Chemical Property Calculation through JavaScript and Applications in QSAR

Department of Chemistry, University of Wisconsin-Milwaukee, Milwaukee, WI 53201, USA
Molecules 1999, 4(1), 16-27; https://doi.org/10.3390/40100016
Submission received: 30 January 1998
/
Accepted: 1 July 1998
/
Published: 8 February 1999
Abstract
:The inorganic property (I) and organic property (O) values of general organic groups are re-proposed here. Both I and O values of drug and biological molecules or groups can be calculated based on their common group values. The calculation can be performed easily on-line through JavaScript. Similar calculation can be done for the drug and biological molecular group electronegativity (X) according to the author's published paper. The calculation of lipophilicity (π or logP) parameter of (macro)molecules (like proteins) can also be performed on-line through JavaScript. Two equations expressed with I and O are provided here to define the hydrophobicity of each amino acid. The correlations of inorganic property and organic property values with other parameters are also discussed. These calculated parameters combined with other parameters can be used for QSAR studies in some drug molecules.
Introduction
Quantitative structure-activity relationship (QSAR) correlation has been widely applied in biological activities over several decades. Many new descriptors (parameters) have been developed [1,2,3,4,5]. Six main types of molecular descriptors were introduced: constitutional, geometrical, topological, electrostatic, quantum-chemical, and thermodynamic descriptors, the calculations of these descriptors using different packages were introduced by Katritzky and coworkers [1]. Inorganic property and organic property values [6,7], and group electronegativity [8] were also calculated solely on the basis of intrinsic structural information of the molecular species under consideration. Obviously the group electronegativity belongs to the electrostatic descriptors, and reflects the characteristics of the partial charge of the group. According to the author's previous work [8], group electronegativity is highly correlated with proton chemical shift in R-H molecules (R is the group). The calculation of inorganic property and organic property values of organic molecules as well as group electronegativity were calculated through JavaScript [9], which is a powerful language used in the Internet [10]. Other parameters (like π or logP value) can also be calculated through JavaScript. The correlation of inorganic property and organic property values of organic molecules or groups with other parameters are analyzed here. The usage of these descriptors in QSAR studies of some drugs is also discussed.
Calculation
Before introducing the calculation of inorganic property and organic property values and group’s electronegativity using JavaScript, the author first gives a simple example: It is common for chemists to calculate the molecular weight of organic or biological molecules (amino acids or nucleic acids). The common atoms in organic or biological molecules are carbon (C), hydrogen (H), oxygen (O), nitrogen (N), sulfur (S), phosphorus (P), and others (like chloride (Cl), fluoride (F), bromide (Br), water (W or w) etc.). To calculate the molecular weight of organic or biological molecules using JavaScript, the users only need to input the type of atoms (either lower case or upper case for the first symbol of atoms, such as: for chloride, input "Cl" or "cl"; for carbon, input "C" or "c") and the numbers of the atoms. There are a total of six rows to input a maximum of six types of atoms in one calculation, which are sufficient for common organic or biological molecules. The molecular weight of some metal organic molecules can also be calculated (for iron, input "Fe" or "fe" in one of the six rows; for cobalt, input "Co" or "co"). The molecular weight calculation is shown at the URL of http://www.unibas.ch/mdpi/ecsoc/e0002/mwcalcf.htm. The calculation for logP is similar to that for molecular weight, the symbols of groups can be obtained from the references [11,12], and the appendix or the source codes. Modification of the symbols can be done by the readers at their own convenience. For large and complicated groups of organic and drug molecules, the structure of the groups can be shown on screen, and the numbers of groups are needed to input from the user to calculate the organic or inorganic property values of the groups. Both the numbers of sub-groups or atoms and the values of their electronegativity are needed to calculate the electronegativity of large groups. Other parameters can also be calculated using JavaScript, in a similar way to the calculation of logP.
| Molecular Weight Calculating Spreadsheet |
| http://www.unibas.ch/mdpi/ecsoc/e0002/mwcalcf.htm |
| logP Calculating using JavaScript |
| http://www.unibas.ch/mdpi/ecsoc/e0002/logpcalc.htm |
| Inorganic property and organic property values of groups. Calculation of Inorganic property value of molecules or groups using JavaScript. Calculation of Organic property value of molecules or groups using JavaScript. |
| Http://www.unibas.ch/mdpi/ecsoc/e0002/calinorg.htm |
| Calculation of Group Electronegativity using JavaScript. |
| Http://www.unibas.ch/mdpi/ecsoc/e0002/calelecg.htm |
Results
The inorganic property and organic property values of common organic or biological molecule groups are listed in Table 1. The author calculated the inorganic and organic property values of 50 common groups, and analyzed their correlation with other parameters, lipophilicity (π), polar constant (F), molar refractivity (Mr), resonance constant (R), Hammett meta constant (σm), and para constant (σp). All the parameters can be seen in Table 2, and the correlations among them can be seen in Table 3. A plot of inorganic property value versus organic property value of molecule groups is shown in Figure 1. It is clear that two sets of molecule groups can be divided: one set is the hydrophobic molecule groups with π (pi) > 0.1, the other set is the hydrophilic molecule groups with π (pi) < 0.1.
