
Identification of Potential Nematicidal Compounds against the Pine Wood Nematode, Bursaphelenchus xylophilus through an In Silico Approach


Abstract
:1. Introduction
2. Results and Discussion
2.1. Target–Template Alignment for Homology Modeling
2.2. Homology Modeling
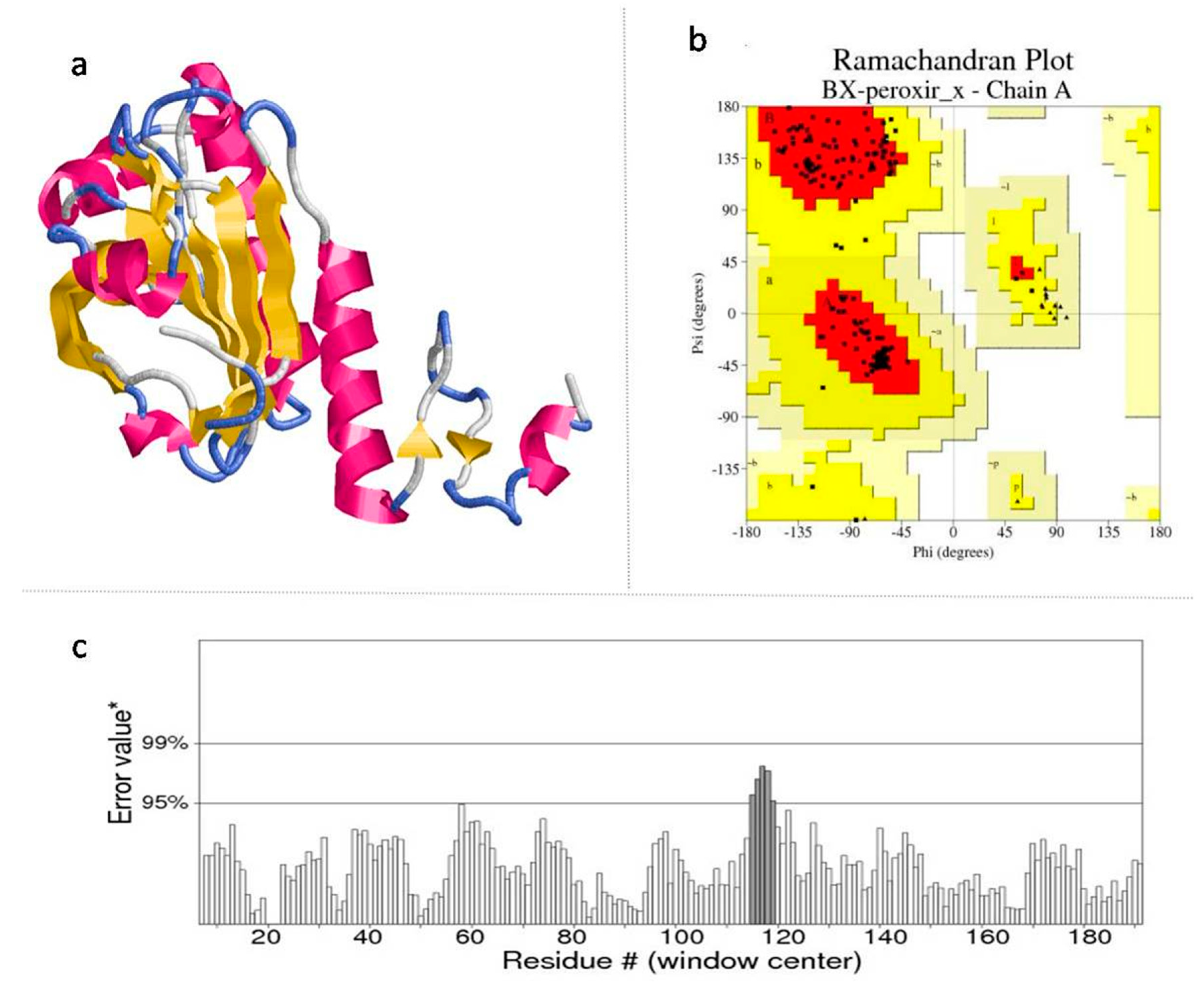
2.3. Model Validation
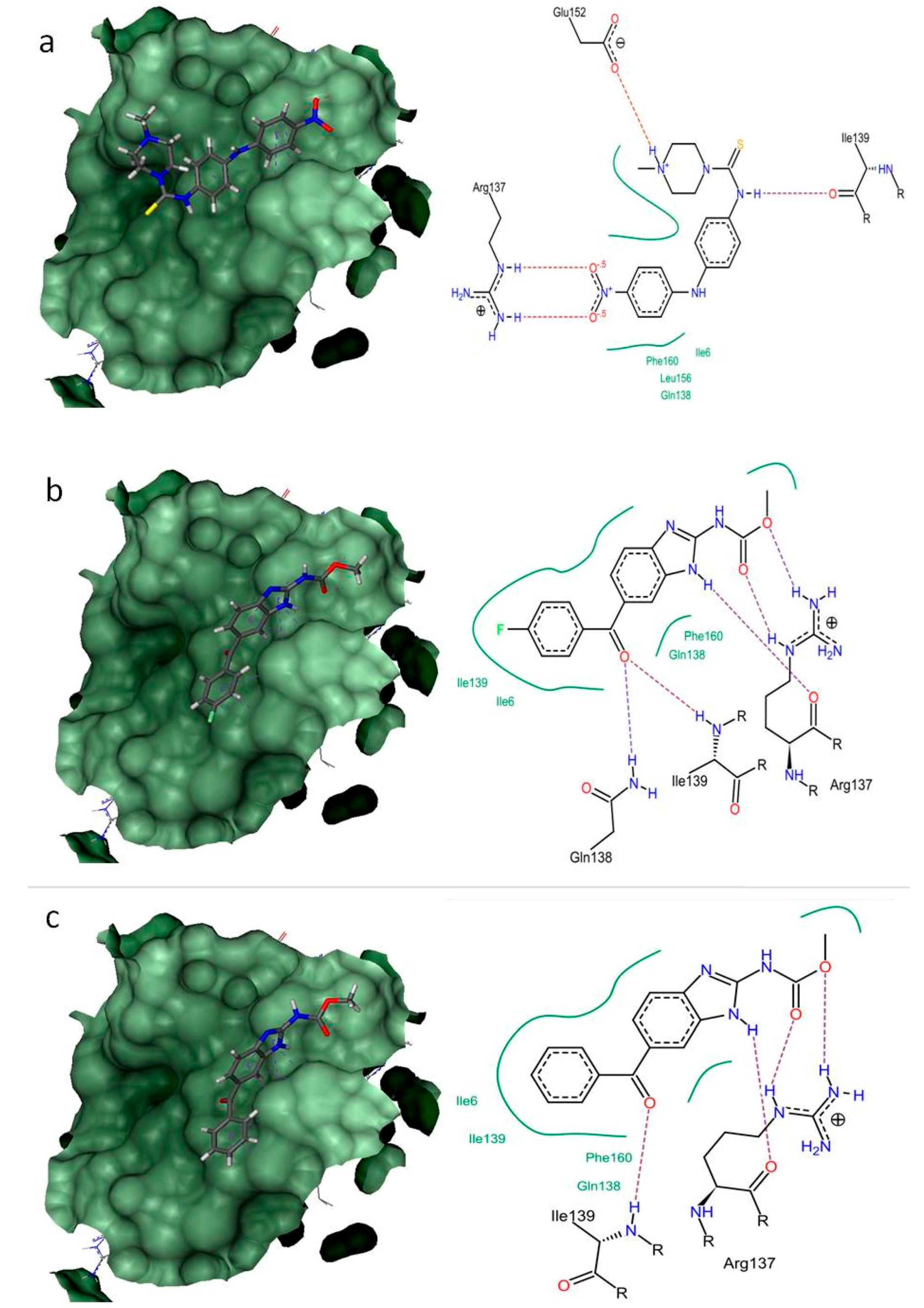
2.4. Structure-Based Virtual Screening
2.5. Density Functional Theory Analysis
3. Materials and Methods
3.1. Sequence Analysis for Potential Templates
3.2. Homology Modeling
3.3. Model Validation
3.4. Structure-Based Virtual Screening
3.5. Docking Interactions
3.6. Electronic Structure Study of Selected Screening Compounds
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, X.; Cheng, F.; Xu, R.; Xie, B. Genetic variation in the invasive process of Bursaphelenchus xylophilus (Aphelenchida: Aphelenchoididae) and its possible spread routes in China. Heredity 2008, 100, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, T.A.; Webster, J.M. Distribution of pine wilt disease with respect to temperature in North America, Japan, and Europe. Can. J. For. Res. 1987, 17, 1050–1059. [Google Scholar] [CrossRef]
- Mamiya, Y. The life history of the pine wood nematode, Bursaphelenchus lignicolus. Jpn. J. Nematol. 1975, 5, 16–25. [Google Scholar]
- Leal, I.; Green, M.; Allen, E.; Humble, L.; Rott, M. Application of a real-time PCR method for the detection of pine wood nematode, Bursaphelenchus xylophilus, in wood samples from lodgepole pine. Nematology 1988, 9, 351–362. [Google Scholar] [CrossRef]
- Inácio, M.L.; Nobrega, F.; Vieira, P.; Bonifacio, L.; Naves, P.; Sousa, E.; Mota, M. First detection of Bursaphelenchus xylophilus associated with Pinus nigra in Portugal and in Europe. For. Pathol. 2015, 45, 235–238. [Google Scholar] [CrossRef]
- Akbulut, S.; Stamps, W.T. Insect vectors of the pinewood nematode: A review of the biology and ecology of Monochamus species. For. Pathol. 2012, 42, 89–99. [Google Scholar] [CrossRef]
