Facile and Stereoselective Synthesis of Non-Racemic 3,3,3-Trifluoroalanine
by
Marcello Crucianelli
1,*,
Natalia Battista
1,
Pierfrancesco Bravo
2,3,
Alessandro Volonterio
2 and
Matteo Zanda
2,* 
1
Dipartimento di Chimica, Ingegneria Chimica e Materiali, Università di L'Aquila, Via Vetoio, I-67010 Coppito, Italy
2
Dipartimento di Chimica del Politecnico, via Mancinelli 7, I-20131 Milano, Italy
3
C.N.R. - Centro di Studio sulle Sostanze Organiche Naturali, via Mancinelli 7, I-20131 Milano, Italy
*
Authors to whom correspondence should be addressed.
Molecules 2000, 5(12), 1251-1258; https://doi.org/10.3390/51201251
Submission received: 6 October 2000
/
Accepted: 7 November 2000
/
Published: 18 December 2000
Abstract
:A highly stereoselective enantiodivergent synthesis of non-racemic 3,3,3-trifluoroalanine 7 is reported. The methodology is based on the reduction, by 9-BBN or DIBAH, of the chiral sulfinimine 3 derived from ethyl trifluoropyruvate, followed by acidic hydrolysis of the resulting diastereomeric sulfinamides 4 and 5.
Introduction
The rapidly expanding interest in the field of peptidomimetics is prompting organic chemists to develop novel and efficient stereocontrolled approaches to rare and unnatural amino acids. An extremely intriguing class of unnatural amino acids is represented by those incorporating one or more fluorine atoms [1]. This interest stems from the peculiar biomedicinal and pharmaceutical properties of fluorinated substrates [2], as well as from the considerable synthetic challenges connected with the preparation of these molecules in stereodefined, non-racemic form [3]. 3,3,3-Trifluoroalanine (7)
(Scheme 2) and its derivatives have attracted a remarkable deal of interest as suicide inhibitors of a number of pyridoxal-phosphate dependent enzymes [4]. Incorporation of 7 into small peptides has been achieved, and some of the resulting oligomers have been found to possess interesting biological activity [5]. Although several preparations of racemic 7 have been described following the seminal