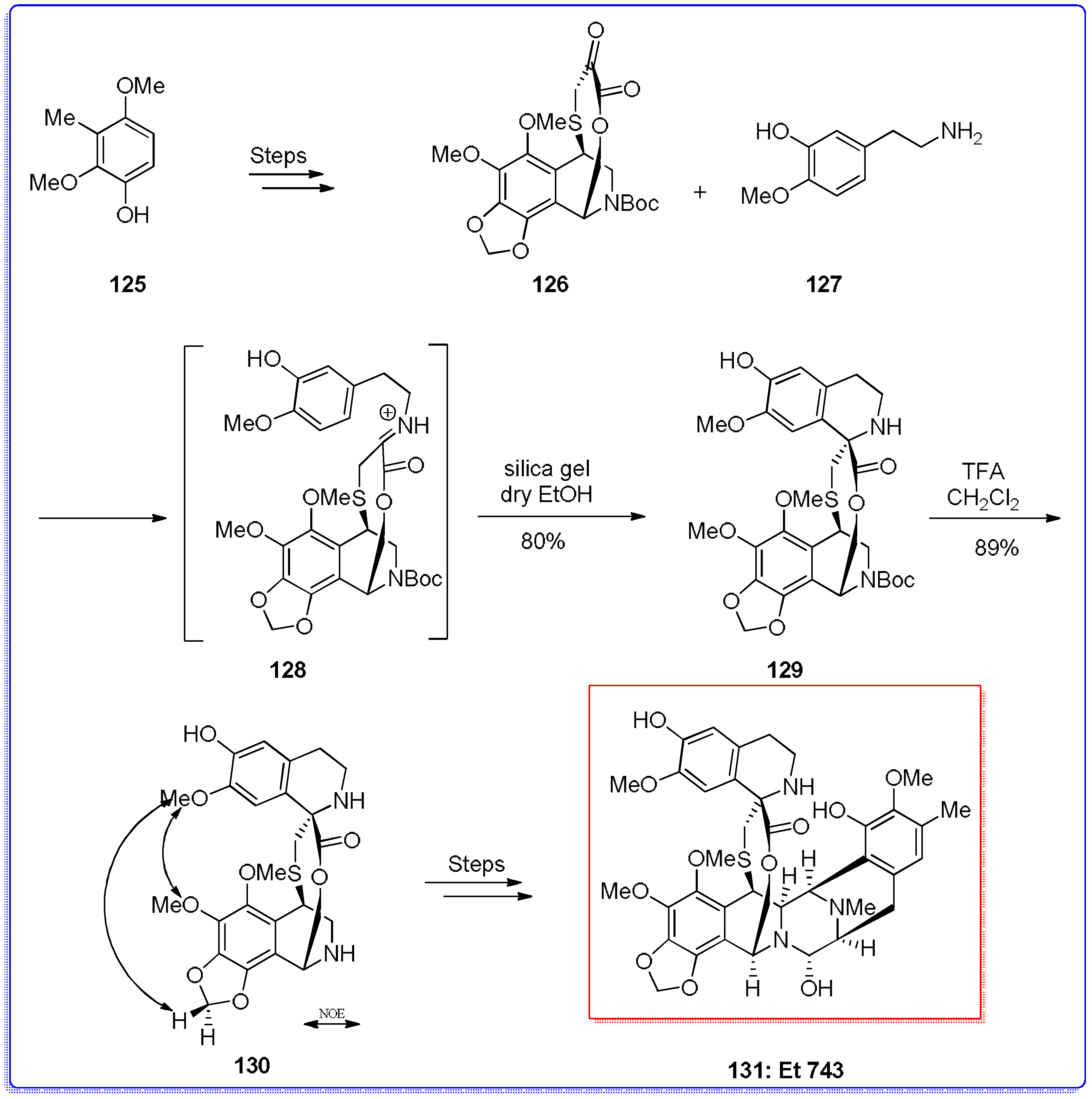
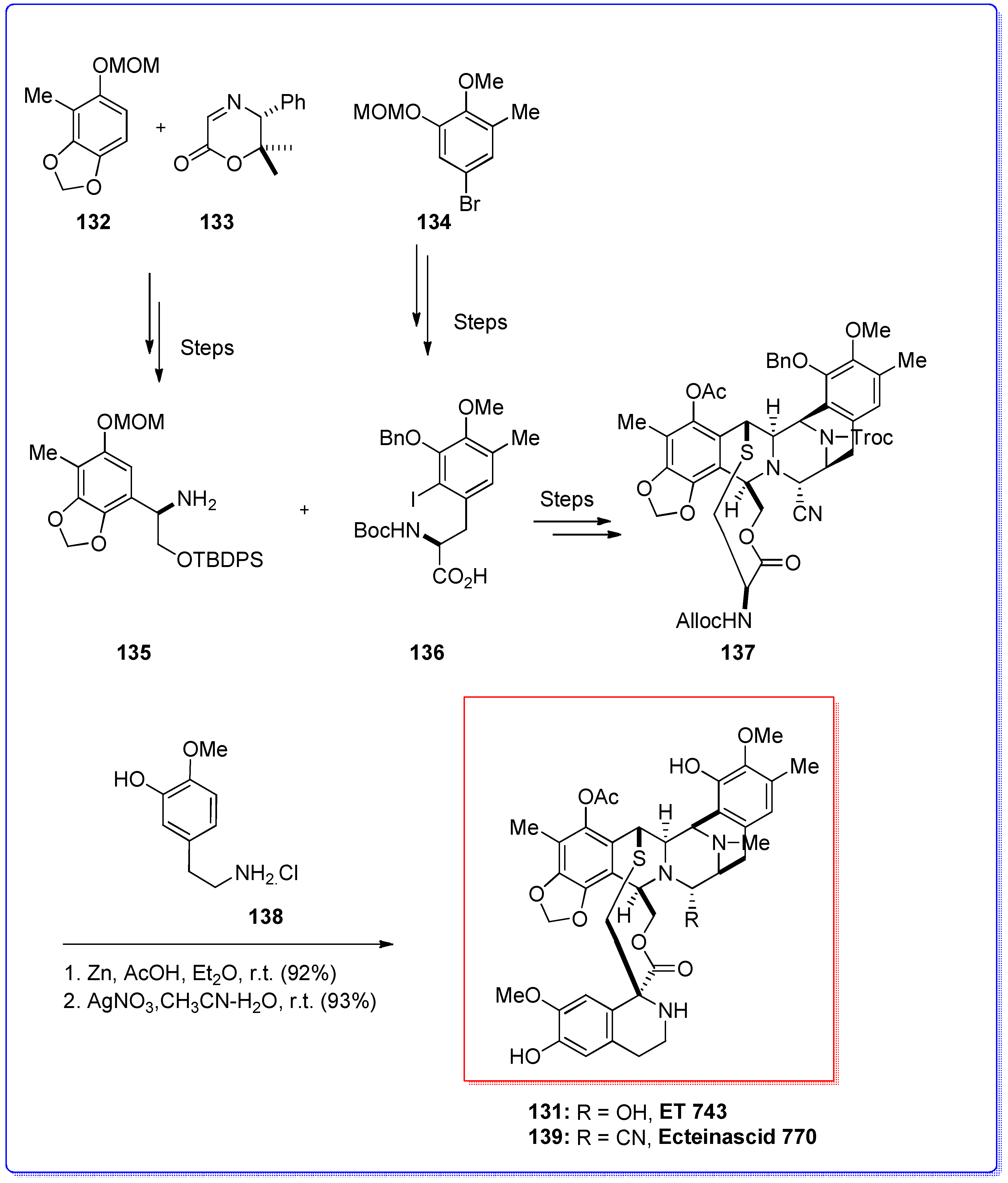
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759a, 759b, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Sakai, R.; Rinehart, K.L.; Guan, Y.; Wang, A. Additional antitumor Ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11456–11460. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.M.; Miranda, N.F. Biosynthetic studies of Ecteinascidins in the Marine Tunicate Ecteinascidia turbinata. J. Nat. Prod. 1995, 58, 1618–1621. [Google Scholar]
- Takahashi, F.; Kubo, A. Structure of Vineomycin B2. J. Antibiot. 1977, 30, 1015–1020. [Google Scholar] [CrossRef]
- Takahashi, K.; Tomita, F. DC-52, a novel antitumor antibiotic. 2. Isolation, physico-chemical characteristics and structure determination. J. Antibiot. 1983, 36, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Jares-Erijiman, E.A.; Manzanares, I.; Silva Elipe, M.V.; Rinehart, K.L. Ecteinascidins: Putative biosynthetic precursors and absolute stereochemistry. J. Am. Chem. Soc. 1996, 118, 9017–9020. [Google Scholar] [CrossRef]
- Zewail-Foote, M.; Hurley, L.H. Ecteinascidin 743: A minor groove alkylator that bends DNA toward the major groove. J. Med. Chem. 1999, 42, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective total synthesis of Ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203. [Google Scholar] [CrossRef]
- Zhou, B.; Guo, J.; Danishefsky, S.J. Studies directed to the total synthesis of Et 743 and analogues Thereof: An expeditious route to the ABFGH subunit. Org. Lett. 2002, 4, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. Total synthesis of Ecteinascidin 743. J. Am. Chem. Soc. 2002, 124, 6552–6554. [Google Scholar] [CrossRef] [PubMed]
