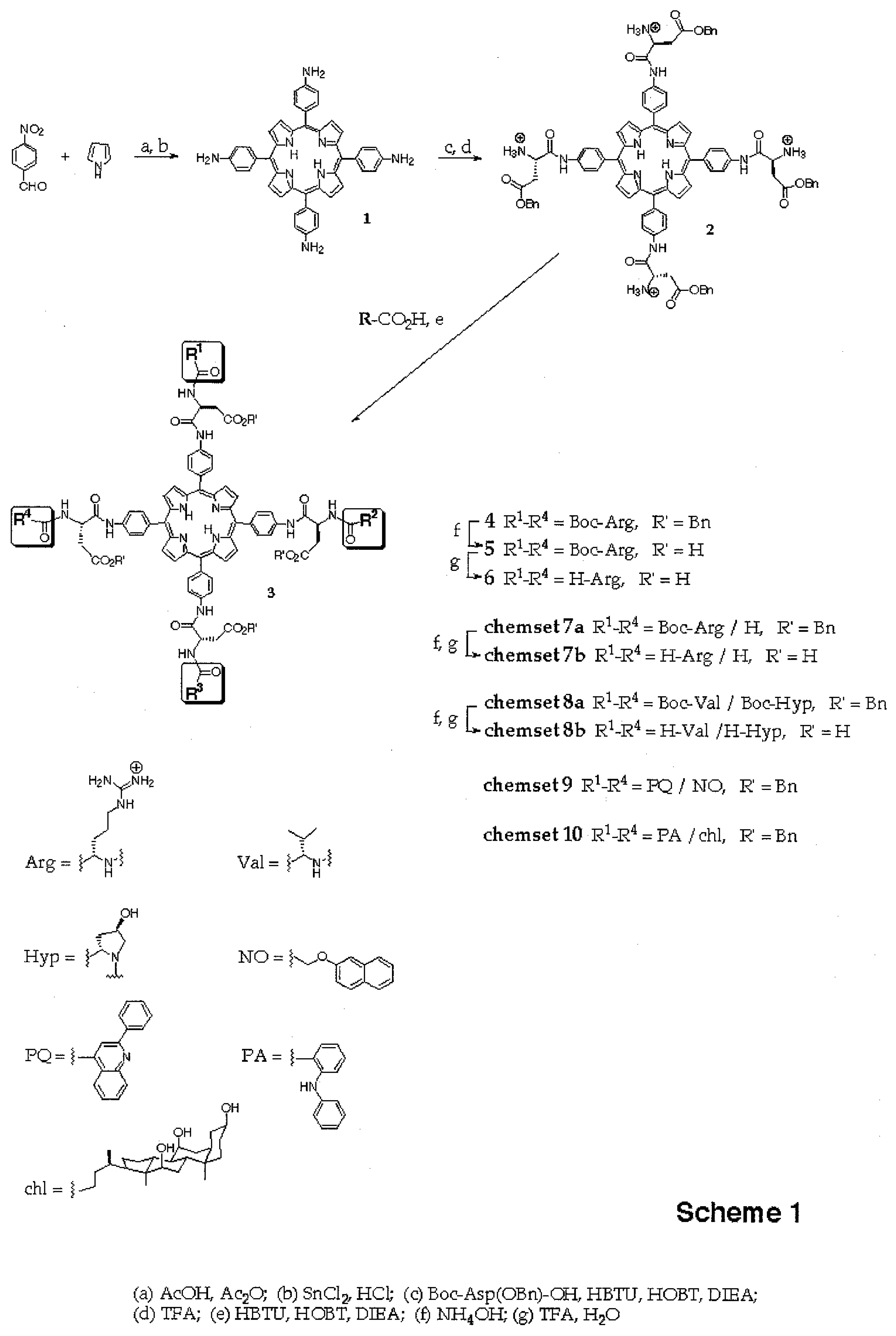
Introduction
Combinatorial synthesis has established itself as a tool for the preparation of libraries of structurally related compounds in an efficient manner [
1]. The most efficient way of synthesizing a combinatorial library would, of course, involve a single reaction, in which a mixture of all required building blocks reacts to produce the desired products. This approach has proven impractical for several reasons. One is the challenge associated with performing a work-up and purification of the product mixture produced in a massively combinatorial one-pot synthesis. Another, perhaps more fundamental reason is the unequal reactivities of building blocks, which leads to non-statistically distributed product mixtures, unless the stability of the products is identical (reaction performed under thermodynamic control) or the reactivities of the building blocks employed are equal (reaction performed under kinetic control). As a result, most combinatorial syntheses are performed on solid supports [
2,
3] and in a format in which no more than one building block reacts with its substrate(s) in any given reaction mixture. This research note reports on a modest exercise in the realm of competitive, mixed condensation reactions leading to combinatorial libraries in a one-pot procedure.