work of Steglich [6], a very few syntheses of non-racemic 7 are available [7], and only recently its absolute configuration has been clarified [8]. To date, a straightforward method for the synthesis of non-racemic 7 from readily available or commercial starting materials, such as trifluoropyruvic esters, is lacking [9]. In this paper we describe the successful accomplishment of this goal.
Results and Discussion
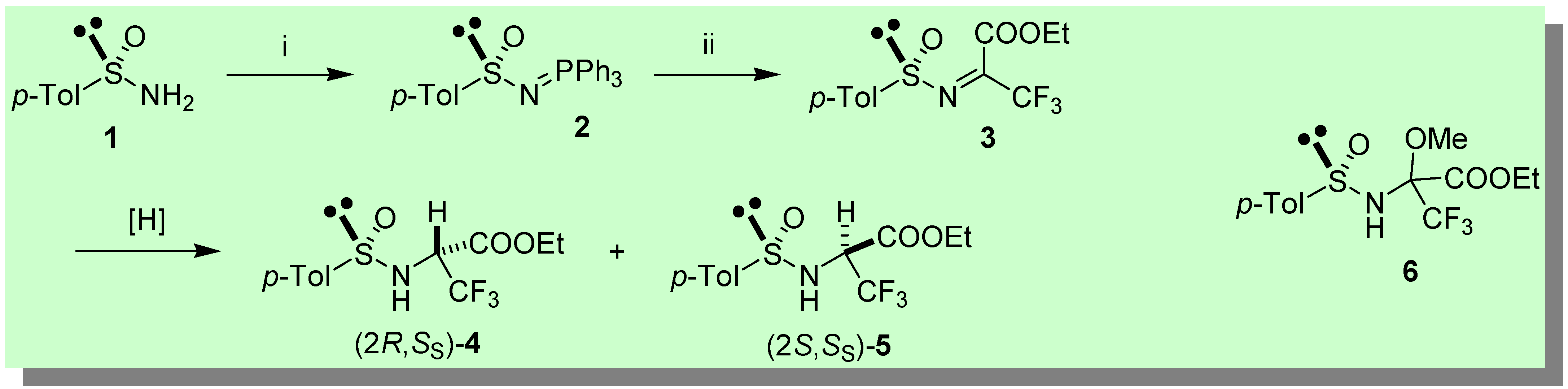
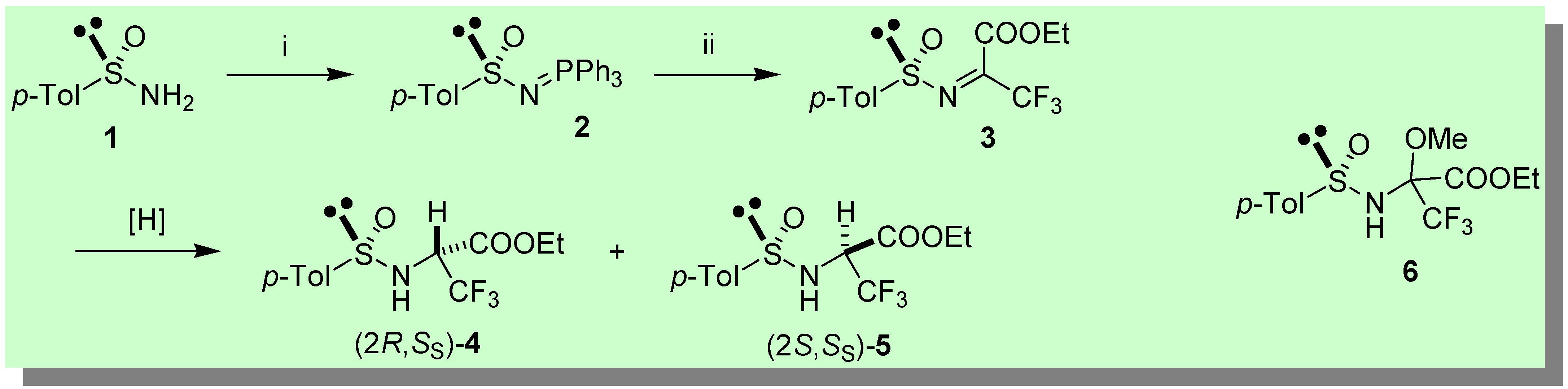
The chiral Staudinger reagent (S)-2 (e.e. > 95%) [10a-d] (Scheme 1) was synthesized by reaction of the Davis sulfinamide (S)-1 [11] with DEAD/PPh3 (92%) [12]. Next, a high yielding Staudinger (aza-Wittig) reaction of (S)-2 with ethyl trifluoropyruvate was performed in benzene freshly distilled from Na (ca. 90 min. at 40 °C), providing (S)-3 [10f]. After evaporation of the solvent, the crude reaction mixture containing the highly electrophilic sulfinimine (S)-3 was treated with a variety of reducing agents (Table 1).
The most interesting results were achieved with DIBAH (entry 1) and 9-BBN [13] (entry 2) which produced with stereodivergent outcomes the diastereomeric sulfinamides 4 and 5, respectively. In the latter case (9-BBN), the reduction occurred with excellent stereoselectivity (20:1) and overall yield (78%). Although DIBAH provided lower stereocontrol (4:1) and yield (52%), the fact that both diastereomers 4 and 5 are readily accessible from the same enantiomeric sulfinimine (S)-3 makes this method remarkably attractive. DIBAH reduction occurred with lower stereocontrol in favour of 4 (2:1) upon pre-complexation of (S)-3 with ZnBr2. Complex mixtures were obtained with K- and L- Selectride® and LiAlH4 in THF as reducing agents. An undesired side-reaction took place upon treatment of (S)-3 with NaBH4 in methanol (entry 4), namely the addition of methanol across the C=N bond. Thus, a diastereomeric mixture of adducts 6 (Scheme 1) was obtained, whereas the reduction products 4,5 could be neither isolated nor detected [14].