- Burk, M.J.; Feaster, J.E.; Nugent, W.A.; Harlow, R.L. Preparation and use of C2-symmetric bis(phospholanes): Production of. Alpha.-amino acid derivatives via highly enantioselective hydrogenation reactions. J. Am. Chem. Soc. 1993, 115, 10125–10138. [Google Scholar] [CrossRef]
- Buckley, T.F.; Rapoport, H. Mild and simple biomimetic conversion of amines to carbonyl compounds. J. Am. Chem. Soc. 1982, 104, 4446–4450. [Google Scholar] [CrossRef]
- Cuevas, C.; Perez, M.; Martin, M.J.; Chicharro, J.L.; Fernandez-Rivas, C.; Flores, M.; Francesch, A.; Gallego, P.; Zarzuelo, M.; Calle, F.; et al. Synthesis of Ecteinascidin Et-743 and phthalascidin Pt-650 from Cyanosafracin B. Org. Lett. 2000, 2, 2545–2548. [Google Scholar] [CrossRef] [PubMed]
- Suwanborirux, K.; Charupant, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A.; Saito, N. Ecteinascidins 770 and 786 from the Thai tunicate Ecteinascidia Thurstoni. J. Nat. Prod. 2002, 65, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Whaley, H.A.; Patterson, E.L.; Dann, M.; Shay, A.J.; Porter, J.N. Isolation and characterization of Lemonomycin, a new antibiotic. Antimicrob. Agents Chemother. 1964, 8, 83–86. [Google Scholar]
- He, H.; Shen, B.; Carter, G.T. Structural Elucidation of Lemonomycin, a potent antibiotic from Streptomyces candidus. Tetrahedron Lett. 2000, 41, 2067–2071. [Google Scholar] [CrossRef]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef] [PubMed]
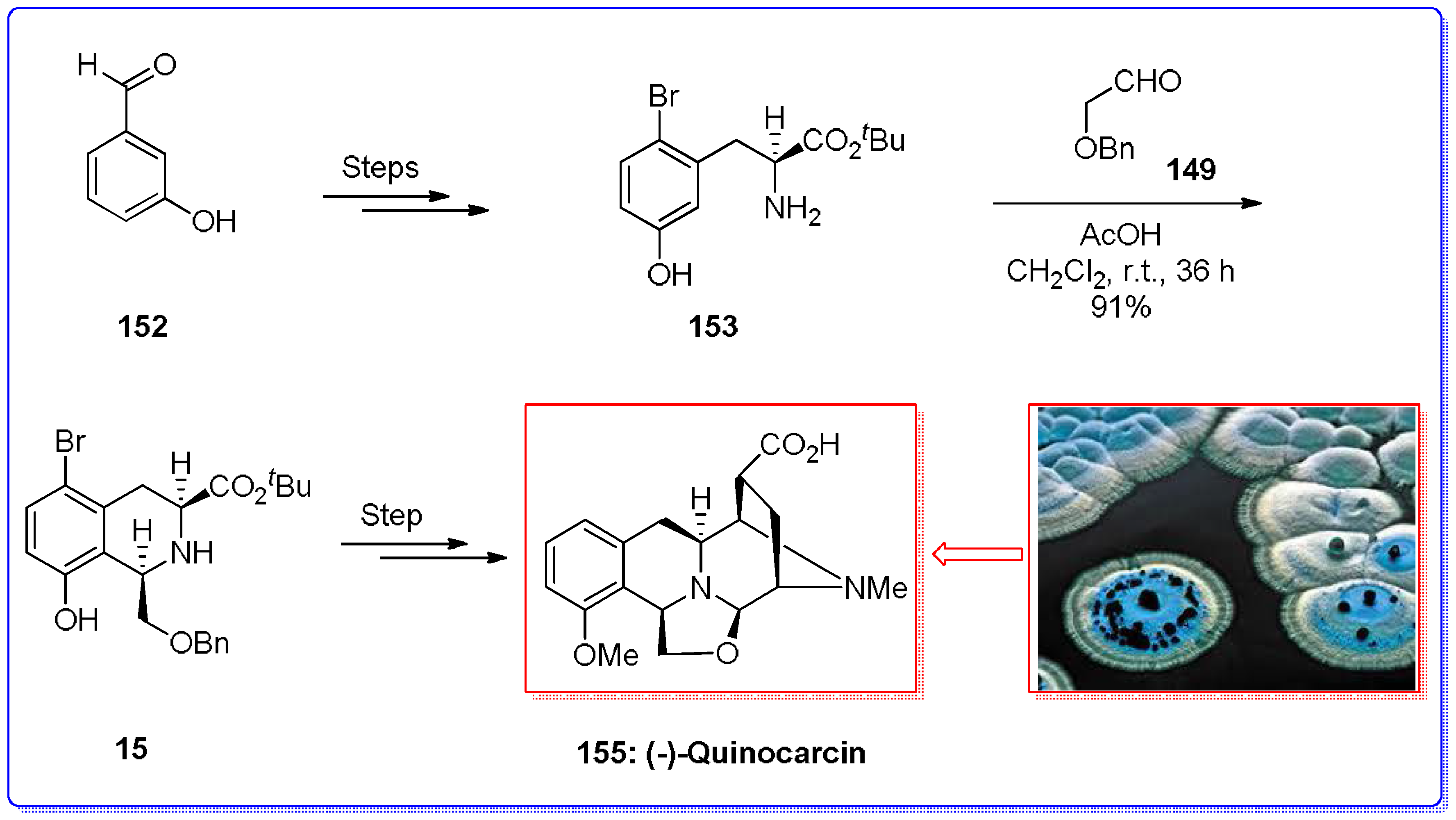
- Garner, P.; Ho, W.B.; Shin, H. The asymmetric synthesis of (−)-quinocarcin via a 1,3-dipolar cycloadditive strategy. J. Am. Chem. Soc. 1993, 115, 10742–10753. [Google Scholar] [CrossRef]
- Maurer, P.J.; Knudsen, C.G.; Palkowitz, A.D.; Rapoport, H. Alpha.-amino acids as chiral educts for asymmetric products. Chirospecific syntheses of methyl l-sibirosaminide and its C-3 epimer from l-allothreonine. J. Org. Chem. 1985, 50, 325–332. [Google Scholar] [CrossRef]