From Table 3, it is shown that there are high correlations among π and I or O (see the equations below), and between Mr and O (r = 0.837). It is also shown that F has high correlation with σm and σp; σp has high correlation with R and σm; X has some correlation with F (r = 0.637); F, R, σm, σp, and X have less correlation with I or O (see Table 3).
The inorganic property and organic property values of twenty amino acids are also calculated (see Table 4). The correlation between inorganic and organic property values of these 20 amino acids is only 0.054. The correlation of logP value of twenty amino acid and inorganic property and organic property values can be seen from Table 4 and from the following equations:
- logP = 5.0863 - 0.01618 I
- n = 20, r = 0.7420, s = 1.0423, F = 22.04
- logP = -1.5130 + 0.01710 O
- n = 20, r = 0.5256, s = 1.3226, F = 6.87
- logP = 3.3275 - 0.01685 I + 0.01847 O
- n = 20, r = 0.9337, s = 0.5729, F = 57.78
Applications
Like logP, most hydrophobicity scales described in reference
[12] are highly correlated with inorganic property and organic property values
of the twenty amino acids. So, the hydrophobic scale can also be defined by I and O values. Two equations can be obtained:
| H1 = I/O - 3, if H1 < 0, the amino acid is hydrophobic, otherwise hydrophilic; H2 = I - O - 160, if H2 < 0, the amino acid is hydrophobic, otherwise hydrophilic. |
| http://www.unibas.ch/mdpi/ecsoc/e0002/aa_io.htm |
A plot of O vs I can be seen from Figure 2. As shown in Figure 1, two sets of amino acids can be seen; one set is more hydrophilic with π < 0.0, the other set is more hydrophobic with π > 0.0.
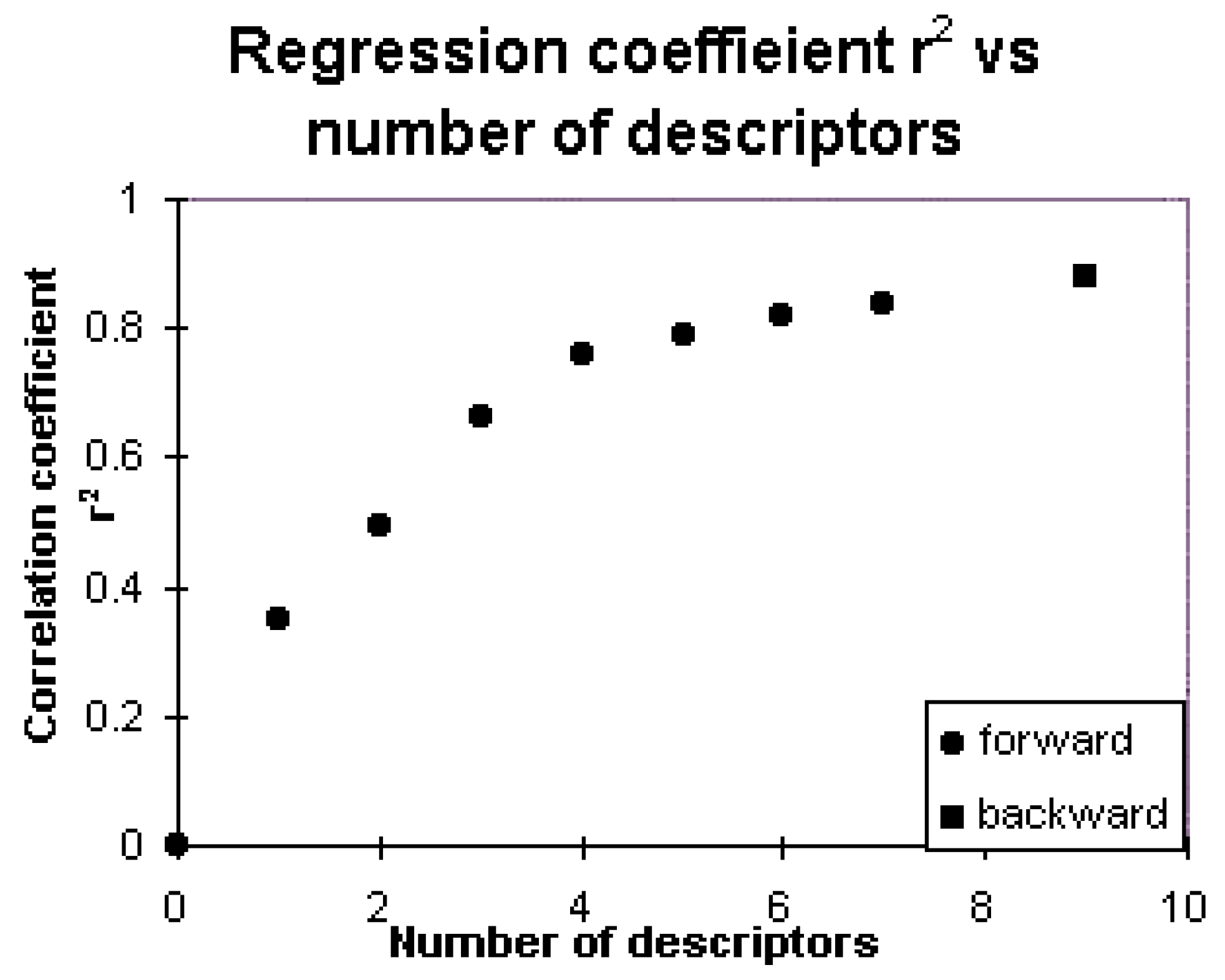
A new group electronegativity scale has been proposed in the reference [8], and its comparison with other scales of group electronegativity has been discussed. The application of group electronegativity, inorganic property and organic property values of drug molecules have been successfully used in QSAR studies of some drug molecules [13,14,15,16,17]. To further test the usefulness of group electronegativity, inorganic property and organic property values, the common data set used in recent published papers was selected and tested (the detail data set and descriptions can be seen in literature [18,19]). Maddalena and Johnston [18] used ten final descriptors out of 6×7 = 42 descriptors (π7, Mr1, Mr2, Mr6, F7, F2, R1, σm3, σn8, μ1), found that using artificial neural network methods which gave a high value of correlation coefficients for both training (0.938) and cross-validation (0.896) to perform similar study. Sung-Sau So and Martin Karplus [19] used six descriptors ( π7, F7, Mr1, σm2, π6, Mr8), which gave QSAR results as good or even better than those calculated using higher dimensions by genetic QSAR neural networks [20]. The author does not focus on the methods for QSARs, but focuses on the parameters proposed here. The electronegativity for each of six groups was calculated, as well as the sum of the inorganic property and organic property values, and the sum of the lipophilicity (π) value. The thirteen parameters used in the references [18,19] were also used here. Therefore, in total, twenty-two descriptors were used here for fifty-seven benzodiazepines QSAR studies, using the Minitab program (a common statistical program). By using forward selection method and selecting up to seven descriptors, no proposed parameters were selected, but most of them are among the best ten alternative descriptors, which means that they can be substituted for other descriptors if they are not available. By using a backward elimination method, the proposed parameters are useful in nine-descriptor-QSAR equation (see below). Table 5 gives the summary, and Figure 3 shows the plot of the correlation (r2) versus number of descriptors used in the equation.