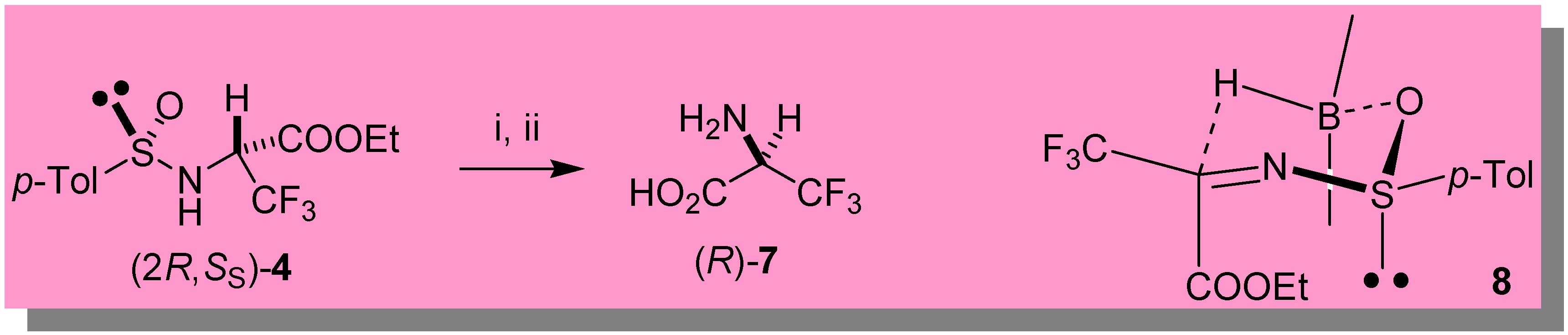
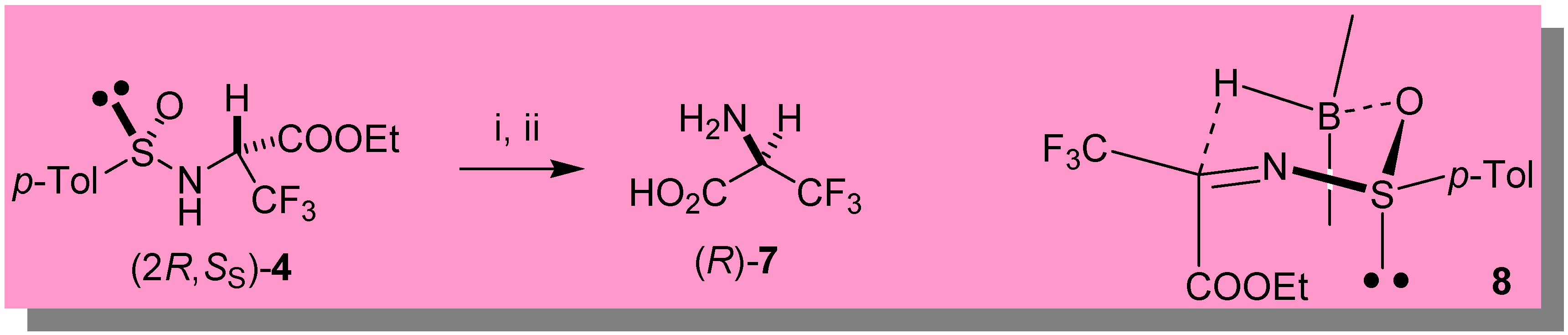
A reasonable transition state (TS) 8 (Scheme 2) accounting for the high stereoselectivity observed with 9-BBN is based on the hypothesis that the sulfinimine (S)-3 is geometrically homogeneous and thermodynamically stable with the sulfinyl and the bulky trifluoromethyl group in trans position with respect to the C=N bond, as strongly suggested by theoretical ab initio calculations supported by NMR spectroscopy [15]. In line with the previously proposed TS for highly stereoselective 9-BBN reductions of ketone derived sulfinimines [16], the boron atom should coordinate the sulfinyl oxygen giving rise to a chair-like TS. As a consequence, the hydride predominantly attacks the Re face of the C=N bond producing the diastereomeric sulfinamide 5 with overwhelming preference.