- Evans, D.A.; Hu, E.; Tedrow, J.S. An aldol-based approach to the asymmetric synthesis of l-callipeltose, the deoxyamino sugar of l-callipeltoside A. Org. Lett. 2001, 3, 3133–3136. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.R.; Cruz, E.J.; Stoltz, B.M. The total synthesis of (−)-lemonomycin. J. Am. Chem. Soc. 2003, 125, 15000–15001. [Google Scholar] [CrossRef] [PubMed]
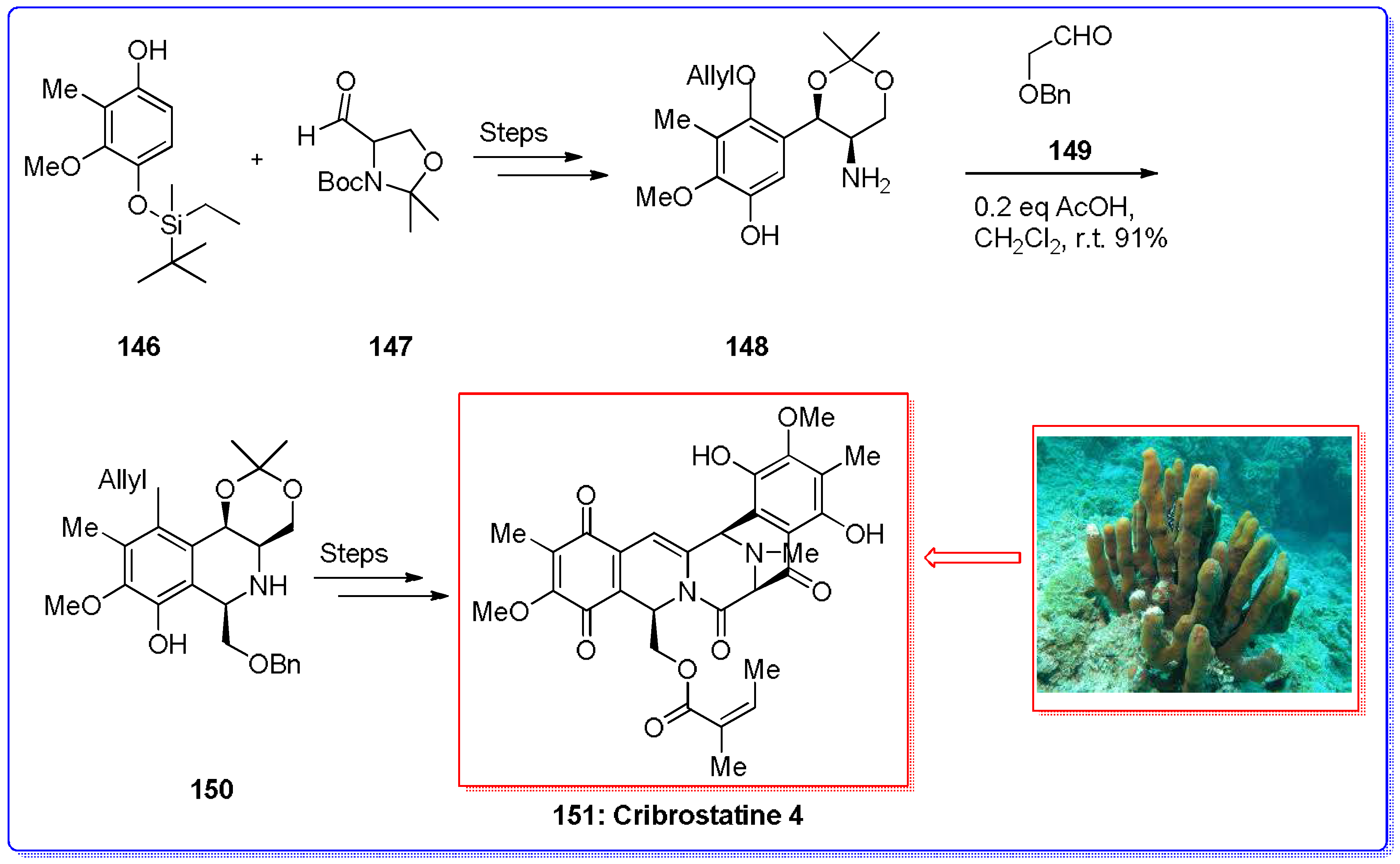
- Pettit, G.R.; Knight, J.C.; Collins, J.C.; Herald, D.L.; Pettit, R.K.; Boyd, M.R.; Young, V.G. Antineoplastic agents 430. Isolation and structure of Cribrostatins 3, 4, and 5 from the republic of Maldives Cribrochalina species. J. Nat. Prod. 2000, 63, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Sakai, H.; Suwanborirux, K.; Pummangura, S.; Kubo, A. 13C-NMR spectral assignment of 5-hydroxy-1,5-imino-3-benzazocin-4,7,10-trione derivatives: The revised structure of Renieramycin H. Heterocycles 2001, 55, 21–28. [Google Scholar] [CrossRef]
- Parameswaran, P.S.; Naik, C.G.; Kamat, S.Y.; Pramanik, B.N. Renieramycins H&L two novel alkaloids from the sponge Halidona Cribricutis dendy. Indian J. Chem. Sect. B 1998, 120, 10272–10273. [Google Scholar]
- Chen, J.; Chen, X.; Zhu, J. Decomposition of the polymerized phases fullerenes nanotubes carbon nanostructures. Angew. Chem. Int. Ed. 2006, 118, 8196–8200. [Google Scholar] [CrossRef]
- Garner, P.; Park, J.M. The synthesis and configurational stability of differentially protected. Beta.-hydroxy-.Alpha.-amino aldehydes. J. Org. Chem. 1987, 52, 2361–2364. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, J. Total synthesis of the marine natural product (−)-cribrostatin 4 (Renieramycin H). Angew. Chem. Int. Ed. 2007, 46, 3962–3965. [Google Scholar] [CrossRef] [PubMed]