Porphyrins are one class of compounds whose combinatorial chemistry has not been explored extensively. Despite the widespread use of porphyrins and porphyrinoids as catalysts, photosensitizers, and receptors, few reports have appeared on the combinatorial synthesis of porphyrin libraries [
4,
5]. This may be due, in part, to synthetic challenges that arise when preparing such libraries. Mixed condensation reactions, such as a Rothemund cyclization with a mixture of aldehyde starting materials, produce product mixtures that can be difficult to separate from the invariably formed oligopyrrolic side products [
6]. Derivatizing on the level of a fully assembled porphyrin core scaffolds can also be challenging, since analyzing the product mixtures with the traditional spectroscopic tools of synthetic organic chemistry is non-trivial. In preliminary reports, the synthesis of para-substituted tetraphenylporphyrin libraries and the selection of a DNA-binding library member via spectrometrically monitored selection experiment (SMOSE) [
5] had been presented [
7,
8]. The difficulties in predicting relative reactivities of building blocks or performing reactions under thermodynamic control has so far limited the scope of combinatorial library syntheses. Here we report how coupling reactions employing a porphyrin derivative can be used to determine the relative reactivity of activated carboxylic acids in amide bond formation. For mixed couplings with building blocks of not too unequal reactivity, this information can be used to adjust the ratio of reactants. For the synthesis of six component libraries from two carboxylic acids of very different reactivity, a onepot reaction is presented that yields near-statistical product distributions.
Results and Discussion
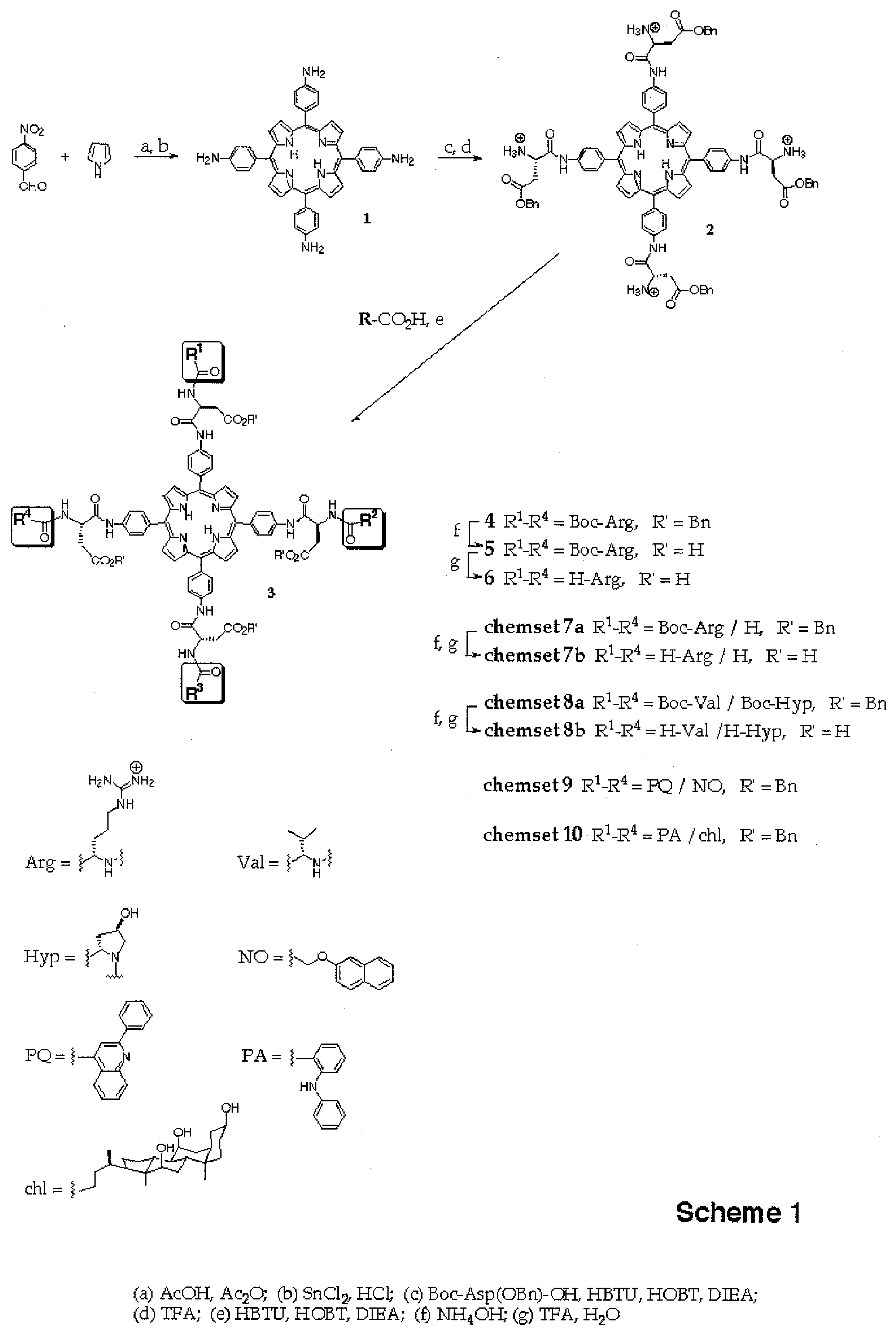
The synthesis of 5,10,15,20-tetrakis(aminophenyl)porphyrin
1 (
Scheme 1) followed literature protocols [
9]. The amino groups of the phenyl substituents were aminoacylated with
L-Boc-Asp(OBn)- OH and the resulting fully protected porphyrin derivative was column-purified and then treated with TFA/CH
2Cl
2 (1:1) to give key intermediate
2 as its tetrakis[trifluoroacetate] salt after precipitation from diethylether. Compound
2 was used in coupling reactions employing carboxylic acid building blocks activated with a mixture of HBTU (0.9 equivalents relative to the acid) and HOBT (1.0 equivalent relative to the acid) in DMF. In order to induce
in situ neutralization of the ammonium groups of
2 and to accelerate activation, the reaction mixtures also contained diisopropyl amine (DIEA). For Boc-Arg-OH this was 0.5 equivalents DIEA/acid to prevent polymerization and for all other carboxylic acid building blocks, 2 equivalents DIEA per acid were used.