logIC50 = 2.9561 - 0.64431XR7 - 0.34616 XR2′ + 0.6575 XR8 - 0.012646 O - 0.30804 π7
+ 0.16273 MR1 + 1.1648 π6 + 0.10732 MR8 + 2.6698 R1
+ 0.16273 MR1 + 1.1648 π6 + 0.10732 MR8 + 2.6698 R1
- n = 57, r2 = 0.878, s = 0.2779, F = 37.58
Discussion
It is known that inorganic property and organic property values are good intrinsic descriptors, which reflect the inorganic property and organic property of the organic and biological molecules or groups. Only carbon hydrates give pure organic property values. Other organic molecules and biological molecules with nitrogen and oxygen atoms have both inorganic and organic property values. Urea (NH2)2CO and carbon dioxide CO2 still have organic property values of 20. An organic molecule or a biological molecule with pure inorganic property value and without organic property value has never been found. According to the inorganic property and organic property values of the common groups shown in Table 1, it is found that most groups, even sulfur (S) or chloride (Cl), also have partial organic property values. The correlation of inorganic property and organic property values of 20 amino acids with other amino acid or residue parameters (like logP) are also extensively studied by the author. The difference between the inorganic property and organic property values, or their ratios, of the amino acids, can be used to identify which amino acids are hydrophobic or hydrophilic. Table 1 cannot cover all the possible groups (e.g. group). The author assigns the inorganic property and organic property values of this group as 10 and 50, respectively. The advantage of using group electronegativity is that all the group electronegativities can be calculated.
The usage of inorganic property and organic property values of drug and biological molecules is that they can replace the π or Mr descriptors, which may be useful in some QSAR studies [21]. The group electronegativity can provide an additional descriptor for each variable group in the drug molecules (for example, in benzodiazepine/GABAA receptors, there are six variable groups (R7, R1, R2′, R6′, R3, R8), and these six group electronegativities Xi can be used as additional descriptors for QSAR studies).
Conclusions
The inorganic and organic property values reflect the inorganic and organic properties of the organic or biological molecules or groups, which mainly reflect the hydrophilic or hydrophobic characteristics. Group electronegativity reflects the electrostatic properties of the groups. JavaScript is an easy tool for chemists to calculate the molecular weight, inorganic property and organic property values, and the group electronegativity of organic or biological molecules, as well as other types of descriptor (like logP). The proposed descriptors are useful in QSAR studies of high-dimensional and large-number-sample systems.
Appendix
Appendix I. Hydrophobic Fragmental Constant of Groups and Amino Acid Residues
The Hydrophobic Fragmental Constant of Groups and Amino Acid Residues can be seen at the URL http://www.unibas.ch/mdpi/ecsoc/e0002/app.htm.