With the diastereomeric sulfinamides 4 and 5 in hand, both enantiomers of 3,3,3-trifluoroalanine (7) are easily accessible, as demonstrated in the preparation of (R)-7 from 4 (obtained by reduction of 3 with DIBAH), which was treated with 10% HCl (reflux, overnight) [6d] followed by ion exchange chromatography with DOWEX-50W. We were also able to prepare (R)-7 one-pot in 38% overall yield from (2R,RS)-5 (obtained from the enantiomeric iminophosphorane (R)-2), by submitting the crude 9-BBN reduction mixture to hydrolysis with 10% HCl, followed by the usual ion-exchange purification. The stereochemistry of (R)-7 was assessed by comparison of its optical power with the literature values for non-racemic 3,3,3-trifluoroalanine [7,8]. Although we could not measure directly the e.e. of (R)-7, its [α]20D + 7.5 (c 0.18, MeOH) suggests a value higher than 70% [17].
In summary, we have developed an extremely facile, straightforward, stereoselective and enantiodivergent approach to non-racemic 3,3,3-trifluoroalanine, which is now readily available for further biochemical studies and incorporation into peptidomimetics.
Experimental
General
Chemical shifts (δ) are reported in parts per million (ppm) of the applied field. Coupling constants (J) are reported in Hertz. Me4Si was used as internal standard ( δH and δC = 0.00) for 1H and 13C nuclei, while C6F6 was used as external standard (δF = −162.90) for 19F nuclei. Peak multiplicities are abbreviated: singlet, s; doublet, d; triplet, t; quartet, q; multiplet, m; etc. Anhydrous solvents were obtained by distillation from sodium (THF, benzene) or from calcium hydride (dichloromethane, diisopropylamine). In all other cases commercially available reagent-grade solvents were employed without purification. Grignard reagents were purchased from Sigma/Aldrich/Fluka Company. Reactions performed in dry solvents were carried out under nitrogen atmosphere. Melting points are uncorrected and were obtained on a capillary apparatus. Analytical thin-layer chromatography (TLC) was routinely used to monitor reactions. Plates precoated with E. Merck silica gel 60 F254 of 0.25 mm thickness were used. Merck silica gel 60 (230-400 ASTM mesh) was employed for flash chromatography (FC).
Synthesis of N-p-Tolylsulfinyl-Imino-Triphenylphosphorane (S)-2
To a solution of (S)-1 (1.93 g, 12.35 mmol) and PPh3 (3.24 g, 12.35 mmol) in dry THF (50 mL) at 0 °C, neat DEAD (1.95 mL, 12.35 mmol) was added dropwise with stirring. The resulting dark-red mixture was allowed to warm to r.t. in 40 min., then the solvent was removed in vacuo. The iminophosphorane (S)-2 was obtained in pure form by FC (n-Hex/AcOEt 3:7) as a yellowish sticky oil (4.75g, 92%): [α]20D + 7.6 (c 0.83, CHCl3); 1H-NMR (CDCl3, 400 MHz): δ 7.8-7.71 (m, 6H), 7.67-7.64 (m, 2H), 7.58-7.53 (m, 3H), 7.49-7.43 (m, 6H), 7.2 (d, J = 8.3, 2H), 2.34 (s, 3H). 13C-NMR (CDCl3, 62.86 MHz): δ 149.8 (d, J = 24), 139.3, 133.0 (d, J = 10.2), 132.4 (d, J = 2.8), 129.0, 128.7 (d, J = 12), 128.6 (d, J = 99.8), 125.0, 21.3. 31P-NMR (CDCl3, 162 MHz) δ 27.8 (s) (H3PO4 as external standard). Anal. Calcd for: C25H22NOPS: C, 72.27; H, 5.34; N, 3.37. Found: C, 72.59; H, 4.99; N, 3.60. Enantiomeric iminophosphorane (R)-2, analogously obtained from (R)-1, had [α]20D − 8.1 (c 0.95, CHCl3).