- Futai, K. Pine wood nematode, Bursaphelenchus xylophilus. Annu. Rev. Phytopathol. 2013, 51, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Moens, M.; Mota, M.; Li, H.; Kikuchi, T. Bursaphelenchus xylophilus: Opportunities in comparative genomics and molecular host-parasite interactions. Mol. Plant Pathol. 2008, 9, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Mamiya, Y.; Enda, N. Transmission of Bursaphelenchus lignicolus (Nematoda: Aphelenchoididae) by Monochamus alternatus (Coleoptera: Cerambycidae). Nematologica 1972, 18, 159–162. [Google Scholar] [CrossRef]
- James, R.; Tisserat, N.; Todd, T. Prevention of pine wilt of Scots pine (Pinus sylvestris) with systemic abamectin injections. Arboric. Urban For. 2006, 32, 195–201. [Google Scholar]
- Gopal, R.M.; Pomroy, W.E.; West, D.M. Resistance of field isolates of Trichostrongylus colubriformis and Ostertagia circumcincta to ivermectin. Int. J. Parasitol. 1999, 29, 781–786. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.-M.; Park, C.G. Bursaphelenchus xylophilus is killed by homologues of 2-(1-undecyloxy)-1-ethanol. Sci. Rep. 2016, 6, 29300. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G.; Roush, R.T.; Liu, N. Selection of high-level abamectin resistance from field-collected house flies. Musca domestica. Experientia 1991, 47, 282–291. [Google Scholar]
- Argentine, J.A.; Clark, J.M. Selection for abamectin resistance in Colorado potato beetle (Coleoptera: Chrysomelidae). Pestic. Sci. 1990, 28, 17–24. [Google Scholar] [CrossRef]
- Lasota, J.A.; Dybas, R.A. Avermectins, a novel class of compounds: Implications for use in arthropod pest control. Annu. Rev. Entomol. 1991, 36, 91–117. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Takanashi, T.; Tsai, I.J.; Assefa, S.A.; Cock, P.J.; et al. Genomic Insights into the Origin of parasitism in the emerging Plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011, 7, e1002219. [Google Scholar] [CrossRef] [PubMed]
- Khanna, V.; Ranganathan, S. In silico approach to screen compounds active against parasitic nematodes of major socioeconomic importance. BMC Bioinform. 2011, 12, S25. [Google Scholar] [CrossRef] [PubMed]
