Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
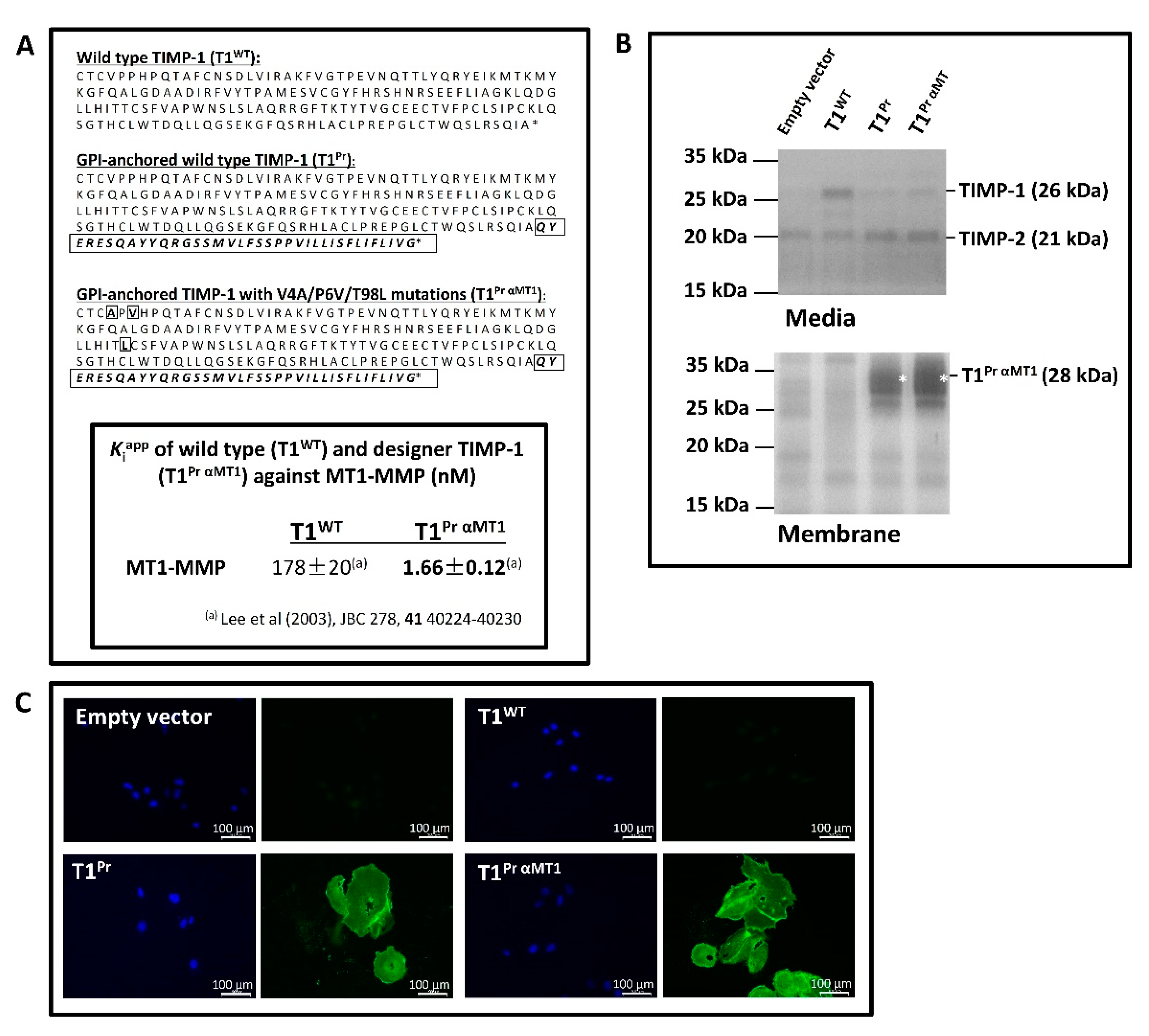
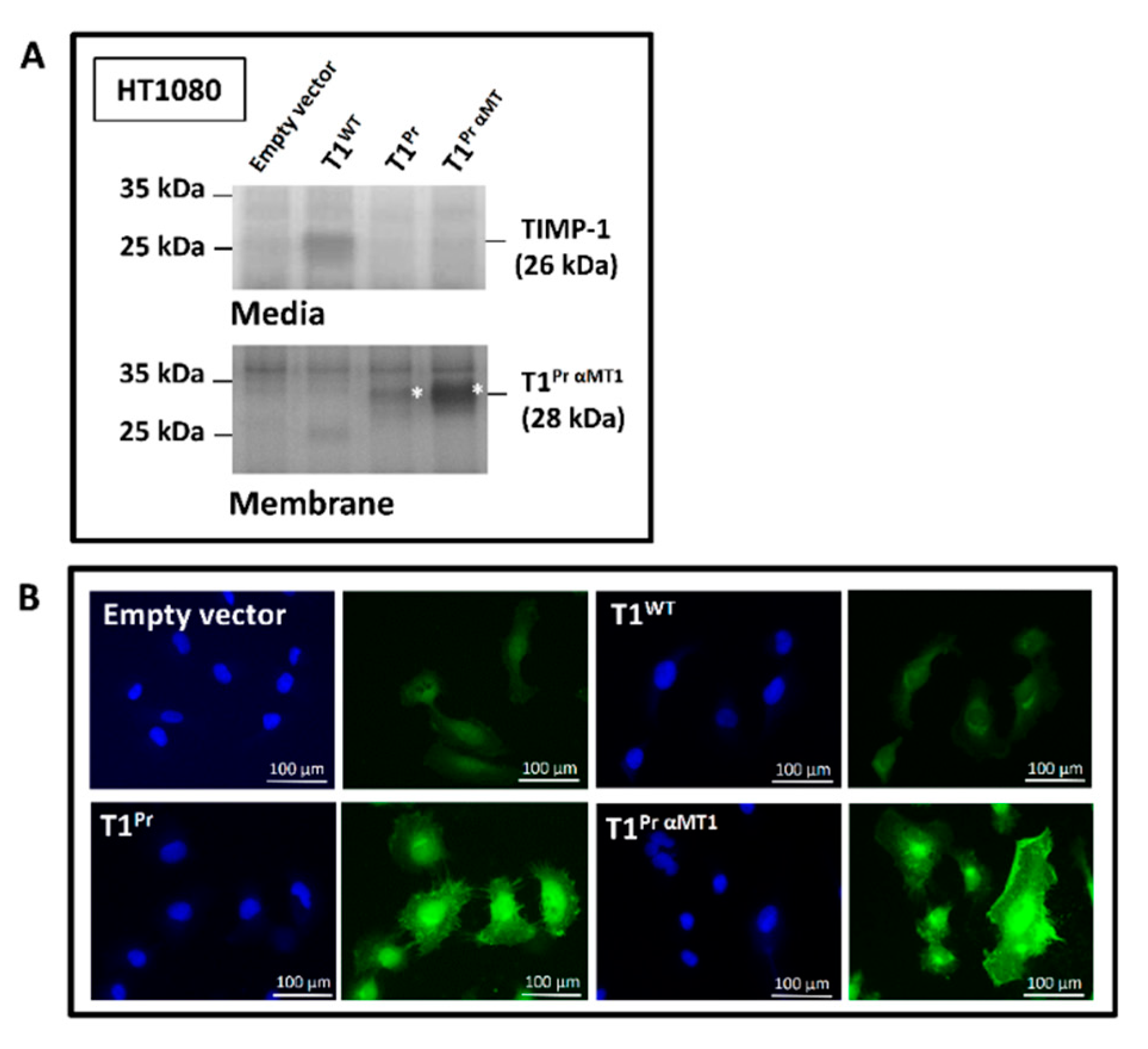
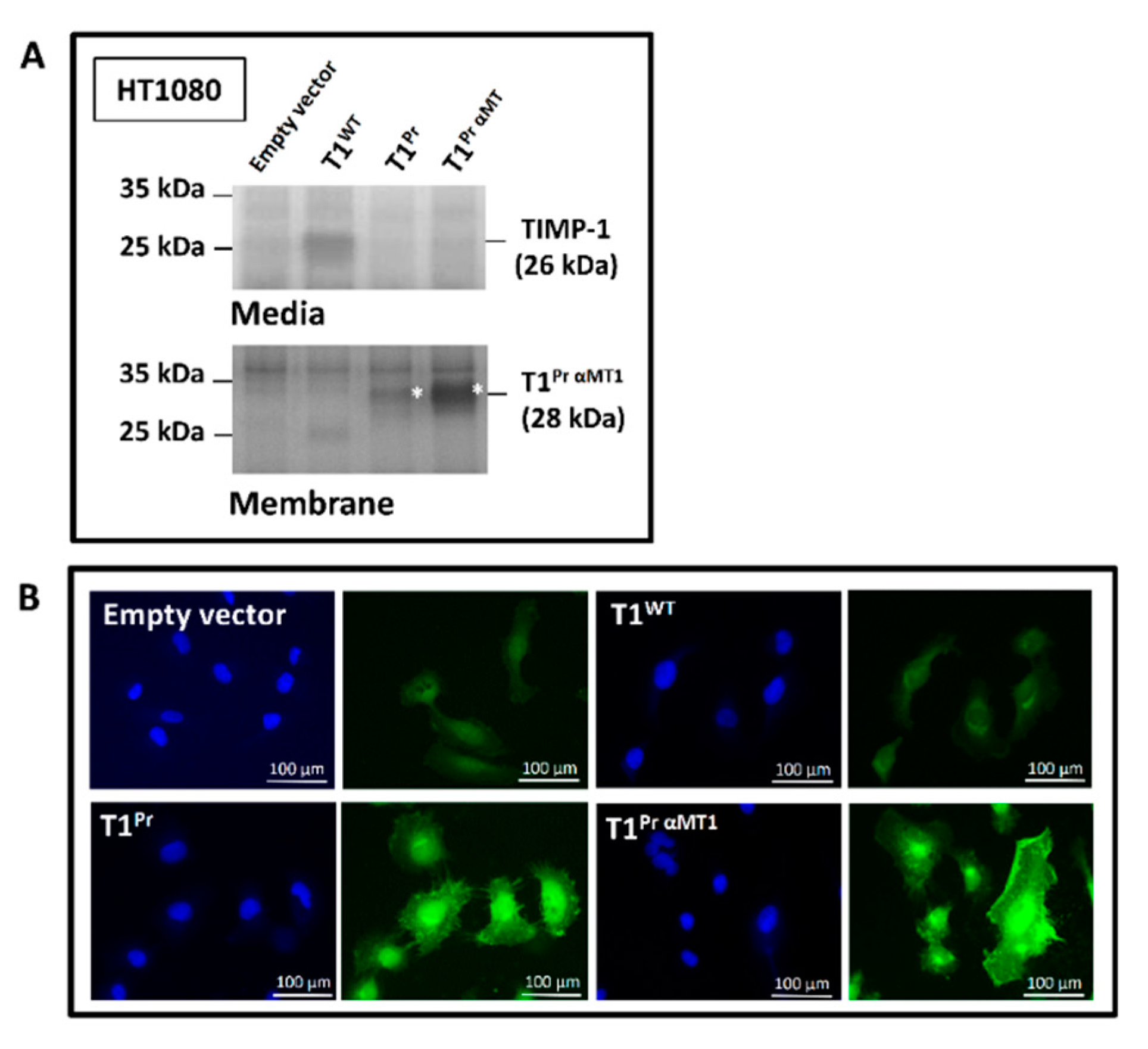
2.1. “T1Pr αMT1”: A Membrane-Tethered, High-Affinity Designer TIMP-1 Tailored Specifically for MT1-MMP Inhibition
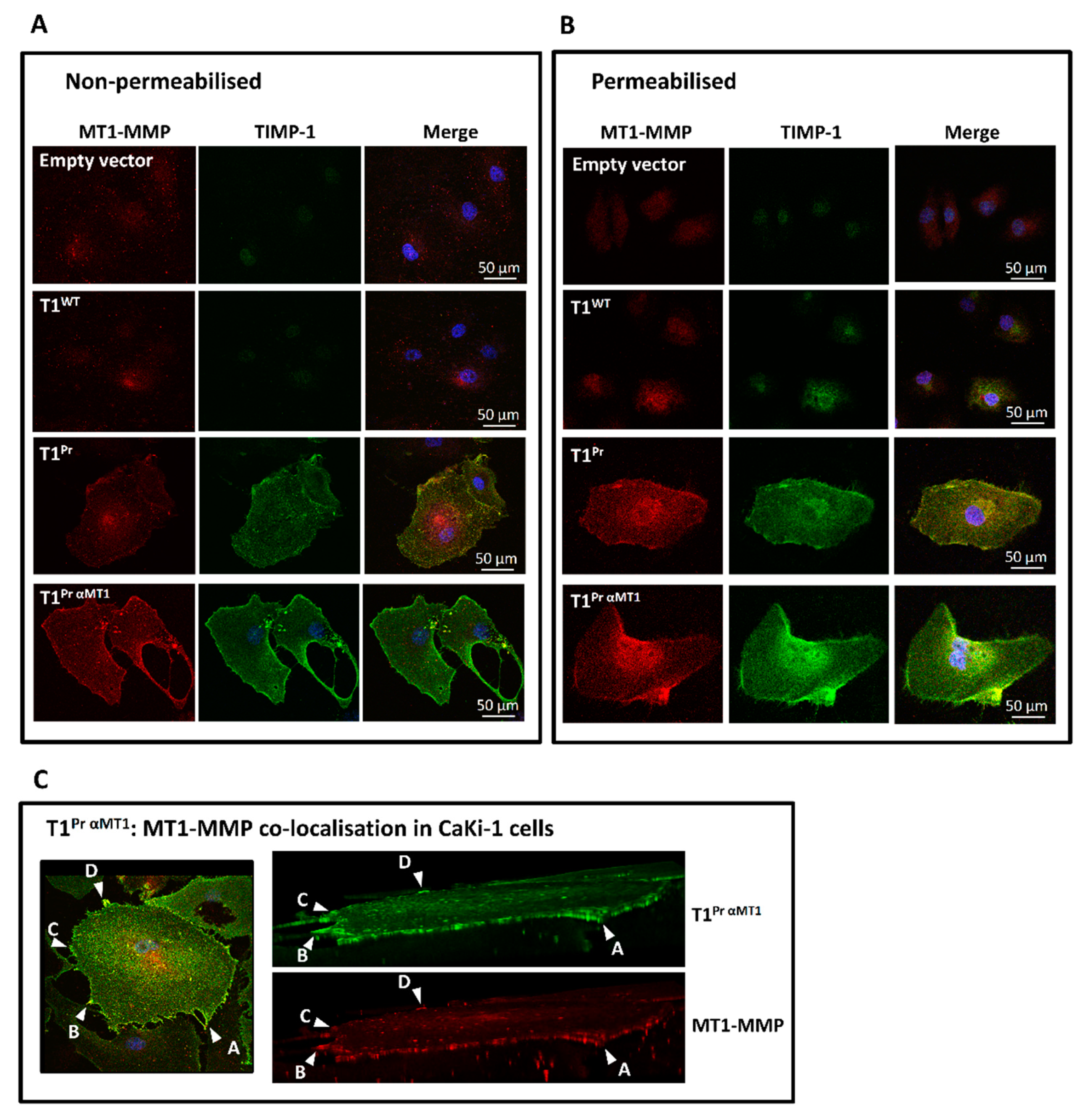
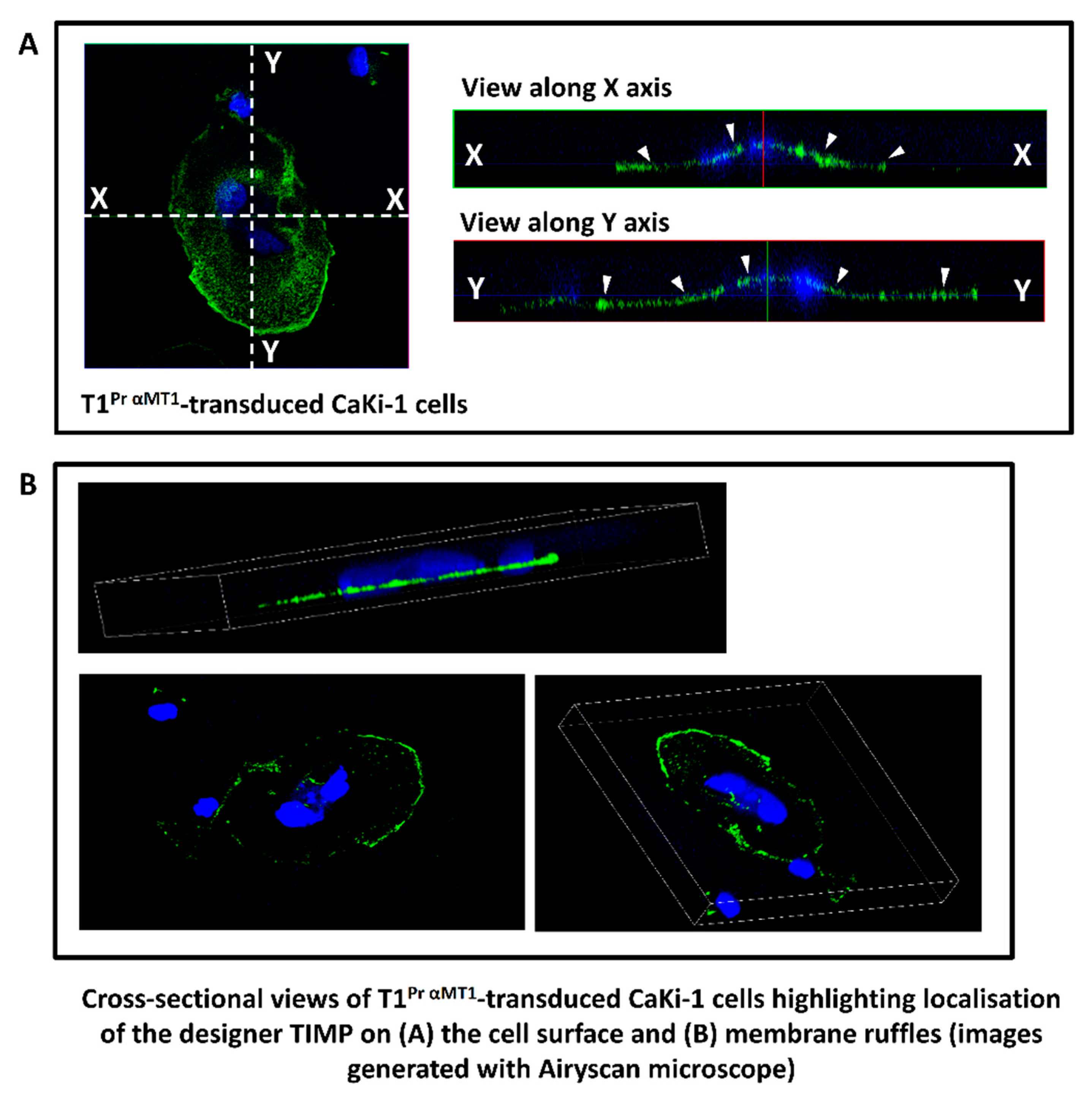
2.2. Localisation of T1Pr αMT1 on CaKi-1 Cell Surface and Membrane Ruffles
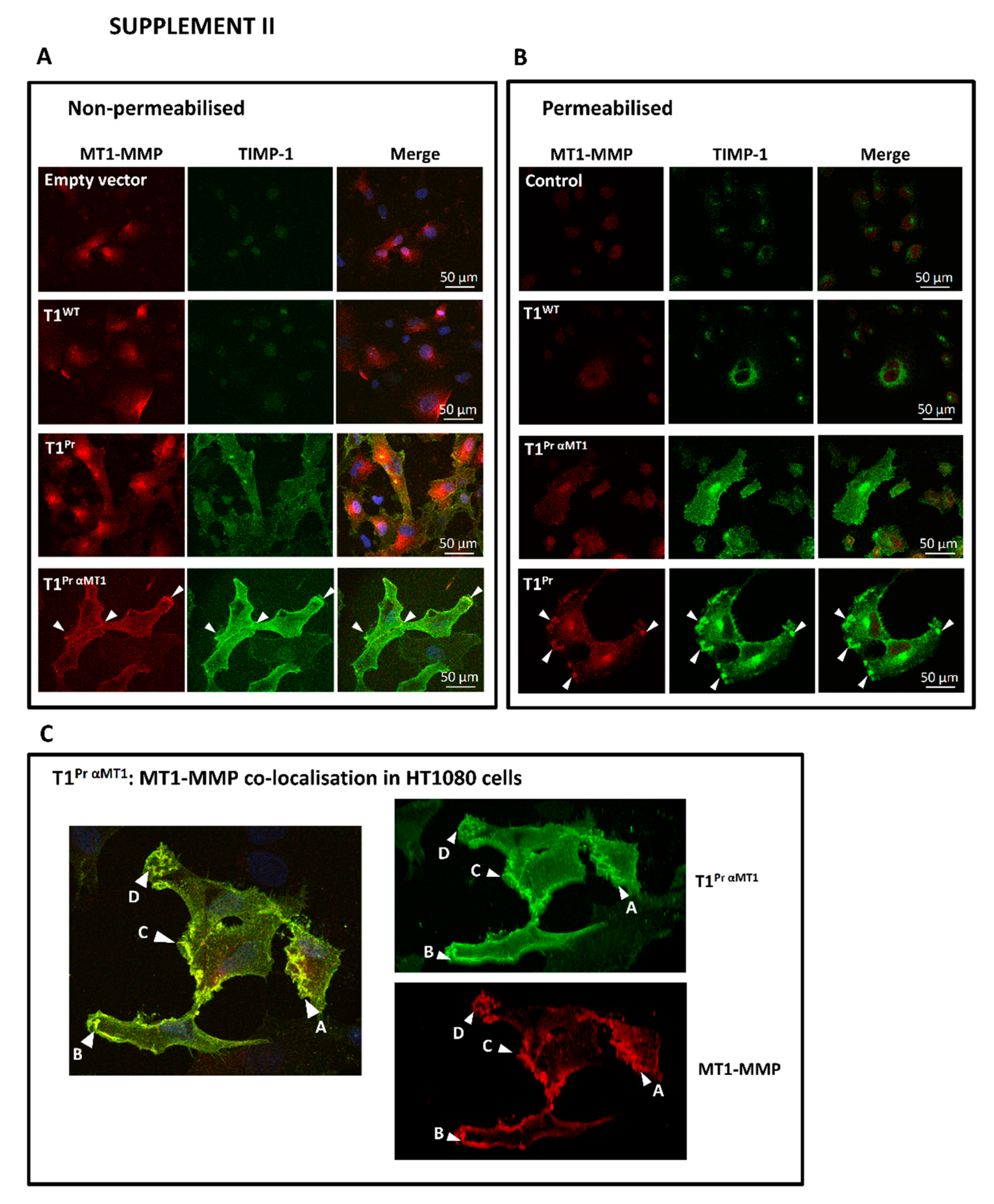
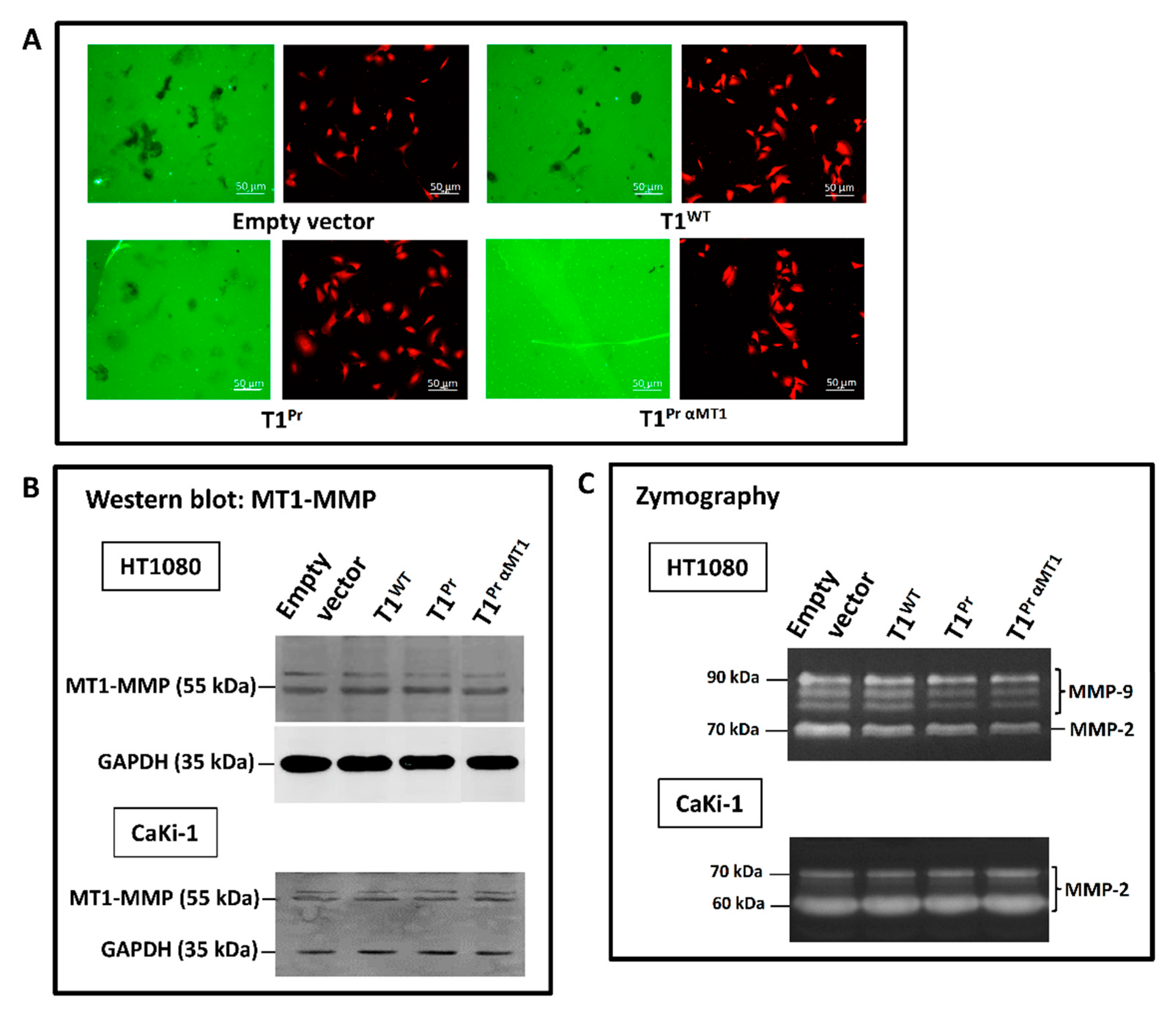
2.3. Superb Co-Localisation of T1Pr αMT1 with Cellular MT1-MMP on the Cell Surface and Membrane Ruffles of CaKi-1 and HT1080 Cells
2.4. T1Pr αMT1 Abolishes MT1-MMP-Mediated Gelatinolytic Capability of HT1080 in Cell-Based Setting
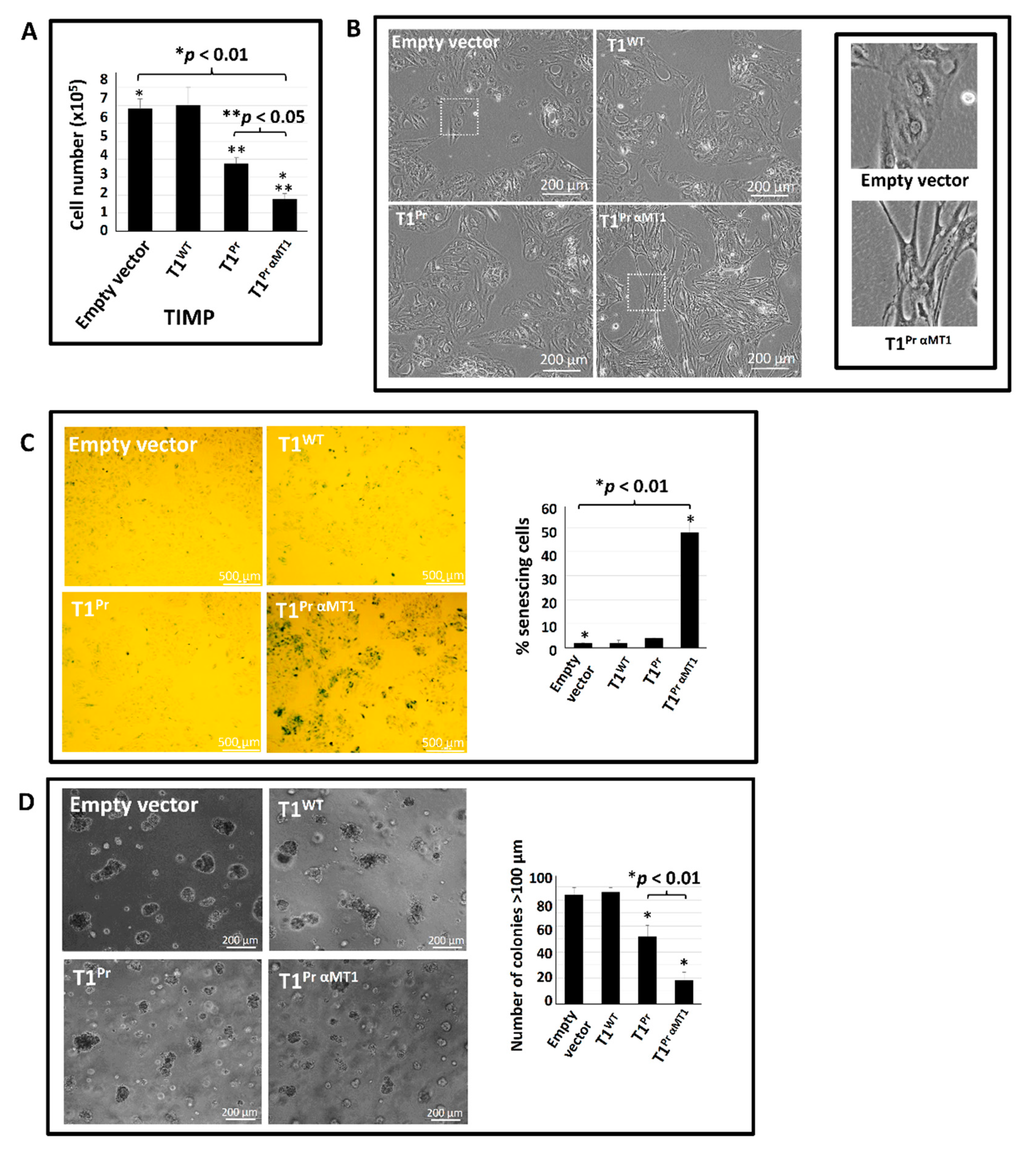
2.5. T1Pr αMT1 Expression Triggers Cellular Senescence in CaKi-1 Cells