References
- Katritzky, A. A.; Rachwal, P.; Law, K. W.; Karelson, M.; Lobanov, V. S. Prediction of polymer glass transition temperatures using a general quantitative structure-property relationship treatment. J. Chem. Inf. Comput. Sci. 1996, 36, 879–884. [Google Scholar] [CrossRef]
- Bravi, G.; Gancia, E.; Zaliani, A. MS-WHIM, New 3D Theoretical Descriptors Derived from Molecular Surface Properties: A Comparative 3D QSAR Study in A Series of Steroids. J. Computer-aided Mol. Design 1997, 11, 79. [Google Scholar] [CrossRef]
- Estrada, E.; Ramirez, A. Edge Adjacency Relationships and Molecular Topographic Descriptors. Definition and QSAR Applications. J. Chem. Inf. Comput. Sci. 1996, 36, 837. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-Chemical Descriptors in QSAR/QSPR Studies. Chem Rev. 1996, 96, 1027. [Google Scholar]
- Katrizky, A.P.; Gordeeva, E.V. Traditional Topological Indices vs. Electronic, Geometrical, and Combined Molecular Descriptors in QSAR/QSPR Research. J. Chem. Inf. Comput. Sci. 1993, 33, 835. [Google Scholar] [CrossRef]
- Fujita, M. Organic Analysis; Kakoya bookstore: Tokyo, 1930; p. 33. (in Japanese) [Google Scholar]
- Yuan, L.B.; Ding, Y. Organic Conceptional Diagram and Its Application in Surface Chemistry. Huaxue Tongbao 1988, (2), 19–21. [Google Scholar]
- Wu, H. Study of Group Electronegativities in Biological and Drug Molecules. Zhongguo Kexue Jishu Daxue Xuebao 1990, 20, 517–24. [Google Scholar]
- JavaScript. See http://home.netscape.com/eng/mozilla/3.0/handbook/javascript/index.html.
- Mosley, D.H.; André, J-M. Use of JavaScript in Simple Quantum Chemical Applications. ECCC3. November 1996. at http://hackberry.chem.niu.edu/ECCC3/.
- Rekker, R.F. The Hydrophobic Fragmental Constant, Its Derivation and Application, A Means of Characterizing Membrane Systems; Elsevier: Amsterdam, 1977. [Google Scholar]
- Black, S.D.; Mould, D.R. Development of Hydrophobicity Parameters to Analyze Proteins Which Bear Post- or Cotranslational Modifications. Anal. Biochem. 1991, 193, 72–82. [Google Scholar] [CrossRef]
- Wu, H.; Wen, Y. Computer Aided Anthracycline Anticancer Drugs Design. In 2nd WATOC World Congress, Toronto, 8–14 July 1990.
- Wu, H.; Wen, Y. Quantitative Structure-Activity Relationship Studies of Lincomycin Derivates. In 2nd Symposium on Molecular Mechanics and Drug Design of China, Shanghai, P.R. China, November 1989.
- Wu, H.; Wen, Y. Quantitative Structure-Activity Relationship Studies of Leucomycin and Clindamycin Derivates. In 2nd Symposium on Molecular Mechanics and Drug Design of China, Shanghai, P.R. China, November 1989.
- Wu, H.; Wen, Y. Structure Activity Studies of Podophyllotoxin Derivates. In 2nd Symposium on Molecular Mechanics and Drug Design of China, Shanghai, P.R. China, November 1989.
- Wu, H.; Wen, Y. Structure Activity Studies of Erythromycin Derivates. In 2nd Symposium on Molecular Mechanics and Drug Design of China, Shanghai, P.R. China, November 1989.
- Maddalena, D.J.; Johnston, G.A.R. Prediction of Receptor Properties and Binding Affinity of Ligands to Benzodiazepine/GABAA Receptors Using Articial Neural Networks. J. Med. Chem. 1995, 38, 715–724. [Google Scholar]
- So, S-S.; Karplus, M. Genetic Neural Networks for Quantitative Structure-Activity Relationships: Improvements and Application of Benzodiazepine Affinity for Benzodiazepine/GABAA Receptors. J. Med. Chem. 1996, 39, 5246–5256. [Google Scholar]
- So, S-S.; Karplus, M. Evolution Optimization in Quantitative Structure-Activity Relationship: An Application of Genetic Neural Networks. J. Med. Chem. 1996, 39, 1521–1530. [Google Scholar]
- Gao, H.; Hansch, C. QSAR of P450 Oxidation: On the Value of Comparing Kcat and Km with Kcat/Km. Drug Metabolism Rev. 1996, 28, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: Not available.
Figure 1.
Organic property value versus inorganic property value of 50 common groups.