Reduction with DIBAH: synthesis of 3,3,3-trifluoro-2-(toluene-4-sulfinyl)amino-propionic acid ethyl esters 4,5
To a solution of iminophosphorane (S)-2 (0.2 g, 0.48 mmol) dissolved in freshly distilled benzene (1 mL),neat ethyl trifluoropyruvate (82 mg, 0.48 mmol) was added dropwise. The mixture was warmed to 40 °C for ca. 2 hours. After a rapid evaporation of the solvent, the crude containing the sulfinimine (S)-3 was redissolved in 1 mL of freshly distilled THF and the resulting yellow solution cooled down to − 70 °C. A 1.0 M n-hexane solution of DIBAH (0.58 mL, 0.58 mmol) was added dropwise while stirring. After 15 min. the reaction was quenched at − 70 °C with a saturated solution of ammonium chloride, filtered over a Celite pad, then extracted with ethyl acetate and the collected organic layers dried over anhydrous sodium sulfate. Purification by FC (n-hexane/ethyl acetate from 80:20 to 70:30) afforded a 80:20 mixture of two diastereomers 4,5 with an overall yield of 52%.
5: (Rf>): m.p. (isopropyl ether) 116-118 °C (dec.); [α]20D + 252.5° (c 0.56, CHCl3). 1H-NMR (CDCl3, 250 MHz) δ 7.58 (d, J = 8.3, 2H), 7.34 (d, J = 8.3, 2H), 5.48 (br d, J = 10, 1H), 4.39-4.25 (m, 2H), 4.14-4.01 (m, 1H), 2.43 (s, 3H), 1.32 (t, J = 7, 3H). 19F-NMR (CDCl3, 235.19 MHz) δ − 73.2 (d, J = 7.4). 13C-NMR (CDCl3, 62.86 MHz) δ 165.08, 141.26, 137.99, 128.72, 125.32, 121.49 (q, J = 281.7), 62.30, 52.42 (q, J = 32), 20.35, 12.76. Anal Calcd. for: C12H14F3NO3S: C, 46.60; H, 4.56; N, 4.53. Found: C, 46.70; H, 4.45; N, 4.78.
4: (Rf <): oil; [α]20D + 119.7° (c 0.33, CHCl3). 1H-NMR (CDCl3, 250 MHz) δ 7.61 (d, J = 8.1, 2H), 7.34 (d, J = 8.1, 2H), 5.07 (br d, J = 8.5, 1H), 4.60-4.48 (m, 1H), 4.34-4.22 (m, 2H), 2.43 (s, 3H), 1.29 (t, J = 7.3, 3H). 19F-NMR (CDCl3, 235.19 MHz) δ − 74.14 (d, J = 7.4, 3F).
Reduction with DIBAH and ZnBr2
A solution containing the sulfinimine (S)-3 dissolved in anhydrous THF (1.5 mL), obtained as described above from the same amount of reagents, was treated with zinc bromide (108 mg, 0.48 mmol) and stirred at r.t. for 30 min. The mixture was cooled to − 70 °C, then a 1.0 M n-hexane solution of DIBAH (0.58 mL, 0.58 mmol) was added dropwise while stirring. After 15 min. the reaction was quenched and worked-up as described above. Purification by FC afforded a 2:1 mixture of the two diastereomers 4,5 with an overall yield of 58 %.