- Tomita, F.; Takahashi, K.; Shimizu, K. DC-52, a novel antitumor antibiotic. 1. Taxonomy, fermentation and biological activity. J. Antibiot. 1983, 36, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tomita, F. Dc-52, a novel antitumor antibiotic. J. Antibiot. 1983, 36, 468–470. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Oka, T.; Morimoto, M. Aiititiinior activity of a novel antitumor antibiotic, Quinocarmycin Citrate (KW2152). Cancer Res. 1987, 47, 1516–1522. [Google Scholar] [PubMed]
- Saito, H.; Hirata, T.; Kasai, M.; Fujimoto, K.; Ashizawa, T.; Morimoto, M.; Sato, A. Synthesis and biological evaluation of quinocarcin derivatives: Thioalkyl-substituted Quinones and Hydroquinones. J. Med. Chem. 1991, 34, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Plowman, J.; Dykers, D.J.; Narayanan, V.L.; Abbott, B.J.; Saito, H.; Hirata, T.; Grever, M.R. Efficacy of the quinocarmycins KW2152 and DX-52-1 against human melanomalines growing in culture and in mice. Cancer Res. 1995, 55, 862–867. [Google Scholar] [PubMed]
- Bunnell, C.A.; Supko, J.G.; Eder, J.P.; Clark, J.W.; Lynch, T.J.; Kufe, D.W.; Shulman, L.N. Phase I clinical trial of 7-cyanoquinocarcinol (DX-52-1) in adult patients with refractory solid Malignancies. Cancer Chemother. Pharmacol. 2001, 48, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Flanagan, M.E.; Tippie, T.N. O2-dependent cleavage of DNA by tetrazomine. Biochemistry 1994, 33, 4086–4092. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Liron, M.; Zhu, J. Asymmetric total synthesis of (−)-quinocarcin. J. Am. Chem. Soc. 2008, 130, 7148–7152. [Google Scholar] [CrossRef] [PubMed]
- Aune, G.J.; Furuta, T.; Pommier, Y. Ecteinascidin 743: A novel anticancer drug with a unique mechanism of action. Anticancer Drug. 2002, 13, 545–555. [Google Scholar] [CrossRef]