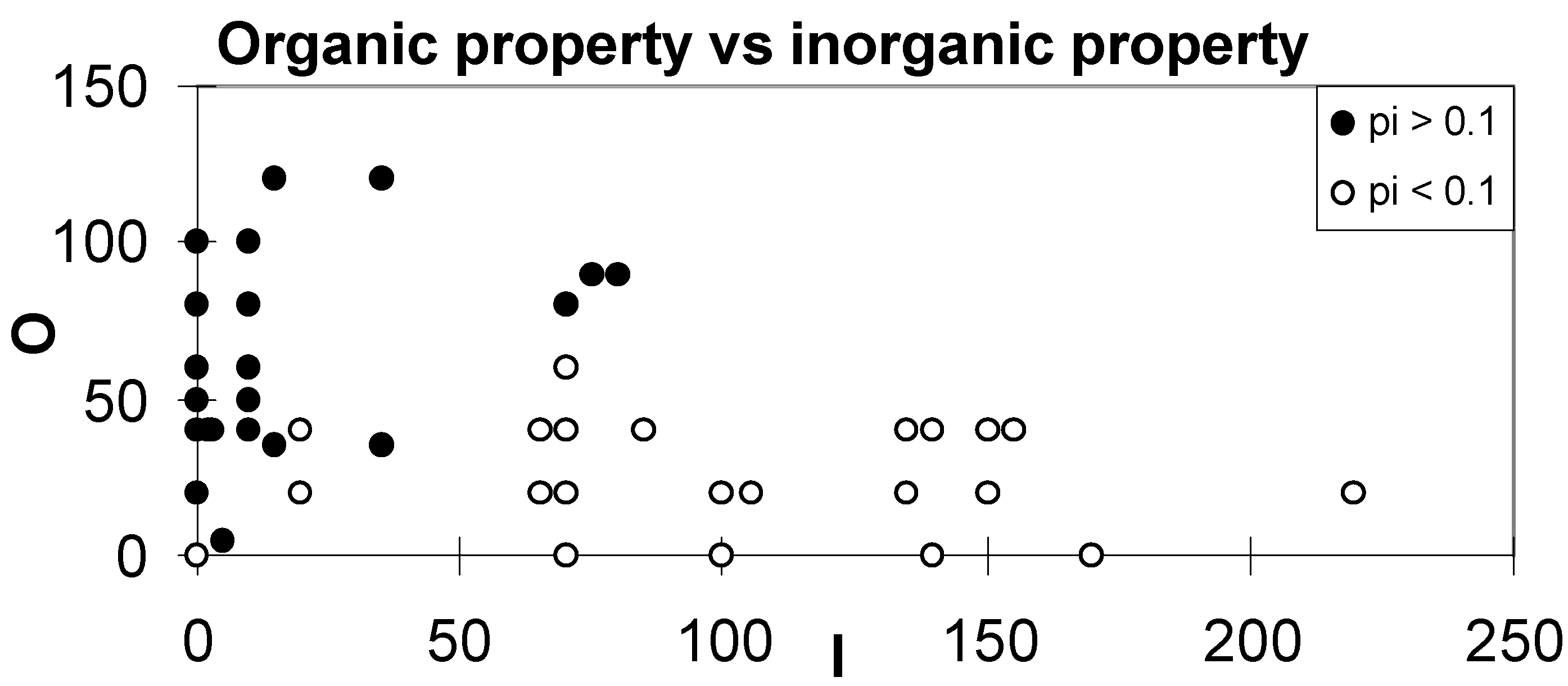
Figure 2.
Organic property value versus inorganic property value of amino acids.
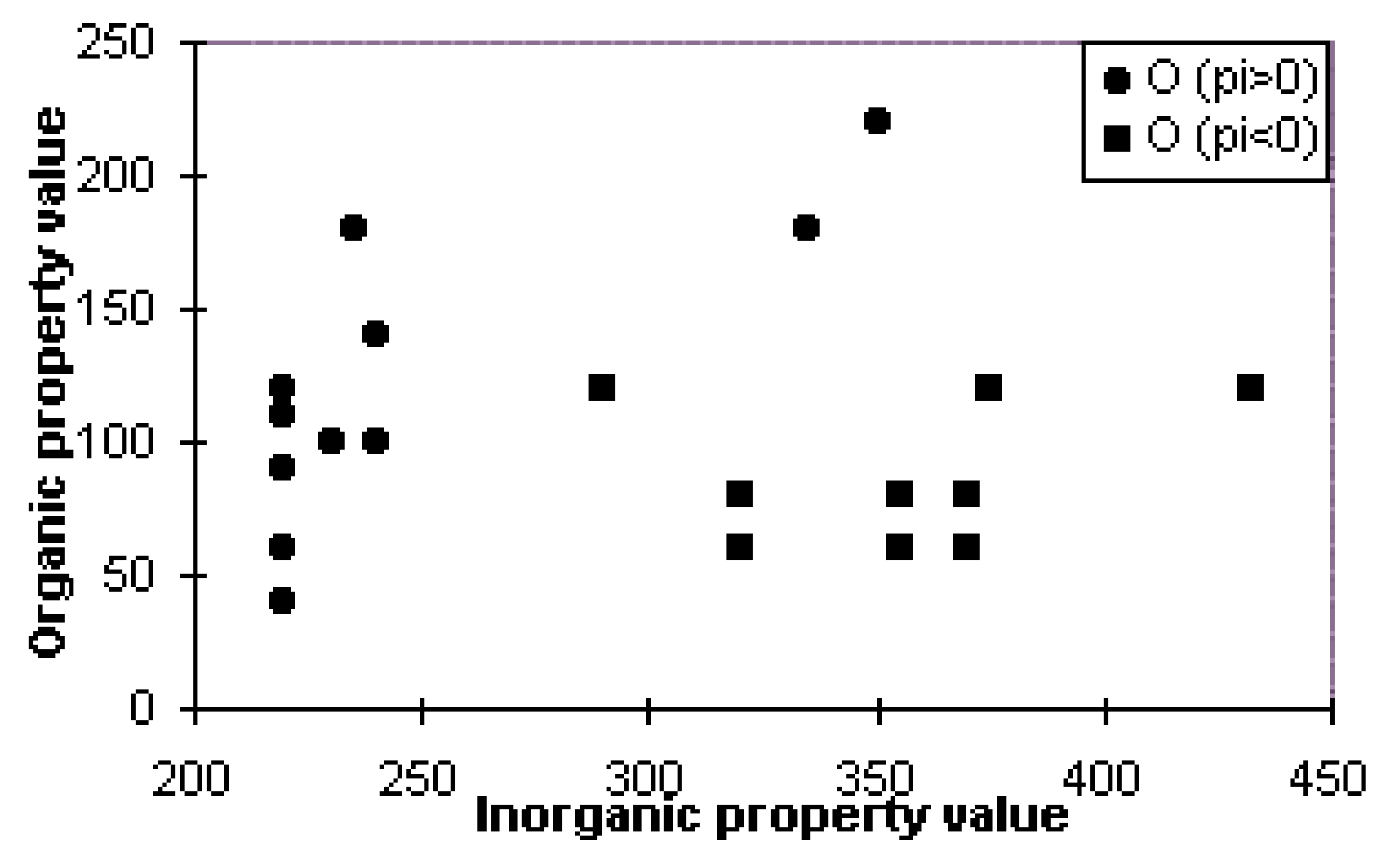
Figure 3.