Reduction with NaBH4
A solution containing the sulfinimine (S)-3 dissolved in distilled methanol (1.5 mL), obtained as described above, was cooled to − 70 °C, then NaBH4 (22 mg, 0.58 mmol) was added in one portion. After 10 min. the reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The collected organic layers were dried over anhydrous sodium sulfate and the resulting crude mixture purified by FC (n-hexane/ethyl acetate from 80:20 to 70:30) affording a 2:1 mixture of two diastereomers 6 (overall yield 66%) resulting from addition of methanol across the C=N bond of (S)-3.
Reduction with 9-BBN
A solution containing the sulfinimine (S)-3 dissolved in freshly distilled THF (1.5 mL), obtained as described above starting from 0.58 mmol of iminophosphorane (S)-2 and 0.58 mmol of ethyl trifluoropyruvate, was cooled to 0 °C, then a 0.5 M THF solution of 9-BBN (1.27 mL, 0.63 mmol) was added dropwise. The reaction mixture was stirred under nitrogen at the same temperature for ca. 2 hours, then worked-up by adding 5 μL of methanol in order to destroy the excess of 9-BBN [13], then the solvent was removed in vacuo. The crude was redissolved in 1.5 mL of diisopropyl ether, filtered and the resulting solution purified by FC, affording the two diastereomers 4, 5 in 1:20 ratio with an overall yield of 78 %.
Direct synthesis of (R)-(+)-3,3,3-trifluoro-alanine 7
The first part of the synthetic procedure was performed using the same conditions described above for the preparation of the sulfinimine (S)-3, the only exception being the use of 1.13 mmol of the (R)-enantiomer of iminophosphorane 2. After addition of 9-BBN (1.24 mmol), the reaction mixture was stirred under nitrogen (0 °C) for ca. 2 hours, then the solvent evaporated in vacuo. The crude was redissolved in 5 mL of conc. HCl and stirred overnight at reflux [6d]. The reaction mixture was diluted with water and, after addition of diethyl ether (2 mL), vigorously stirred for 1 hour. The two layers were then separated, the organic phase washed with two portions of a 10% solution of HCl, and the collected aqueous phases concentrated in vacuo and loaded in a Dowex 50W-X8 column. Elution with 7.5% aqueous ammonia afforded 61 mg (38% overall yield) of free (R)-3,3,3-trifluoro-alanine 7.
(R)-7: [α]20D+7.5 (c 0.18, MeOH), (lit.[7a] value for (c 0.76, MeOH) of enantio-enriched (R)-7 (e.e. 62%) has been reported = +6.8); mp 205-207 °C (sublimate: lit.[6d] sublimation T > 205 °C) (EtOH); 1H-NMR (D2O) δ 4.32 (1H, q, J = 9.0 Hz); 13C-NMR (D2O) δ 164.69, 122.11 (q, J = 280 Hz), 54.89 (q, J = 30 Hz); 19F-NMR (D2O) δ −69.1 (d, J = 9.0 Hz).
Acknowledgments
We thank M.U.R.S.T. COFIN 1998 (Nuove metodologie e strategie di sintesi di composti di interesse biologico) and C.N.R. - C.S.S.O.N. for financial support.
References and Notes
- Fluorine-containing Amino Acids: Synthesis and Properties; Kukhar, V. P.; Soloshonok, V. A. (Eds.) Wiley: Chichester, 1995.
- Welch, J. T.; Eswarakrishnan, S. Fluorine in Bioorganic Chemistry; Wiley: New York, 1991. [Google Scholar] Filler, R.; Kobayashi, Y.; Yagupolskii, L. M. Biomedical Aspects of Fluorine Chemistry; Elsevier: Amsterdam, 1993. [Google Scholar]
- Uneyama, K. Enantiocontrolled Synthesis of Fluoro-Organic Compounds: Stereochemistry, Challenges and Biomedicinal Targets; Soloshonok, V. A., Ed.; Wiley: Chichester, 1999; pp. 391–418. [Google Scholar] For some of our recent efforts in the field: Bravo, P.; Fustero, S.; Guidetti, M.; Volonterio, A.; Zanda, M. J. Org. Chem. 1999, 64, 8731–8735. Crucianelli, M.; Bravo, P.; Arnone, A.; Corradi, E.; Meille, S. V.; Zanda, M. J. Org. Chem. 2000, 65, 2965–2971. [PubMed]
- See Ref. 2a, pp.54-65.