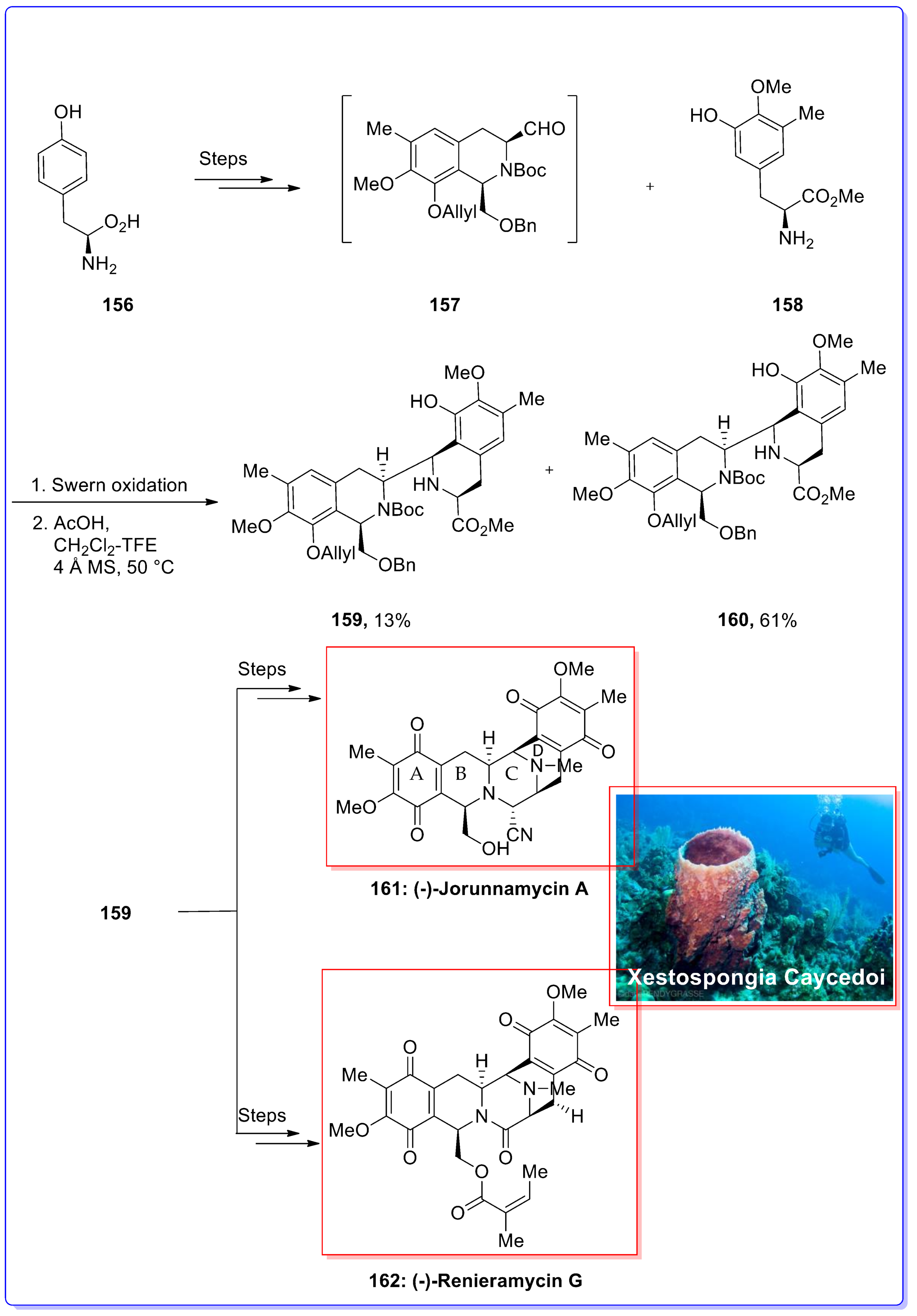
- Charupant, K.; Suwanborirux, K.; Amnuoypol, S.; Saito, E.; Kubo, A.; Saito, N. Jorunnamycins A–C, new stabilized Renieramycin-type bistetrahydroisoquinolines isolated from the Thai Nudibranch Jorunna Funebris. Chem. Pharm. Bull. 2007, 55, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Cavaliere, P.; Wahidulla, S.; Naik, C.G.; Cimino, G. A new antitumor isoquinoline alkaloid from the marine Nudibranch Jorunna Funebris. Tetrahedron 2000, 56, 7305–7308. [Google Scholar] [CrossRef]
- Frincke, J.M.; Faulkner, D.J. Antimicrobial metabolites of the sponge Reniera sp. J. Am. Chem. Soc. 1982, 104, 265–269. [Google Scholar] [CrossRef]
- Lown, J.W.; Joshua, A.V.; Lee, J.S.; Hurley, L.H. Molecular mechanisms of binding and single-strand scission of DNA by the antitumor antibiotics saframycins A and C. Biochemistry 1982, 21, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Oku, N.; Matsunaga, S.; van Soest, R.W.M.; Fusetani, N. Renieramycin J, a highly cytotoxic tetrahydroisoquinoline alkaloid, from a marine sponge Neopetrosia sp. J. Nat. Prod. 2003, 66, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Tanaka, C.; Koizumi, Y.; Suwanborirux, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A. Chemistry of renieramycins. Part 6: Transformation of Renieramycin M into Jorumycin and Renieramycin J including oxidative degradation products, Mimosamycin, Renierone, and Renierol acetate. Tetrahedron 2004, 60, 3873–3881. [Google Scholar] [CrossRef]
- Davidson, B.S. Renieramycin G, a new alkaloid from the sponge Xestospongia caycedoi. Tetrahedron Lett. 1992, 33, 3721–3724. [Google Scholar] [CrossRef]
- Chen, R.; Zhu, D.; Hu, Z.; Zheng, Z.; Chen, X. A new approach to the synthesis of l-3-hydroxy-4-methoxy-5-methyl-phenylalanine derivatives from L-tyrosine. Tetrahedron Asymmetry 2010, 21, 39–42. [Google Scholar] [CrossRef]