The coupling reactions giving porphyrins of general structure
3 (
Scheme 1) were monitored via MALDI-TOF mass spectrometry. For this, aliquots from the reaction mixture were dried
in vacuo to remove DMF, and the residues directly employed for the mass spectrometric analyses. To obtain sufficient signal/noise ratios on a standard mass spectrometer (Bruker BIFLEX II or III), < 10 pmoles total porphyrin were required for the small libraries prepared in this study.
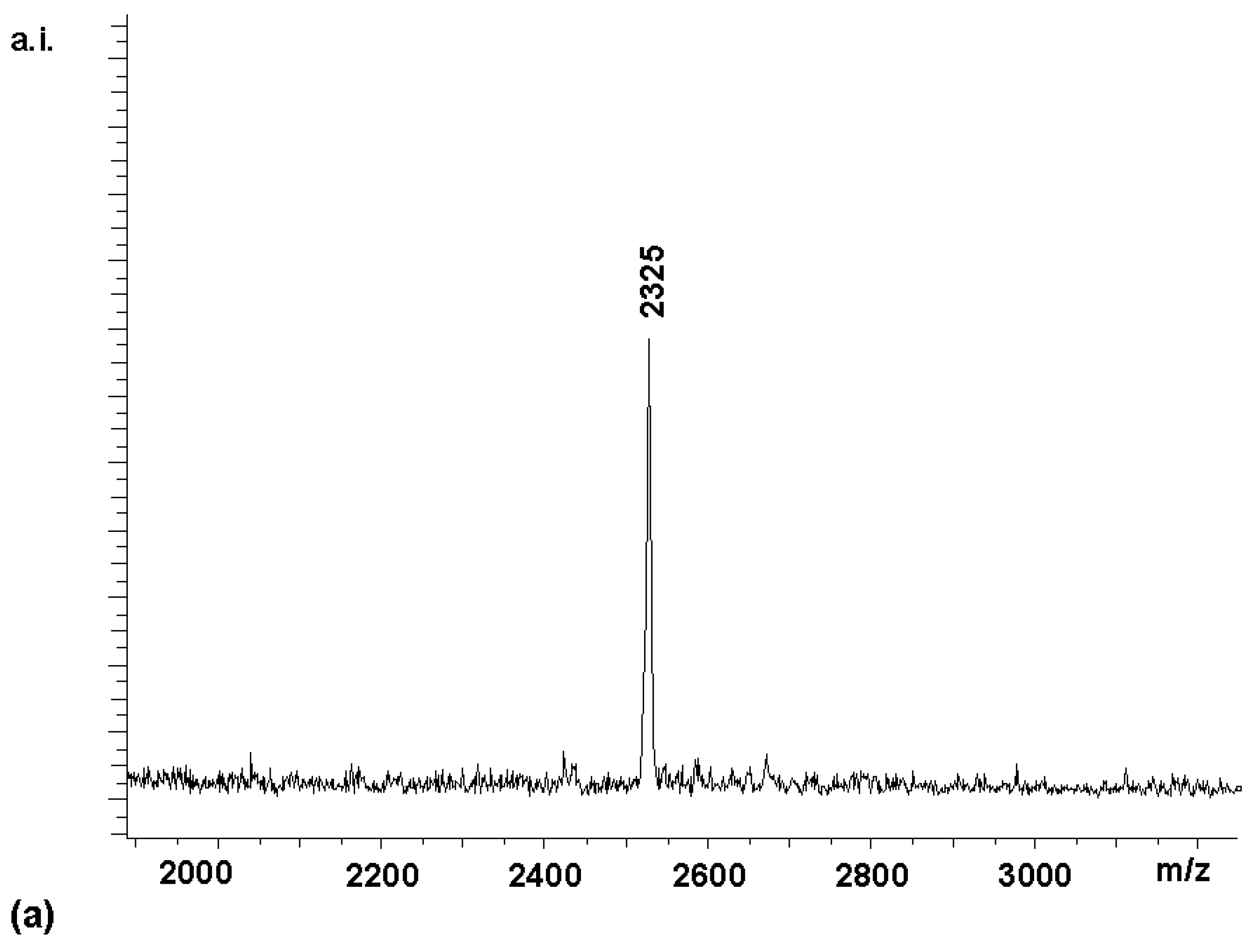
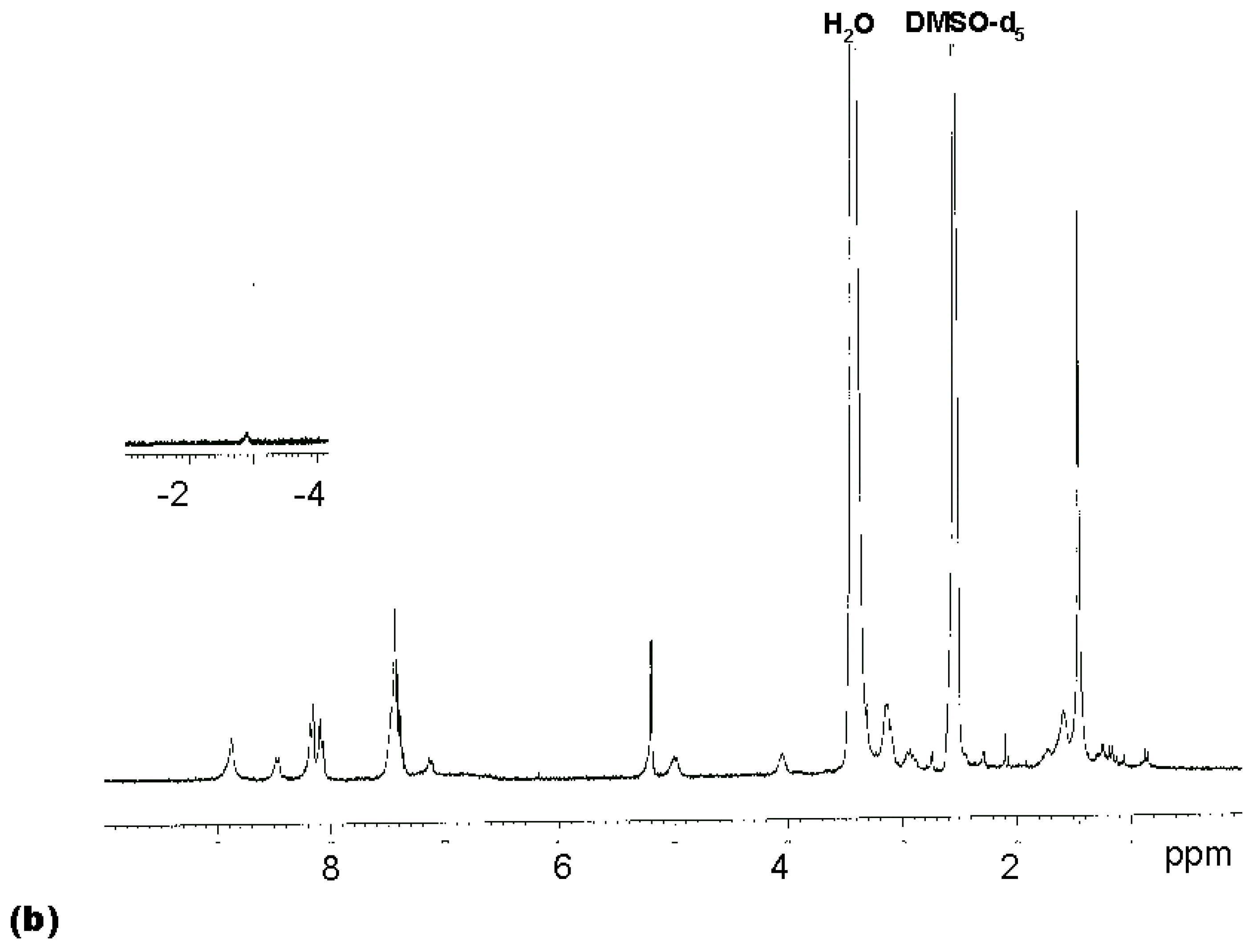
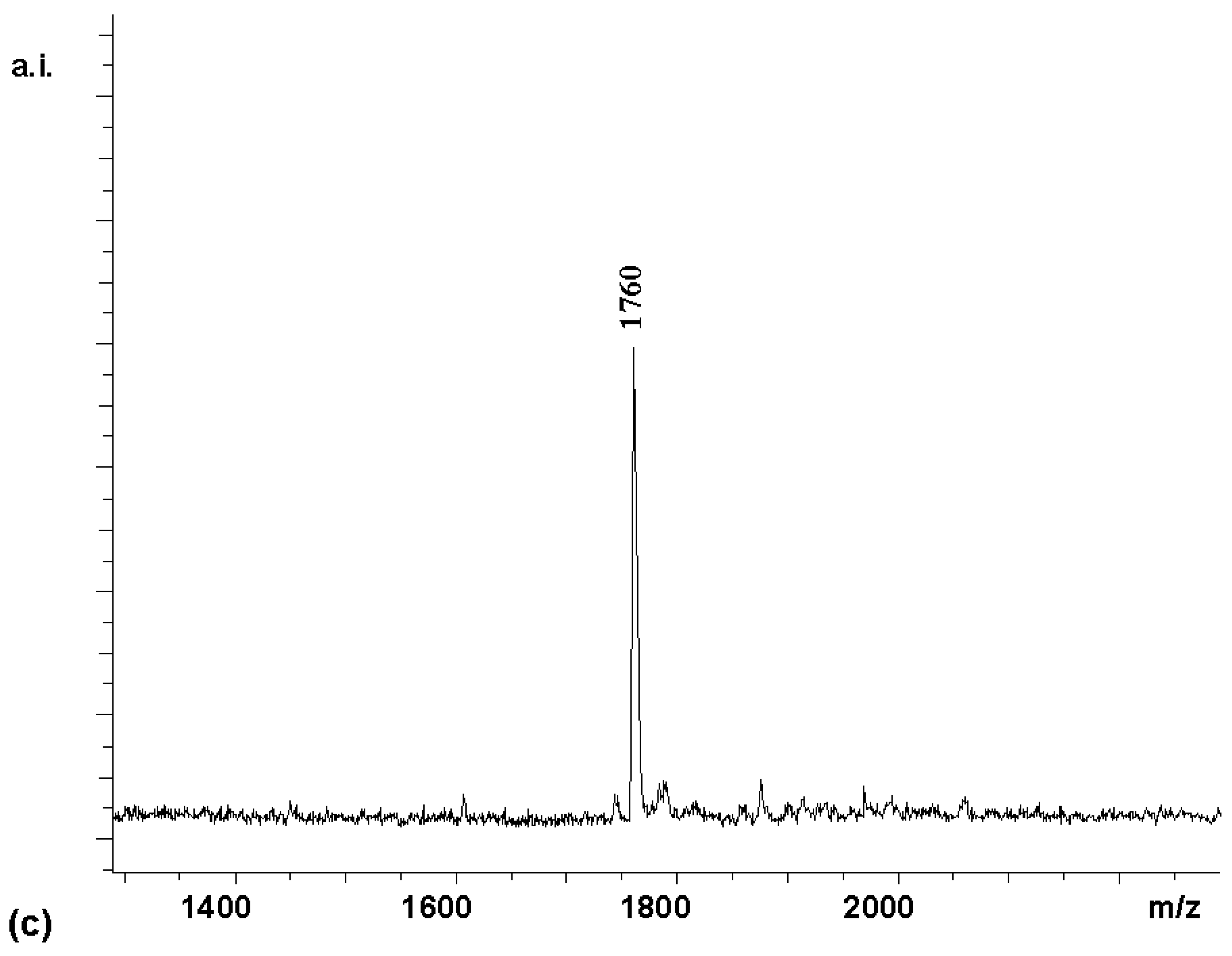