- Bieth, J.; Dimicoli, J. L.; Wermuth, C. G.; Delalande, S. A. Ger. Offen., DE 2806833, 1978; Chem. Abstr. 1979, 90, 72447g. [Google Scholar] Höss, E.; Rudolph, M.; Seymour, L.; Schierlinger, C.; Burger, K. J. Fluorine Chem. 1993, 61, 163–170. and references therein. Osipov, S. N.; Sewald, N.; Kolomiets, A. F.; Fokin, A. V.; Burger, K. Tetrahedron Lett. 1996, 37, 615–618. Dessipri, E.; Tirrell, D. A. Macromolecules 1994, 27, 5463–5470. Bordusa, F.; Dahl, C.; Jakubke, H.-D.; Burger, K.; Koksch, B. Tetrahedron: Asymmetry 1999, 10, 307–313.
- Weygand, F.; Steglich, W.; Fraunberger, F. Angew. Chem. 1967, 79, 822. Weygand, F.; Steglich, W.; Oettmei, W. Chem. Ber. 1970, 103, 818–826. [PubMed]Soloshonok, V. A.; Kukhar, V. P. Tetrahedron 1997, 53, 8307–8314. and references therein. Burger, K.; Hoess, E.; Gaa, K.; Sewald, N.; Schierlinger, C. Z. Naturforsch. 1991, 46b, 361–384. and references therein.
- Sakai, T.; Yan, F.; Kashino, S.; Uneyama, K. Tetrahedron 1996, 52, 233–244. and references therein. Arnone, A.; Bravo, P.; Capelli, S.; Fronza, G.; Meille, S. V.; Zanda, M.; Cavicchio, G.; Crucianelli, M. J. Org. Chem. 1996, 61, 3375, Corrigenda: J. Org. Chem. 1996, 61, 9635.
- Sakai, T.; Yan, F.-Y.; Uneyama, K. Synlett 1995, 753–754. and references therein.
- Uneyama et al. described the enantioselective reduction of N-p-methoxyphenyl-imines of trifluoropyruvate with oxazaborolidine catalysts (see Ref. 7a and 8), producing (R)-7 with 62% e.e.
- Bravo, P.; Crucianelli, M.; Vergani, B.; Zanda, M. Tetrahedron Lett. 1998, 39, 7771–7774. Racemic 2 was also described: Senning, A.; Kelly, P. Naturwissenschaften 1968, 55, 543, Chem. Abstr. 70: 47555v. For an overview of the Staudinger (aza-Wittig) reaction: Staudinger, H.; Meyer, J. Helv. Chim. Acta 1919, 2, 635–646. Molina, P.; Vilaplana, M. J. Synthesis 1994, 1197–1218. Johnson, A. W.; Kasha, W. C.; Starzewsky, K. A. O.; Dixon, D. A. «Iminophosphoranes and Related Compounds». In Ylides and Imines of Phosphorus; New York: Wiley, 1993; pp. 403–483. [Google Scholar] (f) The use of benzene freshly distilled from Na was found to be important for minimizing the racemization of the sulfinimine 3 upon reaction with a variety of Grignard reagents (see Ref. 10a). For this reason the same conditions were used in the present work.
- Davis, F. A.; Reddy, R. E.; Szewczyk, J. M.; Reddy, G. V.; Portonovo, P. S.; Zhang, H.; Fanelli, D.; Reddy, R. T.; Zhou, P.; Carroll, P. J. J. Org. Chem. 1997, 62, 2555–2563. [PubMed] For a review on sulfinimines and sulfinamides: Davis, F. A.; Zhou, P.; Chen, B.-C. Chem. Soc. Rev. 1998, 27, 13–18.