- Wright, B.J.D.; Chan, C.; Danishefsky, S.J. Synthesis and cytotoxic evaluation of some Cribrostatin-ecteinascidin analogues. J. Nat. Prod. 2008, 71, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, W.; Liao, X.; Yan, Z.; Guan, B.; Wang, N.; Liu, Z. Synthesis and cytotoxicity of (−)-renieramycin g analogs. Bioorg. Med. Chem. Lett. 2011, 21, 1419–1421. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.W.; Chen, Y.; Williams, R.M. Asymmetric total syntheses of (−)-jorumycin, (−)-renieramycin G, 3-epi-jorumycin, and 3-epi-renieramycin G. J. Am. Chem. Soc. 2005, 127, 12684–12690. [Google Scholar] [CrossRef] [PubMed]
- Vincent, G.; Williams, R.M. Asymmetric total synthesis of (−)-cribrostatin 4 (Renieramycin H). Angew. Chem. Int. Ed. 2007, 46, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Magnus, P.; Matthews, K.S. Synthesis of the tetrahydroisoquinoline alkaloid (±)-Renieramycin G and A (±)-lemonomycinone analogue from a common intermediate. J. Am. Chem. Soc. 2005, 127, 12476–12477. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, H.; Chen, X. Asymmetric total synthesis of (−)-jorunnamycins A and C and (−)-jorumycin from L-tyrosine. J. Nat. Prod. 2013, 76, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, R.; Chen, X. A rapid and efficient access to renieramycin-type alkaloids featuring A temperature-dependent stereoselective cyclization. Org. Biomol. Chem. 2014, 12, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Hagel, J.M.; Facchini, P.J. Benzylisoquinoline alkaloid metabolism: A century of discovery and a Brave New World. Plant Cell Physiol. 