- Guiliano, D.B.; Hong, X.; McKerrow, J.H.; Blaxter, M.L.; Oksov, Y.; Liu, J.; Ghedin, E.; Lustigman, S. A gene family of cathepsin L-like proteases of filarial nematodes are associated with larval molting and cuticle and eggshell remodeling. Mol. Biochem. Parasitol. 2004, 136, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Ren, J.; Huang, L.; Li, H.; Ye, J. Screening and functional analysis of the peroxiredoxin specifically expressed in Bursaphelenchus xylophilus—The causative agent of pine wilt disease. Int. J. Mol. Sci. 2014, 15, 10215–10232. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Z.; Li, D.; Chen, Q. Identification and characterization of a Bursaphelenchus xylophilus (Aphelenchida: Aphelenchoididae) thermotolerance-related gene: Bx-hsp90. Int. J. Mol. Sci. 2012, 16, 8819–8833. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Koh, Y.H.; Moon, Y.S.; Lee, S.H. Molecular properties of a venom allergen-like protein suggest a parasitic function in the pinewood nematode Bursaphelenchus xylophilus. Int. J. Parasitol. 2012, 42, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.M.; Wang, Q.; Rosa, B.A.; Huang, S.C.C.; Powell, K.; Schedl, T.; Pearce, E.J.; Abubucker, S.; Mitreva, M. Discovery of Anthelmintic Drug Targets and Drugs Using Chokepoints in Nematode Metabolic Pathways. PLoS Pathog. 2013, 9, e1003505. [Google Scholar] [CrossRef] [PubMed]
- Lacey, E. Mode of action of benzimidazoles. Parasitol. Today 1990, 6, 112–115. [Google Scholar] [CrossRef]
- Taylor, D. The Pharmaceutical industry and the future of drug development. In Pharmaceuticals in the Environment; Hester, R.E., Harrison, R.M., Eds.; The Royal Society of Chemistry: London, UK, 2015; pp. 1–33. [Google Scholar]
- Xue, Y.; Shui, G.; Wenk, M.R. TPS1 drug design for rice blast disease in Magnaporthe oryzae. SpringerPlus 2014, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.O.; Krishna, P.B.; Eapen, S.J. Virtual screening and in vitro assay to explore novel inhibitors from black pepper against potential targets of Radopholus similis. Int. J. Comput. Appl. 2014, 86, 35–43. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Honig, B. An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments. J. Mol. Biol. 2000, 301, 691–711. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [PubMed]
- SAVES Server. Available online: http://services.mbi.ucla.edu/SAVES/ (accessed on 28 November 2017).