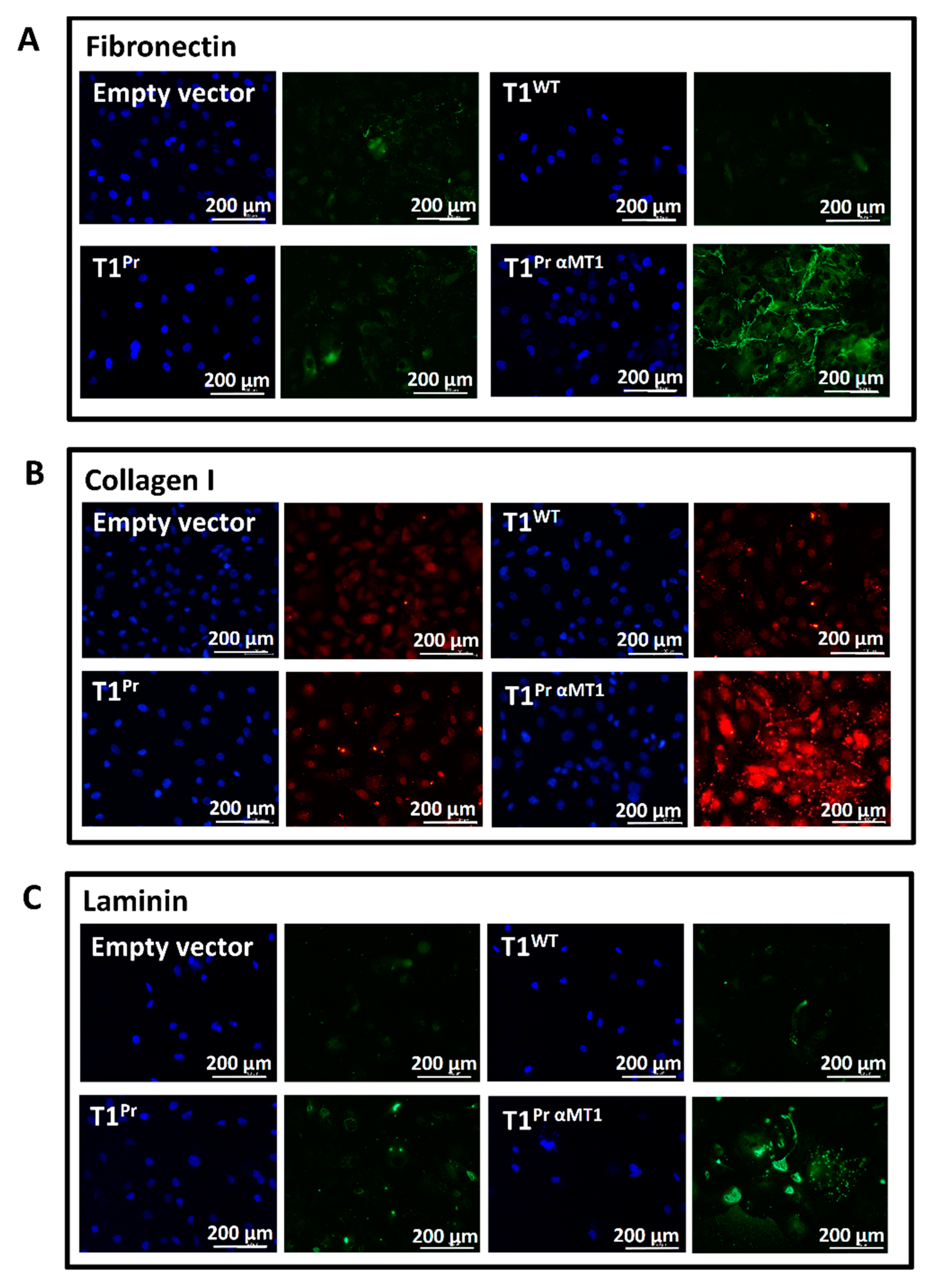
2.6. T1Pr αMT1 Expression Causes Accumulation of Fibronectin, Collagen I and Laminin at the Pericellular Matrices
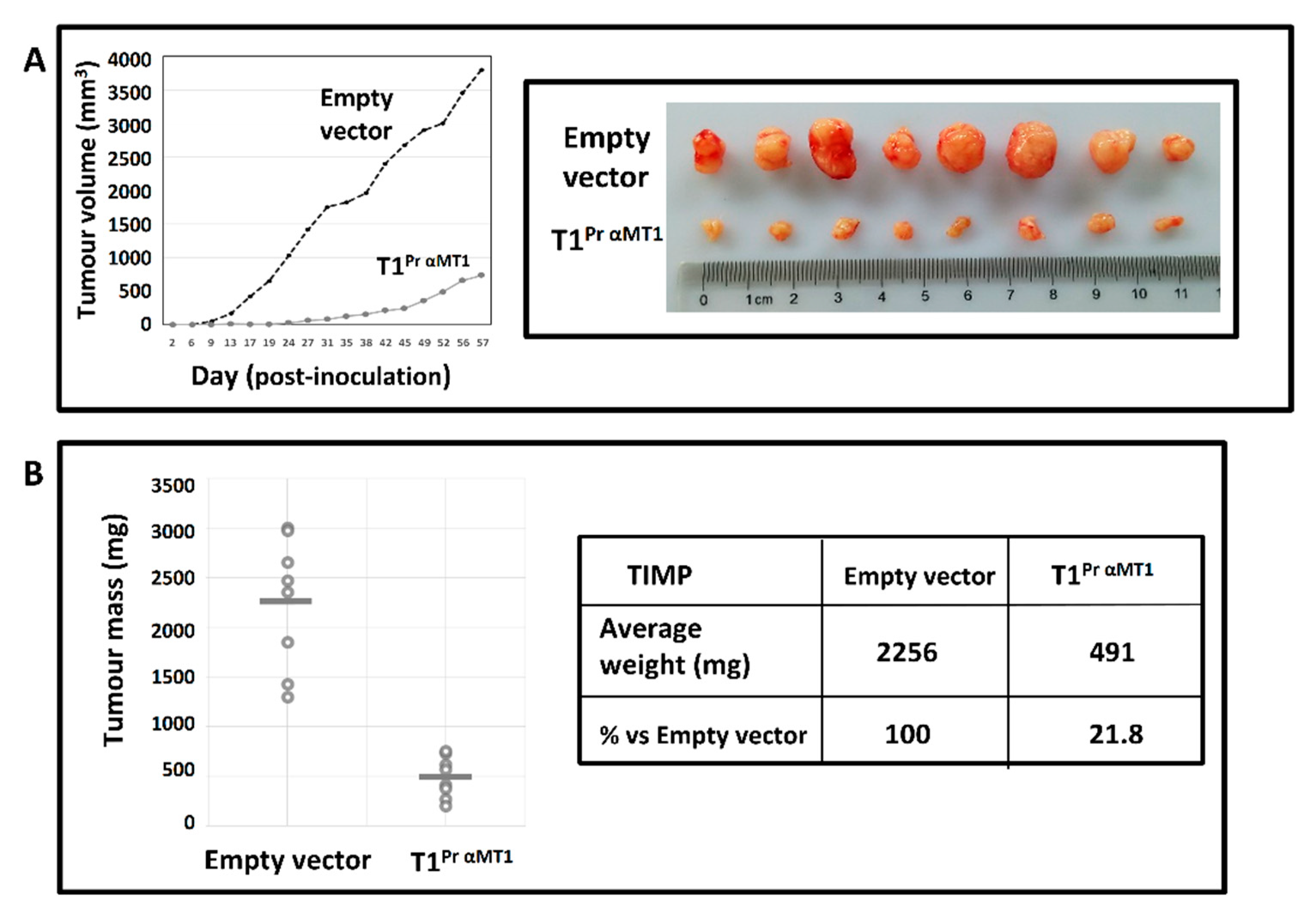
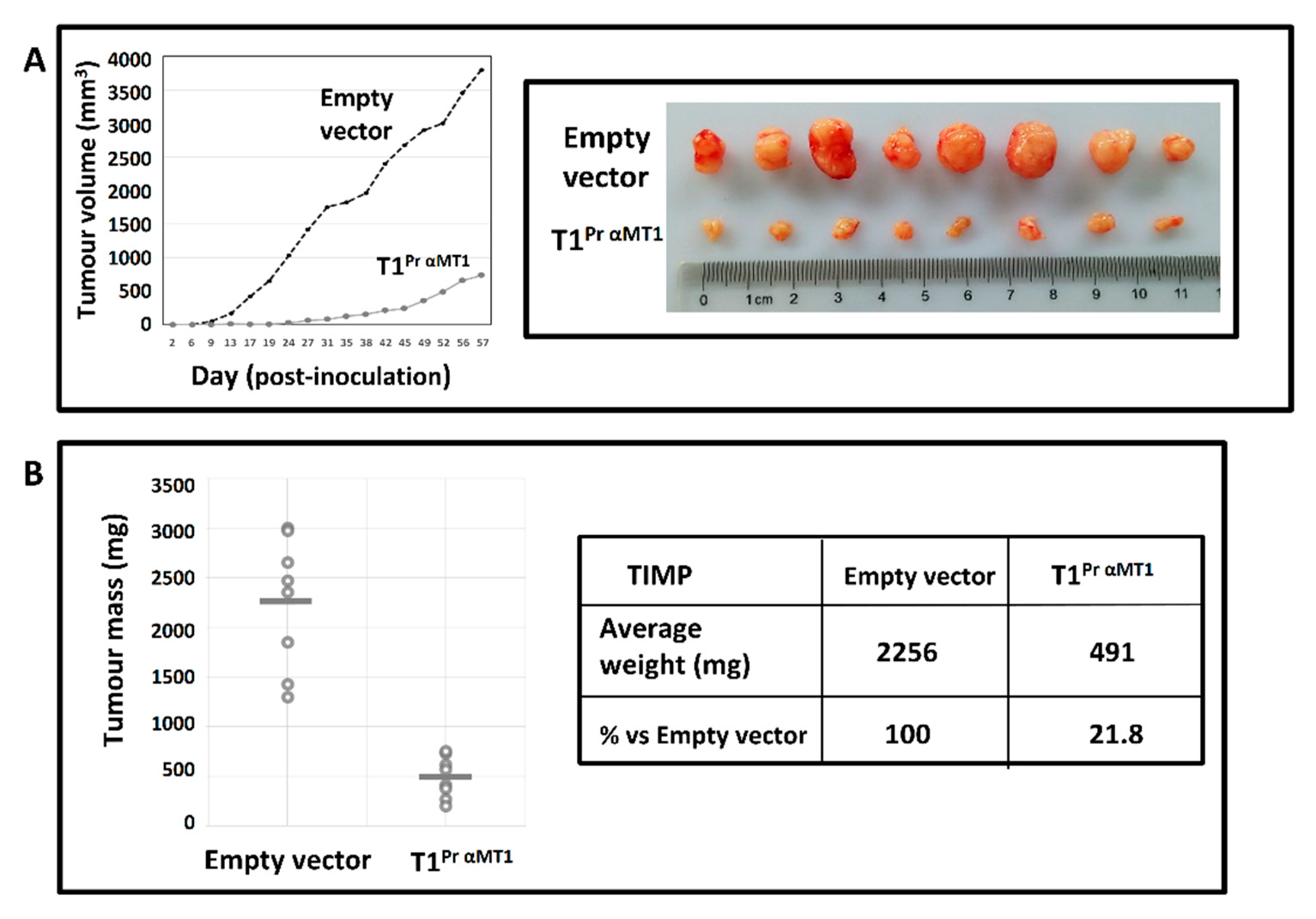
2.7. T1Pr αMT1 Inhibits CaKi-1 Growth in NOD/SCID Xenograft
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cloning, Lentiviral Transduction, Reverse Zymography and Immunofluorescence Microscopy
4.3. Gelatin Degradation Assay
4.4. Cellular Senescence Assay with X-Gal
4.5. Renal Carcinoma Clonogenic Development in Matrigel
4.6. Tumour Development Study in Non-Obese Diabetic/Severe Combined Immuno-Deficient (NOD/SCID) Model
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ECM | extracellular matrix |
| GPI | glycosyl-phosphatidyl inositol |
| MMP | matrix metalloproteinase |
| MT1-MMP | membrane type 1-matrix metalloproteinase |