2013, 54, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, L.; Yang, H.; Qian, W.; Guo, L.; Zhou, S.; Gao, B.; Li, Z.; Zhou, Y.; Jiang, H.; et al. Asymmetric total synthesis and identification of tetrahydroprotoberberine derivatives as new antipsychotic agents possessing a dopamine D(1), D(2) and serotonin 5-HT(1A) multi-action profile. Bioorg. Med. Chem. Lett. 2013, 21, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Gadhiya, S.; Shashikanth, P.; Harding, W.W. A divergent route to 9,10-oxygenated tetrahydroprotoberberine and 8-oxoprotoberberine alkaloids: Synthesis of (±)-isocorypalmine and oxypalmatine. Tetrahedron 2015, 71, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Rasterlli, L.; Capasso, A.; Pizza, L.; De Tommasi, N. New protopine and benzyltetrahydroprotoberberine alkaloids from aristolochia constricta and their activity on isolated Guinea-Pig ileum. J. Nat. Prod. 1997, 60, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Ninomiya, K.; Shimoda, H.; Nishida, N.; Yoshikawa, M. Javaberine A, new TNF-α and nitric oxide production inhibitor, from the roots of talinum paniculatum. Heterocycles 2001, 55, 2043–2050. [Google Scholar]
- Rochfort, S.J.; Towerzey, L.; Carroll, A.; King, G.; Michael, A.; Pierens, G.; Rali, T.; Redburn, J.; Whitmore, J.; Quinn, R.J. Latifolians A and B, novel JNK3 Kinase inhibitors from the Papua new Guinean plant Gnetum latifolium. J. Nat. Prod. 2005, 68, 1080–1082. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Grkovic, T.; Sykes, M.L.; Shelper, T.; Avery, V.M.; Camp, D.; Quinn, R.J.; Davis, R.A. Alkaloids from the Chinese Vine Gnetum montanum. J. Nat. Prod. 2011, 74, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
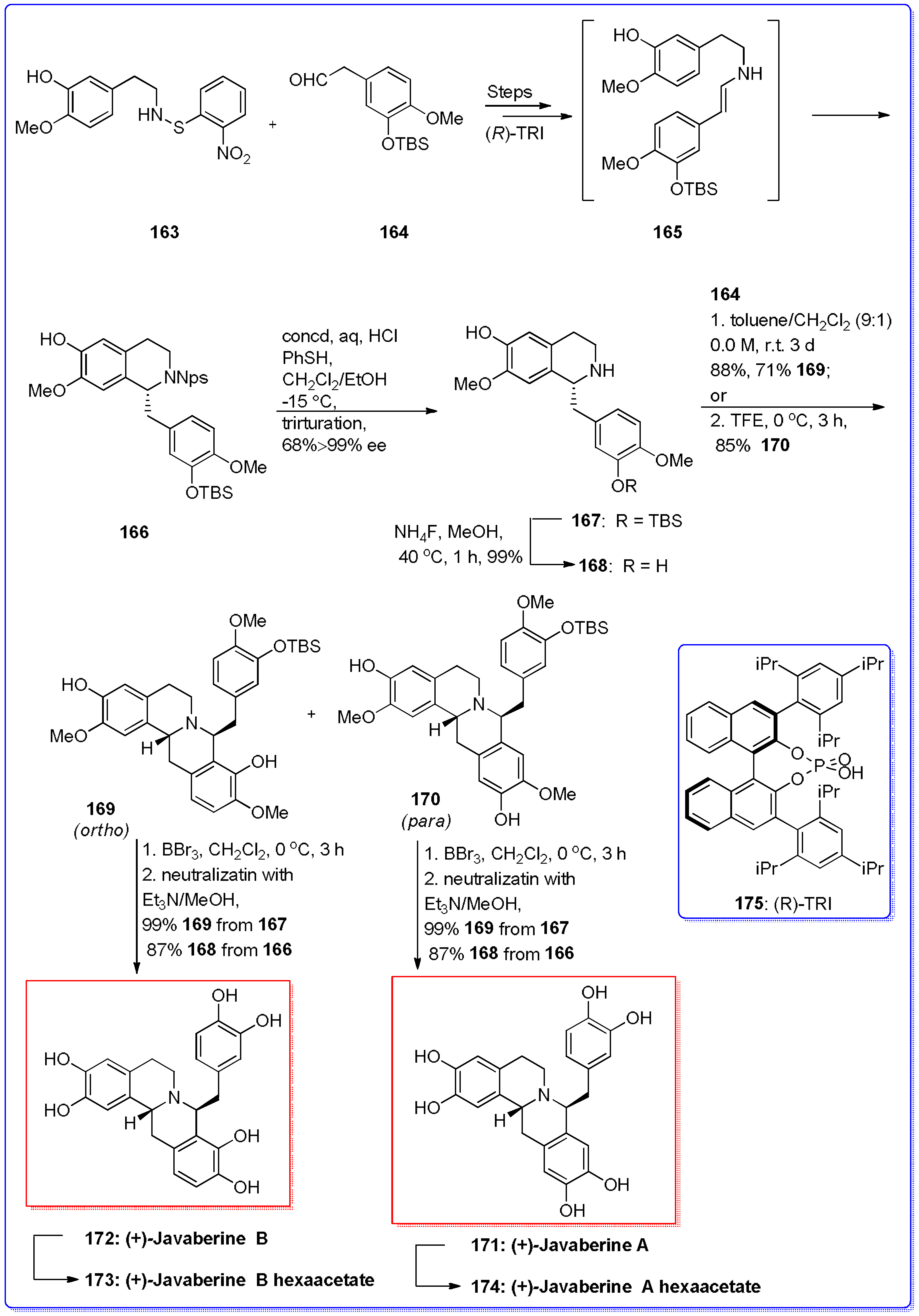
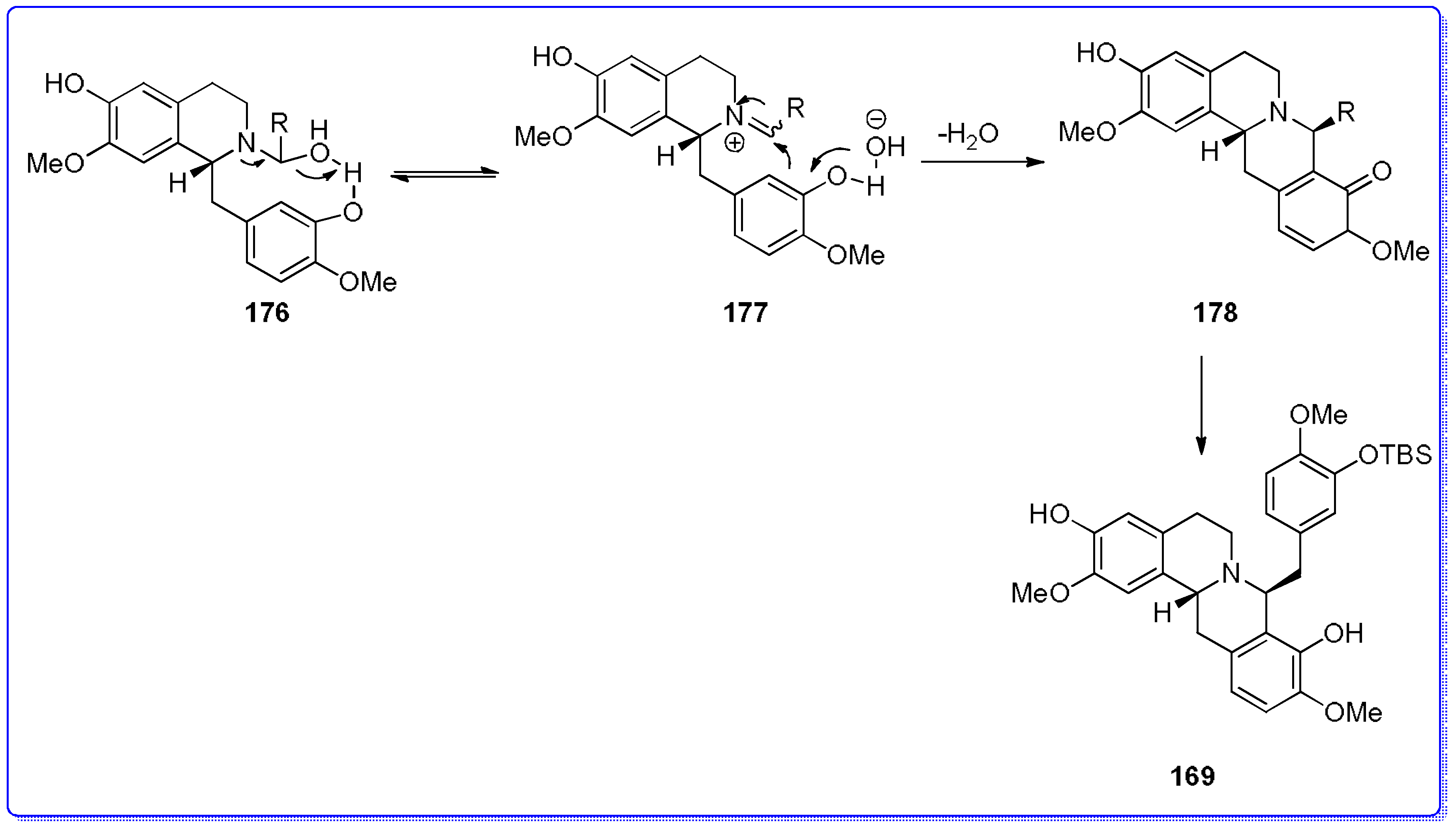
- Ruiz-Olalla, A.; Würdemann, M.A.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Organocatalytic enantioselective Pictet–Spengler approach to biologically relevant 1-benzyl-1,2,3,4-tetrahydroisoquinoline alkaloids. J. Org. Chem. 2015, 80, 5125–5132. [Google Scholar] [CrossRef] [PubMed]