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK a program to check the stereo chemical quality of protein structure, J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Colovos, C.; Yeates, T.O. ERRAT: An empirical atom-based method for validating protein structures. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Poroikov, V.V. PASS: Computerized prediction of biological activity spectra for chemical substances. In Bioactive Compound Design: Possibilities for Industrial Use; BIOS Scientific: Oxford, UK, 1996; pp. 47–56. [Google Scholar]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sharma, O.P.; Pan, A.; Hoti, S.L.; Jadhav, A.; Kannan, M.; Mathur, P.P. Modeling, docking, simulation, and inhibitory activity of the benzimidazole analogue against β-tubulin protein from Brugia malayi for treating lymphatic filariasis. Med. Chem. Res. 2012, 21, 2415–2427. [Google Scholar] [CrossRef]
- Sen, K.D.; Mingos, D.M.P. Chemical Hardness: Structure and Bonding; Springer: Berlin, Germany, 1993. [Google Scholar]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.R.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB viewer (deep view). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Schellhammer, I.; Rarey, M. FlexX-Scan: Fast, structure-based virtual screening. Proteins Struct. Funct. Bioinform. 2004, 57, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Rarey, M. PoseView—Molecular interaction patterns at a glance. J. Cheminform. 2010, 2 (Suppl. 1), P50. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods; Gaussian: Pittsburgh, PA, USA, 1995. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Senet, P. Chemical hardnesses of atoms and molecules from frontier orbitals. Chem. Phys. Lett. 1997, 275, 527–532. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
Sample Availability: All the compounds are available from Pubchem and ZINC database. |




{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Target | Function |
|---|---|---|
| 1. | Cathepsin L-like cystein proteinase (BxCLCP) (UniProt ID: Q6LDJ1) | Post embryonic development |
| 2. | 2-cysteine peroxiredoxin (BxPRX) (UniProt ID: B0LFQ7) | Reproduction and pathogenecity |
| 3. | Heat Shock Protein 90 (BxHSP90) (UniProt ID: A4UU63) | Adapts to different climatic conditions |
| 4. | Venom allergen Protein-3 (BxVAP-3) (UniProt ID: E0WW94) | Invasion parasitic genes |
| 5. | β-Tubulin (BxTUB) (UniProt ID: D1MX18) | Microtubule, mitosis, motility |
| Compound Name (Pubchem Id) | Cathepsin L-Like Cystein Proteinase (BxCLCP) | 2-Cysteine Peroxiredoxin (BxPRX) | Heat Shock Protein 90 (BxHSP90) | Venom Allergen Protein-3 (BxVAP-3) | β-Tubulin (BxTUB) |
|---|---|---|---|---|---|
| Kainic acid (CID 10255) | −17.653 | −18.586 | −11.942 | −12.681 | −24.909 |
| Carbendazim (CID 25429) | −14.879 | −16.525 | −12.365 | −15.173 | −21.44 |
| Naphthalen-2-ol (CID 8663) | −9.5734 | −11.793 | −9.6124 | −11.458 | −12.053 |
| Pyrantel (CID 708857) | −8.0396 | −12.548 | −8.519 | −9.0794 | −12.559 |
| Closantel (CID 42574) | −15.618 | −6.0856 | −8.3846 | −15.835 | −14.155 |