- Bittner, S.; Assaf, Y.; Krief, P.; Pomerantz, M.; Ziemnicka, B. T.; Smith, C. G. J. Org. Chem. 1985, 50, 1712–1718.
- Brown, H. C.; Krishnamurthy, S. J. Org. Chem. 1975, 40, 1864–1865.
- A similar side-reaction has been observed for related fluorine-containing compounds: see Ref. 7b.
- Asensio, A.; Bravo, P.; Crucianelli, M.; Farina, A.; Fustero, S.; Soler, J. C.; Meille, S. V.; Panzeri, W.; Viani, F.; Volonterio, A.; Zanda, M. submitted for publication. See also: Fustero, S.; Navarro, A.; Pina, B.; Asensio, A.; Bravo, P.; Crucianelli, M.; Volonterio, A.; Zanda, M. J. Org. Chem. 1998, 63, 6210–6219. [PubMed]Bravo, P.; Fustero, S.; Guidetti, M.; Volonterio, A.; Zanda, M. J. Org. Chem. 1999, 64, 8731–8735.
- Hua, D.; Lagneau, N.; Wang, H.; Chen, J. Tetrahedron: Asymmetry 1995, 6, 349–352.
- In our hands, free 3,3,3-trifluoroalanine 7 was poorly stable. We observed complete decomposition of solid samples of 7, purified by ion-exchange chromatography, after one week at 4 °C. Racemic 7 is known to undergo progressive elimination of HF at pH > 6: Weygand, F.; Steglich, W.; Oettmeier, W. Chem. Ber. 1970, 103, 1655–1663. [PubMed] Therefore, we cannot rule out that a partial racemization of free 7 might take place during the purification or storage, even if short.
- Sample Availability: Samples of iminophosphoranes (R)- and (S)-2 are available from the authors.
Scheme 1.
Key: i) PPh3, DEAD (92%); ii) CF3COCO2Et, C6H6 freshly distilled from Na, 40 °C (−Ph3PO).
Scheme 2.
Key: i) HCl conc., reflux, overnight. ii) Dowex 50W-X8.

{kind=link}
{kind=link}
| Entry | [H] | Conditions | Yield (%)a | D.r. 4/5 |
|---|---|---|---|---|
| 1 | DIBAH | THF, −70 °C | 52 | 4:1 |
| 2 | 9-BBN | THF, 0 °C | 78 | 1:20 |
| 3 | DIBAH/ZnBr2 | THF, r.t. to –70 °C | 58 | 2:1 |
| 4 | NaBH4 | Methanol, −70 °C | /b,c | /b,c |
a Yields from (S)-2. b We recovered 66% of 6 with 33% d.e. c Very low yields in THF, 0 °C to r.t.
© 2000 MDPI. All rights reserved.
Share and Cite
MDPI and ACS Style
Crucianelli, M.; Battista, N.; Bravo, P.; Volonterio, A.; Zanda, M. Facile and Stereoselective Synthesis of Non-Racemic 3,3,3-Trifluoroalanine. Molecules 2000, 5, 1251-1258. https://doi.org/10.3390/51201251
AMA Style
Crucianelli M, Battista N, Bravo P, Volonterio A, Zanda M. Facile and Stereoselective Synthesis of Non-Racemic 3,3,3-Trifluoroalanine. Molecules. 2000; 5(12):1251-1258. https://doi.org/10.3390/51201251
Chicago/Turabian StyleCrucianelli, Marcello, Natalia Battista, Pierfrancesco Bravo, Alessandro Volonterio, and Matteo Zanda. 2000. "Facile and Stereoselective Synthesis of Non-Racemic 3,3,3-Trifluoroalanine" Molecules 5, no. 12: 1251-1258. https://doi.org/10.3390/51201251