- Mons, E.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Organocatalytic enantioselective Pictet–Spengler reactions for the syntheses of 1-substituted 1,2,3,4-tetrahydroisoquinolines. J. Org. Chem. 2014, 74, 7380–7739. [Google Scholar] [CrossRef] [PubMed]
- Cashaw, J.L.; Davis, V.E.; Mcmurtrey, K.D.; Meyerson, L.R. Kinetics and product distribution in Pictet–Spengler cyclization of tetrahydropapaveroline to tetrahydroprotoberberine alkaloids. J. Org. Chem. 1984, 49, 947–948. [Google Scholar]
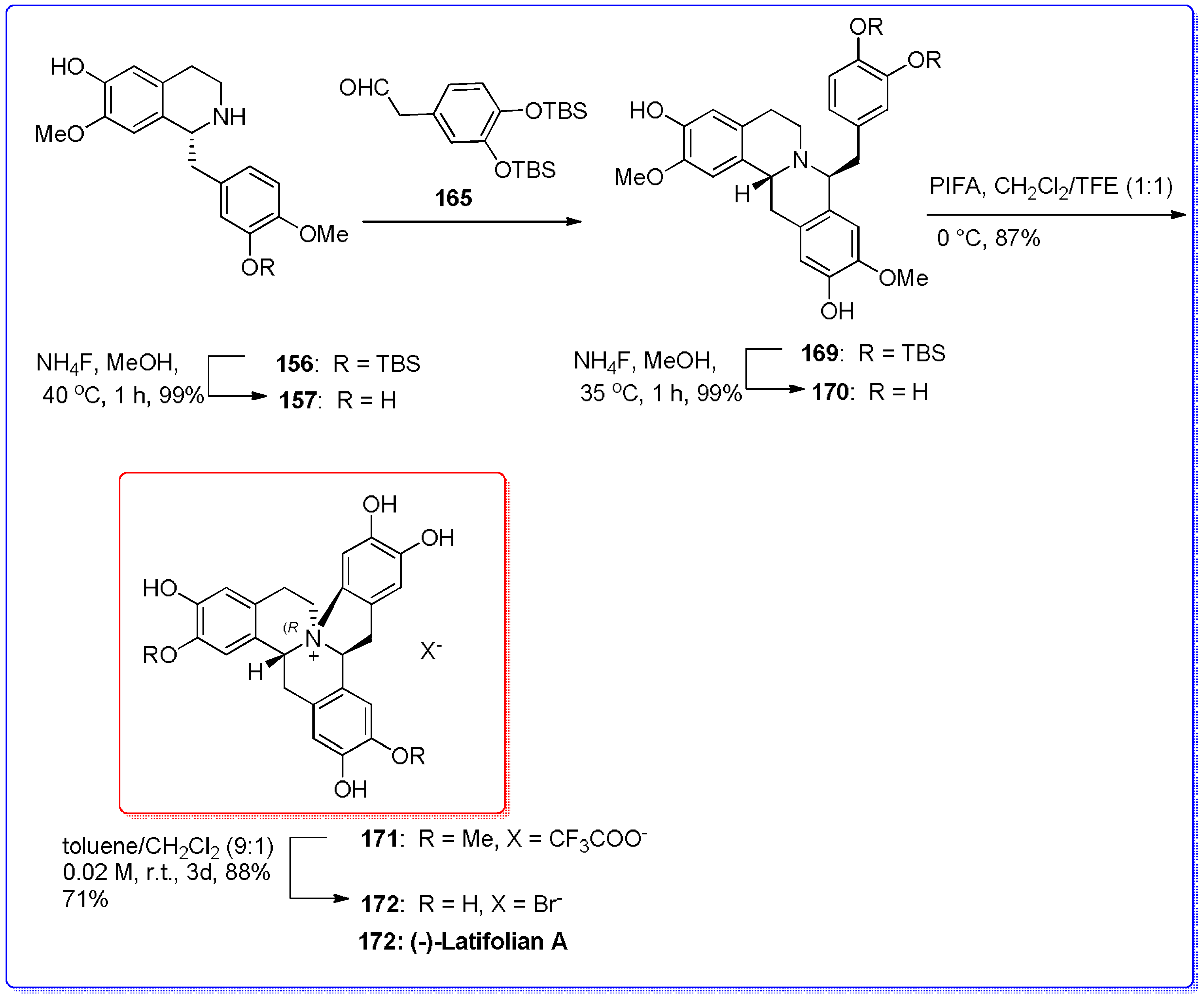
- Kayhan, J.; Wanner, M.J.; Ingemann, S.; van Maarseveen, J.H.; Hiemstra, H. Consecutive Pictet–Spengler condensations toward bioactive 8-benzylprotoberberines: Highly selective total syntheses of (+)-javaberine A, (+)-javaberine B, and (−)-latifolian A. Eur. J. Org. Chem. 2016, 2016, 3705–3708. [Google Scholar] [CrossRef]
































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
M. Heravi, M.; Zadsirjan, V.; Malmir, M. Application of the Asymmetric Pictet–Spengler Reaction in the Total Synthesis of Natural Products and Relevant Biologically Active Compounds. Molecules 2018, 23, 943. https://doi.org/10.3390/molecules23040943
M. Heravi M, Zadsirjan V, Malmir M. Application of the Asymmetric Pictet–Spengler Reaction in the Total Synthesis of Natural Products and Relevant Biologically Active Compounds. Molecules. 2018; 23(4):943. https://doi.org/10.3390/molecules23040943
Chicago/Turabian StyleM. Heravi, Majid, Vahideh Zadsirjan, and Masumeh Malmir. 2018. "Application of the Asymmetric Pictet–Spengler Reaction in the Total Synthesis of Natural Products and Relevant Biologically Active Compounds" Molecules 23, no. 4: 943. https://doi.org/10.3390/molecules23040943