| Thiabendazole (CID 5430) | −12.071 | −15.395 | −12.1 | −13.143 | −16.532 |
| Schaftoside (CID 442658) | −10.435 | −9.6139 | −4.0356 | −5.3029 | −22.876 |
| Mebendazole (CID 4030) | −18.322 | −20.111 | −18.993 | −18.699 | −25.531 |
| Oxfendazole (CID 40854) | −15.653 | −19.8592 | −13.344 | −17.071 | −21.242 |
| Levamisole (CID 26879) | −8.1927 | −12.361 | −6.1674 | −12.326 | −13.724 |
| Tetramizole (CID 3913) | −6.7261 | −10.75 | −5.2535 | −12.811 | −17.188 |
| Coumafos (CID 2871) | −6.7703 | −18.175 | −1.7963 | −5.8927 | −15.065 |
| Amocarzine (CID 5464102) | −18.752 | −30.163 | −22.895 | −19.279 | −27.122 |
| Fenbendazole (CID 3334) | −14.391 | −18.826 | −14.743 | −14.202 | −24.141 |
| Flubendazole (CID 35802) | −19.364 | −23.2623 | −15.053 | −17.962 | −28.058 |
| Potential Targets from B. xylophilus | Best Docked Compounds | ||
|---|---|---|---|
| Amocarzine (CID 5464102) | Flubendazole (CID 35802) | Mebendazole (CID 4030) | |
| Cathepsin L-like cystein proteinase (BxCLCP) (UniProt ID: Q6LDJ1) | #Gln26 *, His27 #Glu28 *, Lys113 Thr206 * | Ile25 *, #Gln62 * #Cys65 *, Gly66 Cys68, Thr206 * His207, Trp230 | #Gln62 *, #Cys65 *, Gly66, #Trp230 * |
| −18.752 | −19.364 | −18.322 | |
| 2-cysteine peroxiredoxin (BxPRX) (UniProt ID: B0LFQ7) | Ile6, Arg137 * Gln138, Ile139 * Leu156, Glu152 * Phe160 | Ile6, Arg137 * #Gln138 *, #Ile139 *, Phe160 | Ile6, Arg137 * Gln138, #Ile139 * Phe160 |
| −30.163 | −23.2623 | −20.111 | |
| Heat Shock Protein 90 (BxHSP90) (UniProt ID: A4UU63) | #Lys332 *, Ala333 Gln334, #Arg337 * Asp338, Ser339 Met342 | #Met331*, Lys332 #Gln334 *, Ala335, #Arg337 * | Met331 *, Lys332 Ala333, Gln334* Ala335, #Arg337 * |
| −22.895 | −15.053 | −18.993 | |
| Venom allergen Protein-3 (BxVAP-3) (UniProt ID: E0WW94) | #Trp95 *, #Pro96 * His97, #Asn160 * | Ala93, Gln94 #Trp95 *, #Asn160 *, Trp161 | Ala93, Gln94 #Trp95 *, #Asn160 * Trp161 |
| −19.279 | −17.962 | −18.699 | |
| β-Tubulin (BxTUB) (UniProt ID: D1MX18) | Gln11, Gly98 * #Asn99 *, Ser138 Gly141, Thr143 * Ser176, #Asp177 * Glu181, Asn204 | Gln11 *, Cys12 #Asn99 *, Gly141 Gly142 *, Thr143 * Asp177, Thr178 Asn204, Tyr222 | Gln11, #Cys12 * Ser138, Gly141 Val169, Ser172 * #Asp177 *, Asn204 * Tyr222 |
| −27.122 | −28.058 | −25.531 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanmugam, G.; Lee, S.K.; Jeon, J. Identification of Potential Nematicidal Compounds against the Pine Wood Nematode, Bursaphelenchus xylophilus through an In Silico Approach. Molecules 2018, 23, 1828. https://doi.org/10.3390/molecules23071828
Shanmugam G, Lee SK, Jeon J. Identification of Potential Nematicidal Compounds against the Pine Wood Nematode, Bursaphelenchus xylophilus through an In Silico Approach. Molecules. 2018; 23(7):1828. https://doi.org/10.3390/molecules23071828
Chicago/Turabian StyleShanmugam, Gnanendra, Sun Keun Lee, and Junhyun Jeon. 2018. "Identification of Potential Nematicidal Compounds against the Pine Wood Nematode, Bursaphelenchus xylophilus through an In Silico Approach" Molecules 23, no. 7: 1828. https://doi.org/10.3390/molecules23071828