| TIMP | tissue inhibitor of matrix metalloproteinase |
Appendix A

References
- Malemud, C.J. Matrix metalloproteinases (MMPs) in health and disease: An overview. Front. Biosci. 2006, 11, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Murphy, G. Matrix metalloproteinases at a glance. J. Cell Sci. 2004, 117, 4015–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Turunen, S.P.; Tatti-Bugaeva, O.; Lehti, K. Membrane-type matrix metalloproteases as diverse effectors of cancer progression. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Bedogni, B. The membrane tethered matrix metalloproteinase MT1-MMP at the forefront of melanoma cell invasion and metastasis. Pharmacol. Res. 2016, 111, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef] [PubMed]
- Sabbota, A.L.; Kim, H.R.; Zhe, X.; Fridman, R.; Bonfil, R.D.; Cher, M.L. Shedding of RANKL by tumor-associated MT1-MMP activates Src-dependent prostate cancer cell migration. Cancer Res. 2010, 70, 5558–5566. [Google Scholar] [CrossRef]
- Thathiah, A.; Carson, D.D. MT1-MMP mediates MUC1 shedding independent of TACE/ADAM17. Biochem. J. 2004, 382, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rosse, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Nishibashi, K.; Itoh, Y.; Yana, I.; Seiki, M. Competitive disruption of the tumor-promoting function of membrane type 1 matrix metalloproteinase/matrix metalloproteinase-14 in vivo. Mol. Cancer Ther. 2005, 4, 1157–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrella, B.L.; Brinckerhoff, C.E. Tumor cell invasion of von Hippel Lindau renal cell carcinoma cells is mediated by membrane type-1 matrix metalloproteinase. Mol. Cancer 2006, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Inki, P.; Jalkanen, M. The