The correlation r2 versus number of descriptors.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Inorganic property and organic property values of some inorganic groups and organic / inorganic groups†
| Inorganic group | Value | Organic / inorganic group | Organic Value | Inorganic value |
|---|---|---|---|---|
| -SO2-NH- | 240 | -OSO3H | 20 | 220 |
| =N-OH | 210 | >SO2 | 40 | 170 |
| -NHCONH- | ||||
| -CO-NH-* | 200 | >SO | 40 | 140 |
| -COOH | 150 | -SCN | 90 | 80 |
| R-C|H(CH2)n-C=OO| | 120 | -NCS | 90 | 75 |
| -COO- (in a ring above) | ||||
| -CO-O-CO- | 110 | -NO2 | 70 | 70 |
| Anthracene or Phenanthrene nuclei | 105 | -As< | 40 | 70 |
| -CN | ||||
| -OH | 100 | -P< | 20 | 70 |
| >Hg (covalent) | 95 | -O- [CH2CH2O] -CH2- | 30 | 60 |
| -NH-NH- | 80 | -NO | 50 | 50 |
| -O-CO-O- | ||||
| -N< (-NH2,-NHPh,-NPh2) | 70 | -O-NO2 | 60 | 40 |
| >CO | 65 | -NC | 40 | 40 |
| -COOPh | 60 | -P=P- | 30 | 30 |
| naphthalene nuclei | -NCO | |||
| >C=NH | 50 | -ONO | 40 | 20 |
| -SH | ||||
| -S- | ||||
| -O-O- | 40 | -I | 80 | 10 |
| -N=N- | 30 | -Br | 60 | 10 |
| -O- | 20 | =S | 50 | 10 |
| Benzene nuclei | 15 | -Cl | 40 | 10 |
| (single aromatic ring) | ||||
| Ring (non aromatic single ring) | 10 | -F | 5 | 5 |
| Triple bond | 3 | Iso-** | −10 | 0 |
| Double bond | 2 | Tert-** | −20 | 0 |
† Reproduced from [7]. * Applied to non-ring compound. ** Applied to end of the compound.
| Group | π | Mr | F | R | σm | σp | I | O | X |
|---|---|---|---|---|---|---|---|---|---|
| F | 0.14 | 0.92 | 0.43 | −0.34 | 0.34 | 0.06 | 5 | 5 | 3.98 |
| Cl | 0.71 | 6.03 | 0.41 | −0.15 | 0.37 | 0.23 | 10 | 40 | 3.16 |
| Br | 0.86 | 8.88 | 0.44 | −0.17 | 0.39 | 0.23 | 10 | 50 | 2.96 |
| I | 1.12 | 13.94 | 0.4 | −0.19 | 0.35 | 0.18 | 10 | 60 | 2.66 |
| H | 0 | 1.03 | 0 | 0 | 0 | 0 | 0 | 0 | 2.20 |
| OH | −0.67 | 2.85 | 0.29 | −0.64 | 0.12 | −0.37 | 100 | 0 | 3.03 |
| SH | 0.39 | 9.22 | 0.28 | −0.11 | 0.25 | 0.15 | 20 | 40 | 2.45 |
| NH2 | −1.23 | 5.42 | 0.02 | −0.68 | −0.16 | −0.66 | 70 | 0 | 2.70 |
| NHOH | −1.34 | 7.22 | 0.06 | −0.4 | −0.04 | −0.34 | 170 | 0 | 2.87 |
| NHNH2 | −0.88 | 8.44 | 0.17 | −0.71 | −0.02 | −0.55 | 140 | 0 | 2.80 |
| CF3 | 0.88 | 5.02 | 0.38 | 0.19 | 0.43 | 0.54 | 15 | 35 | 3.16 |
| OCF3 | 1.04 | 7.86 | 0.38 | 0 | 0.38 | 0.35 | 35 | 35 | 3.35 |
| CN | −0.57 | 6.33 | 0.51 | 0.19 | 0.56 | 0.66 | 70 | 40 | 2.76 |
| NCS | 1.15 | 17.24 | 0.51 | −0.09 | 0.48 | 0.38 | 75 | 90 | 2.85 |
| SCN | 0.41 | 13.4 | 0.36 | 0.19 | 0.41 | 0.52 | 80 | 90 | 2.64 |
| NHCN | −0.26 | 10.14 | 0.26 | −0.18 | 0.21 | 0.06 | 140 | 40 | 2.82 |
| CHO | −0.65 | 6.88 | 0.31 | 0.13 | 0.35 | 0.42 | 65 | 20 | 2.75 |
| CO2H | −0.32 | 6.93 | 0.33 | 0.15 | 0.37 | 0.45 | 150 | 20 | 2.87 |
| CH2Br | 0.79 | 13.39 | 0.1 | 0.05 | 0.12 | 0.14 | 10 | 80 | 2.51 |
| CH2Cl | 0.17 | 10.49 | 0.1 | 0.03 | 0.11 | 0.12 | 10 | 60 | 2.54 |
| CH2I | 1.5 | 18.6 | 0.09 | 0.03 | 0.1 | 0.11 | 10 | 100 | 2.47 |