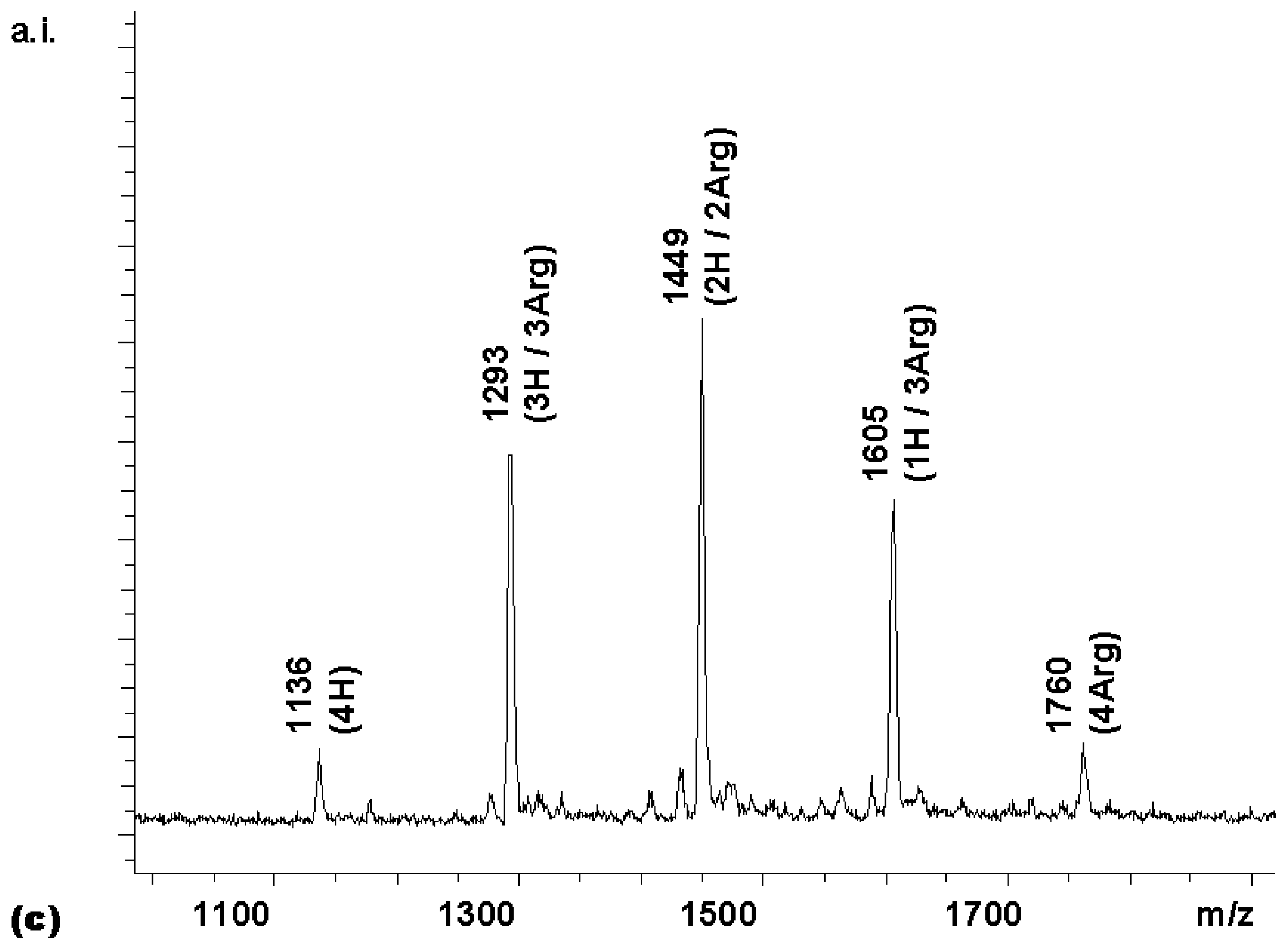
Coupling
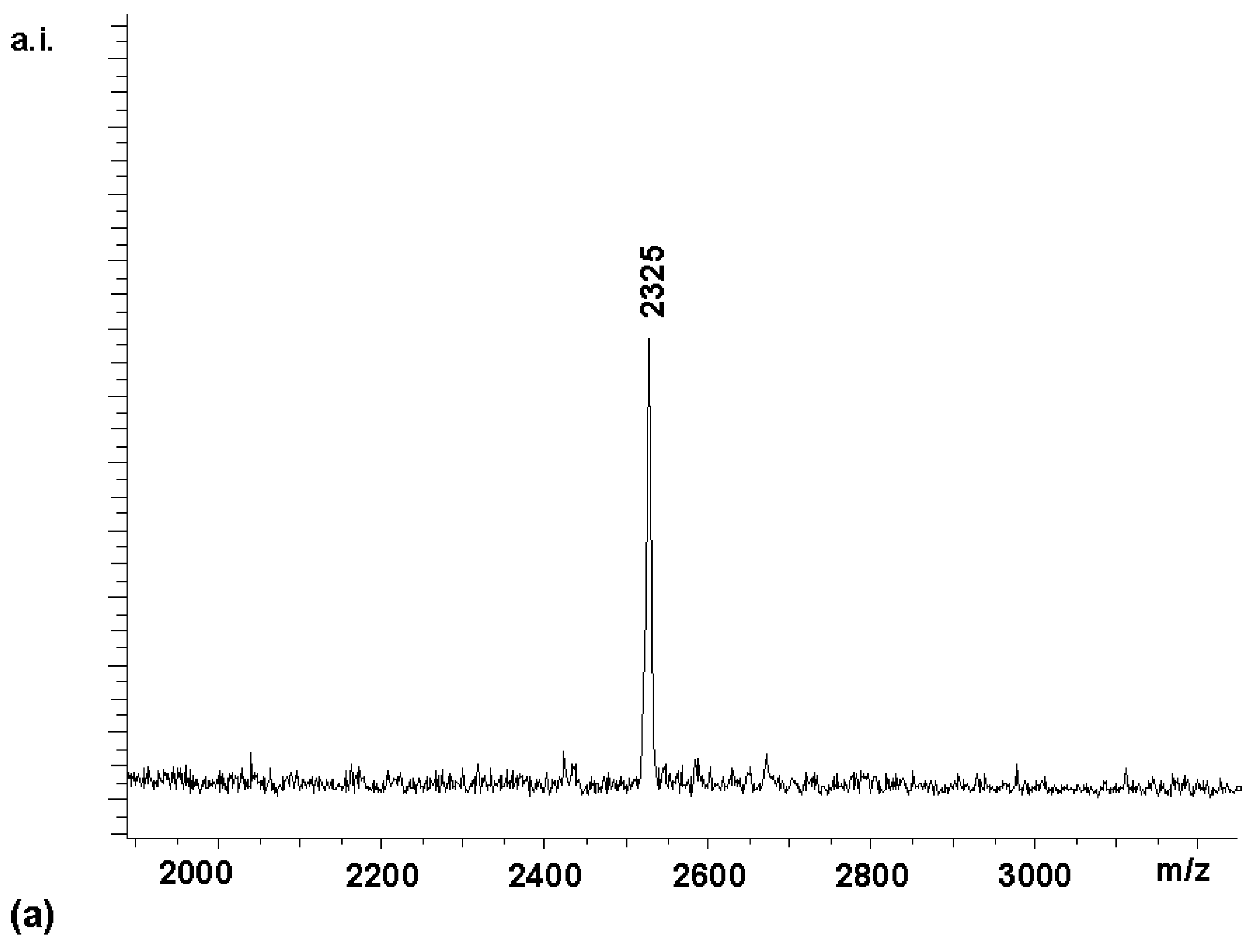
2 with 8 equivalents (2 per amino group) of Boc-Arg-OH for 60 min gave tetrasubstituted compound
4 [7] as the predominant porphyrin species in the MALDI spectrum (
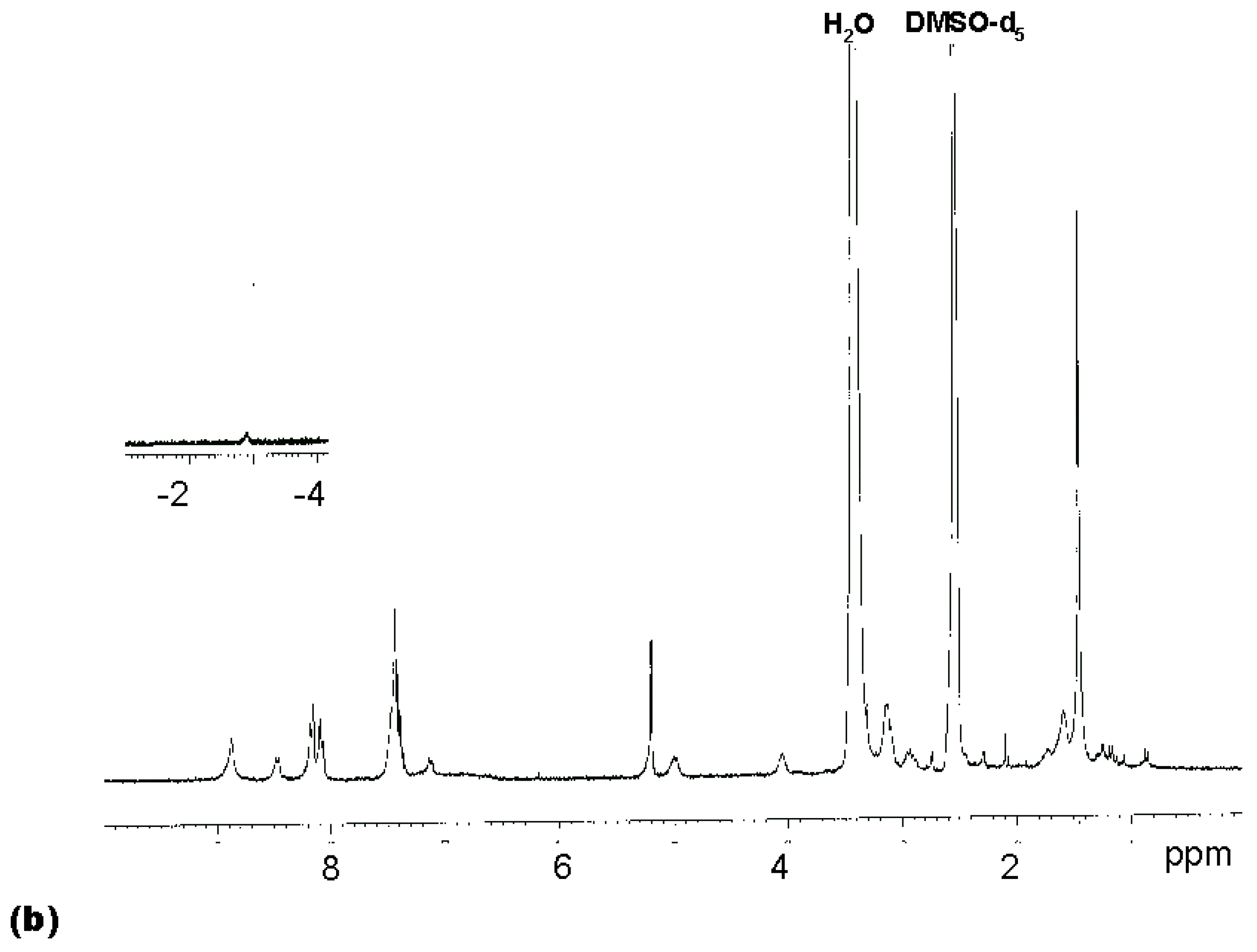