role of syndecan-1 in malignancies. Ann. Med. 1996, 28, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Rapti, M.; Murphy, G. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J. Biol. Chem. 2003, 278, 40224–40230. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Rapti, M.; Knauper, V.; Murphy, G. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J. Biol. Chem. 2004, 279, 17562–17569. [Google Scholar] [CrossRef] [PubMed]
- Stracke, J.O.; Hutton, M.; Stewart, M.; Pendas, A.M.; Smith, B.; Lopez-Otin, C.; Murphy, G.; Knauper, V. Biochemical characterization of the catalytic domain of human matrix metalloproteinase 19. Evidence for a role as a potent basement membrane degrading enzyme. J. Biol. Chem. 2000, 275, 14809–14816. [Google Scholar] [CrossRef]
- Lee, M.H.; Atkinson, S.; Rapti, M.; Handsley, M.; Curry, V.; Edwards, D.; Murphy, G. The activity of a designer tissue inhibitor of metalloproteinases (TIMP)-1 against native membrane type 1 matrix metalloproteinase (MT1-MMP) in a cell-based environment. Cancer Lett. 2010, 290, 114–122. [Google Scholar] [CrossRef]
- Shingarev, R.; Jaimes, E.A. Renal Cell Carcinoma: New Insights and Challenges for a Clinician Scientist. Am. J. Physiol. Renal. Physiol. 2017. [Google Scholar] [CrossRef]
- Merza, H.; Bilusic, M. Current Management Strategy for Metastatic Renal Cell Carcinoma and Future Directions. Curr. Oncol. Rep. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Petrella, B.L.; Vincenti, M.P. Interleukin-1beta mediates metalloproteinase-dependent renal cell carcinoma tumor cell invasion through the activation of CCAAT enhancer binding protein beta. Cancer Med. 2012, 1, 17–27. [Google Scholar] [CrossRef]