| NHCHO | −0.98 | 10.31 | 0.25 | −0.23 | 0.19 | 0 | 135 | 20 | 2.81 |
| CONH2 | −1.49 | 9.81 | 0.24 | 0.14 | 0.28 | 0.36 | 135 | 20 | 2.83 |
| CH=NOH | −0.38 | 10.28 | 0.25 | −0.13 | 0.22 | 0.1 | 220 | 20 | 2.64 |
| CH3 | 0.56 | 5.65 | −0.04 | −0.13 | −0.07 | −0.17 | 0 | 20 | 2.40 |
| NHCONH2 | −1.3 | 13.72 | 0.04 | −0.28 | −0.03 | −0.24 | 105 | 20 | 2.83 |
| OCH3 | −0.02 | 7.87 | 0.26 | −0.51 | 0.12 | −0.27 | 20 | 20 | 3.09 |
| CH2OH | −1.03 | 7.19 | 0 | 0 | 0 | 0 | 100 | 20 | 2.52 |
| NHCH3 | −0.47 | 10.33 | −0.11 | −0.74 | −0.3 | −0.84 | 70 | 20 | 2.74 |
| -C°CH | 0.4 | 9.55 | 0.19 | 0.05 | 0.21 | 0.23 | 3 | 40 | 2.50 |
| NHCOCF3 | 0.08 | 14.3 | 0.36 | −0.21 | 0.3 | 0.12 | 150 | 40 | 2.84 |
| CH2CN | −0.57 | 10.11 | 0.21 | −0.18 | 0.16 | 0 | 70 | 60 | 2.48 |
| -CH=CH2 | 0.82 | 10.99 | 0.07 | −0.08 | 0.05 | −0.02 | 2 | 40 | 2.46 |
| COCH3 | −0.55 | 11.18 | 0.32 | 0.2 | 0.38 | 0.5 | 65 | 40 | 2.78 |
| OCOCH3 | −0.64 | 12.47 | 0.41 | −0.07 | 0.39 | 0.31 | 85 | 40 | 3.22 |
| CO2CH3 | −0.01 | 12.87 | 0.33 | 0.15 | 0.37 | 0.45 | 85 | 40 | 2.88 |
| NHCOCH3 | −0.97 | 14.93 | 0.28 | −0.26 | 0.21 | 0 | 135 | 40 | 2.82 |
| NHCO2CH3 | −0.37 | 16.53 | 0.14 | −0.28 | 0.07 | −0.15 | 155 | 40 | 2.84 |
| CH2CH3 | 1.02 | 10.3 | −0.05 | −0.1 | −0.07 | −0.15 | 0 | 40 | 2.43 |
| CH2OCH3 | −0.78 | 12.07 | 0.01 | 0.02 | 0.02 | 0.03 | 20 | 40 | 2.53 |
| OCH2CH3 | 0.38 | 12.47 | 0.22 | −0.44 | 0.1 | −0.24 | 20 | 40 | 3.10 |
| N(CH3)2 | 0.18 | 15.55 | 0.1 | −0.92 | −0.15 | −0.83 | 70 | 40 | 2.78 |
| CH(CH3)2 | 1.53 | 14.96 | −0.05 | −0.1 | −0.07 | −0.15 | 0 | 50 | 2.46 |
| C3H7 | 1.55 | 14.96 | −0.06 | −0.08 | −0.07 | −0.13 | 0 | 60 | 2.43 |
| C4H9 | 2.13 | 19.61 | −0.06 | −0.11 | −0.08 | −0.16 | 0 | 80 | 2.43 |
| C(CH3)3 | 1.98 | 19.62 | −0.07 | −0.13 | −0.1 | −0.2 | 0 | 60 | 2.49 |
| C6H5 | 1.96 | 25.36 | 0.08 | −0.08 | 0.06 | −0.01 | 15 | 120 | 2.51 |
| C5H11 | 2.67 | 24.26 | −0.06 | −0.08 | −0.08 | −0.16 | 0 | 100 | 2.43 |
| OC6H5 | 2.08 | 27.68 | 0.34 | −0.35 | 0.25 | −0.03 | 35 | 120 | 3.13 |
| N(C2H5)2 | 1.18 | 24.85 | 0.01 | −0.91 | −0.23 | −0.9 | 70 | 80 | 2.80 |
| Descriptors | π | Mr | F | R | σm | σp | I | O |
|---|---|---|---|---|---|---|---|---|
| Mr | 0.601 | |||||||
| F | −0.175 | −0.251 | ||||||
| R | 0.116 | −0.104 | 0.243 | |||||
| σm | −0.105 | −0.250 | 0.934 | 0.574 | ||||
| σp | 0.007 | −0.200 | 0.650 | 0.895 | 0.879 | |||
| I | −0.667 | −0.109 | 0.234 | −0.171 | 0.136 | −0.025 | ||
| O | 0.752 | 0.837 | −0.030 | 0.206 | 0.050 | 0.144 | −0.378 | |
| X | −0.193 | −0.245 | 0.637 | −0.219 | 0.459 | 0.122 | 0.186 | −0.244 |
π = 0.9485 + 0.011901 I n = 50, r = 0.6670, s = 0.78, F = 38.47 π = 0.8795 + 0.02583 O n = 50, r = 0.7521, s = 0.69, F = 62.5 π = 0.1558 + 0.007963 I + 0.02003 O n = 50, r = 0.8581, s = 0.5437, F = 65.60 π = 0.4221 + 0.001135 (I - O) n = 50, r = 0.8202, s = 0.5993, F = 98.65
Table 4.
Amino acid residue logP, amino acid inorganic property and organic property values and residue electronegativity values.
| Amino acid(residue) | logP Value | Inorganic (I) Value | Organic (O) Value | Residue Electronegativity (X) |
|---|---|---|---|---|
| A (Ala) | 0.072 | 220 | 60 | 2.40 |
| R (Arg) | −2.061 | 432 | 120 | 2.43 |
| N (Asn) | −1.003 | 355 | 60 | 2.49 |
| D (Asp) | −1.935 | 370 | 60 | 2.50 |
| C (Cys) | 0.987 | 240 | 100 | 2.44 |
| Q (Gln) | −0.936 | 355 | 80 | 2.49 |
| E (Glu) | −1.868 | 370 | 80 | 2.44 |
| G (Gly) | 0.184 | 220 | 40 | 2.20 |
| H (His) | −1.321 | 375 | 120 | 2.45 |
| I (Ile) | 2.167 | 220 | 120 | 2.46 |
| L (Leu) | 2.167 | 220 | 110 | 2.44 |
| K (Lys) | −0.790 | 290 | 120 | 2.43 |
| M (Met) | 1.246 | 240 | 140 | 2.44 |
| F (Phe) | 2.423 | 235 | 180 | 2.45 |
| P (Pro) | 1.128 | 230 | 100 | / |
| S (Ser) | −0.453 | 320 | 60 | 2.52 |
| T (Thr) | −0.042 | 320 | 80 | 2.45 |
| W (Trp) | 1.878 | 335 | 220 | 2.45 |
| Y (Tyr) | 1.887 | 335 | 180 | 2.45 |
| V (Val) | 1.640 | 220 | 90 | 2.46 |
| Step | 1 | 2 | 3 | 4 | 5 | 6 | 7* |
| Constant | 1.757 | 1.444 | 1.506 | 1.850 | 2.098 | 2.145 | 1.998 |
| σm2 | −2.54 | −2.31 | −1.91 | −1.78 | −1.79 | −1.64 | −1.68 |
| T-ratio | −5.44 | −5.50 | −5.36 | −5.84 | −6.20 | −6.03 | −6.42 |
| MR1 | 0.067 | 0.093 | 0.106 | 0.100 | 0.126 | 0.131 | |
| T-ratio | 3.97 | 6.25 | 8.15 | 7.93 | 8.72 | 9.33 | |
| π (total) | −0.363 | −0.352 | −0.234 | −0.221 | −0.290 | ||
| T-ratio | −5.08 | −5.78 | −3.23 | −3.29 | −4.07 | ||
| F7 | −1.15 | −1.71 | −1.85 | −1.76 | |||
| T-ratio | −4.55 | −5.38 | −6.19 | −6.09 | |||
| π7 | −0.209 | −0.239 | −0.203 | ||||
| T-ratio | −2.69 | −3.29 | −2.85 | ||||
| R1 | 2.33 | 2.36 | |||||
| T-ratio | 3.06 | 3.22 | |||||
| MR6 | 0.116 | ||||||
| T-ratio | 2.30 | ||||||
| s | 0.593 | 0.527 | 0.436 | 0.372 | 0.352 | 0.326 | 0.313 |
| r2 | 0.3496 | 0.4968 | 0.6615 | 0.7578 | 0.7879 | 0.8213 | 0.8387 |
| F | 29.56 | 26.65 | 34.53 | 40.68 | 37.89 | 38.29 | 36.39 |
The next best alternative variables are π6, XR7, XR8, σp8, MR8, O, XR1, F2, XR6.
© 1999 MDPI. All rights reserved.
Share and Cite
MDPI and ACS Style
Wu, H. Chemical Property Calculation through JavaScript and Applications in QSAR. Molecules 1999, 4, 16-27. https://doi.org/10.3390/40100016
AMA Style
Wu H. Chemical Property Calculation through JavaScript and Applications in QSAR. Molecules. 1999; 4(1):16-27. https://doi.org/10.3390/40100016
Chicago/Turabian StyleWu, Hanqing. 1999. "Chemical Property Calculation through JavaScript and Applications in QSAR" Molecules 4, no. 1: 16-27. https://doi.org/10.3390/40100016