- Goold, R.; McKinnon, C.; Rabbanian, S.; Collinge, J.; Schiavo, G.; Tabrizi, S.J. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J. Cell Sci. 2013, 126, 3552–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Zhang, Y.; Liu, J.; Tsigkou, A.; Rapti, M.; Lee, M.H. Ensnaring membrane type 1-matrix metalloproteinase (MT1-MMP) with tissue inhibitor of metalloproteinase (TIMP)-2 using the haemopexin domain of the protease as a carrier: A targeted approach in cancer inhibition. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotary, K.B.; Allen, E.D.; Brooks, P.C.; Datta, N.S.; Long, M.W.; Weiss, S.J. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell 2003, 114, 33–45. [Google Scholar] [CrossRef]
- Duan, J.X.; Rapti, M.; Tsigkou, A.; Lee, M.H. Expanding the Activity of Tissue Inhibitors of Metalloproteinase (TIMP)-1 against Surface-Anchored Metalloproteinases by the Replacement of Its C-Terminal Domain: Implications for Anti-Cancer Effects. PLoS ONE 2015, 10, e0136384. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, B.; Liu, J.; Lee, M.H. Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy. Molecules 2019, 24, 255. https://doi.org/10.3390/molecules24020255
Jiang B, Liu J, Lee MH. Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy. Molecules. 2019; 24(2):255. https://doi.org/10.3390/molecules24020255
Chicago/Turabian StyleJiang, Bingjie, Jian Liu, and Meng Huee Lee. 2019. "Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy" Molecules 24, no. 2: 255. https://doi.org/10.3390/molecules24020255
APA StyleJiang, B., Liu, J., & Lee, M. H. (2019). Targeting a Designer TIMP-1 to the Cell Surface for Effective MT1-MMP Inhibition: A Potential Role for the Prion Protein in Renal Carcinoma Therapy. Molecules, 24(2), 255. https://doi.org/10.3390/molecules24020255