Figure 1a). A satisfactory NMR spectrum of the product was obtained in 72 % yield after precipitation from diethylether (
Figure 1b). This suggested that the amino groups of
2 were not substantially less reactive than those of a typical Merrifield resin bearing amino acid residues, where two equivalents of HBTU-activated amino acids can also be expected to give a dipeptide in high yield after a one hour reaction time. As reported in a preliminary report [
7], two-step deprotection of
4 to give
6 via
5 proceeds uneventfully, allowing the preparation of dipeptidyl-porphyrins via a route based entirely on reactions in solution.
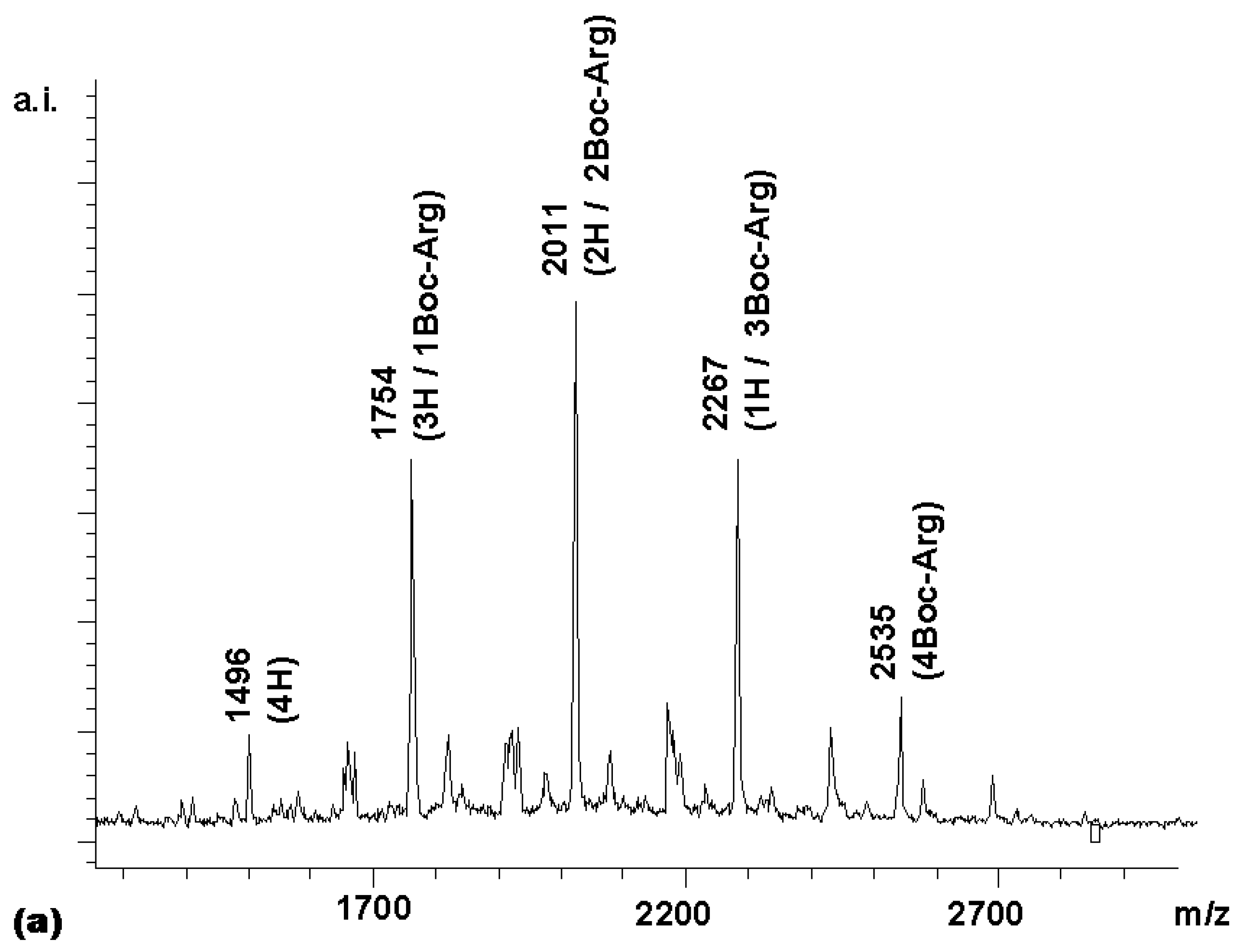
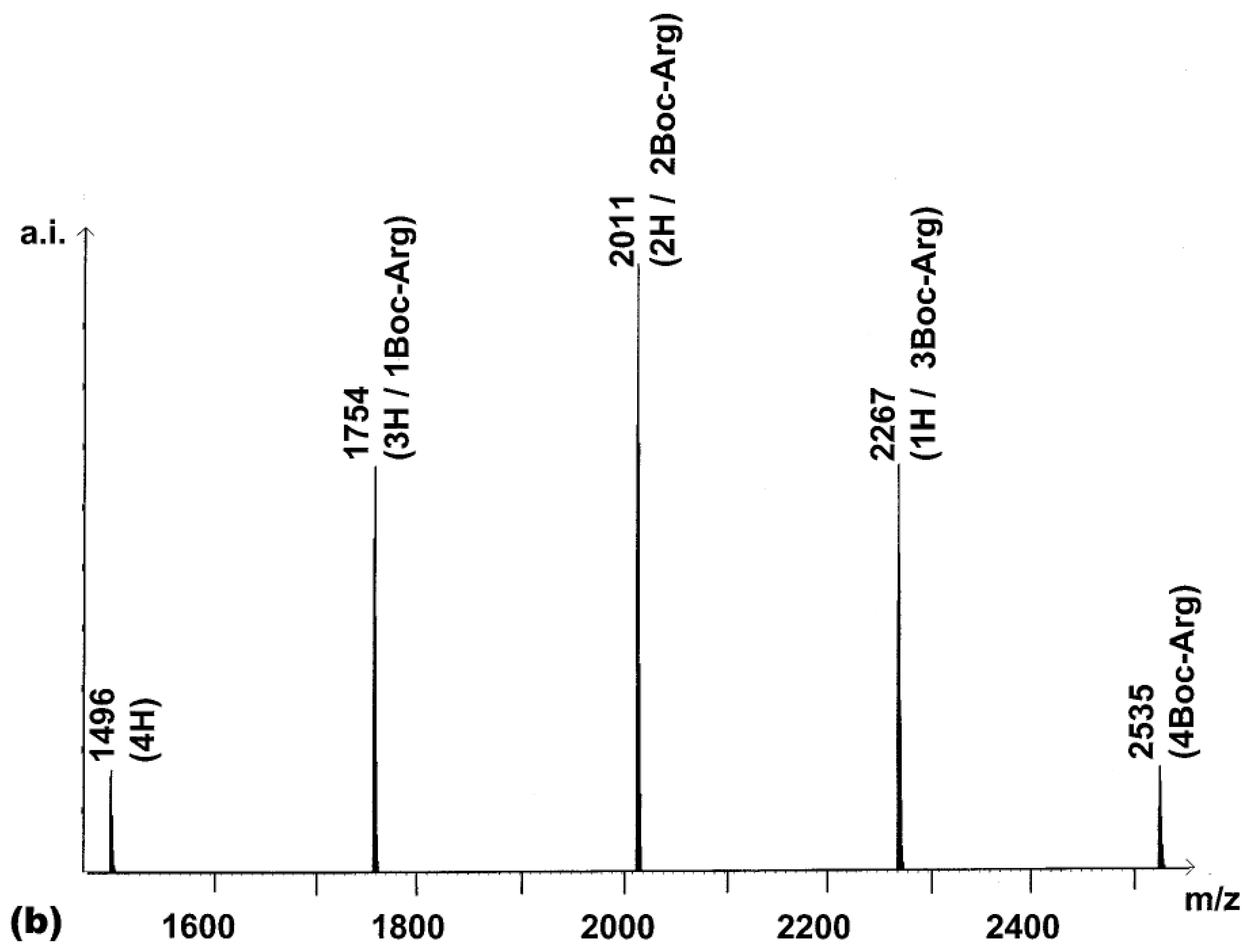
Next, the synthesis of a combinatorial library of porphyrins bearing 0-4 arginine residues (R
1-R
4 in
3 = H or Boc-Arg) was undertaken (
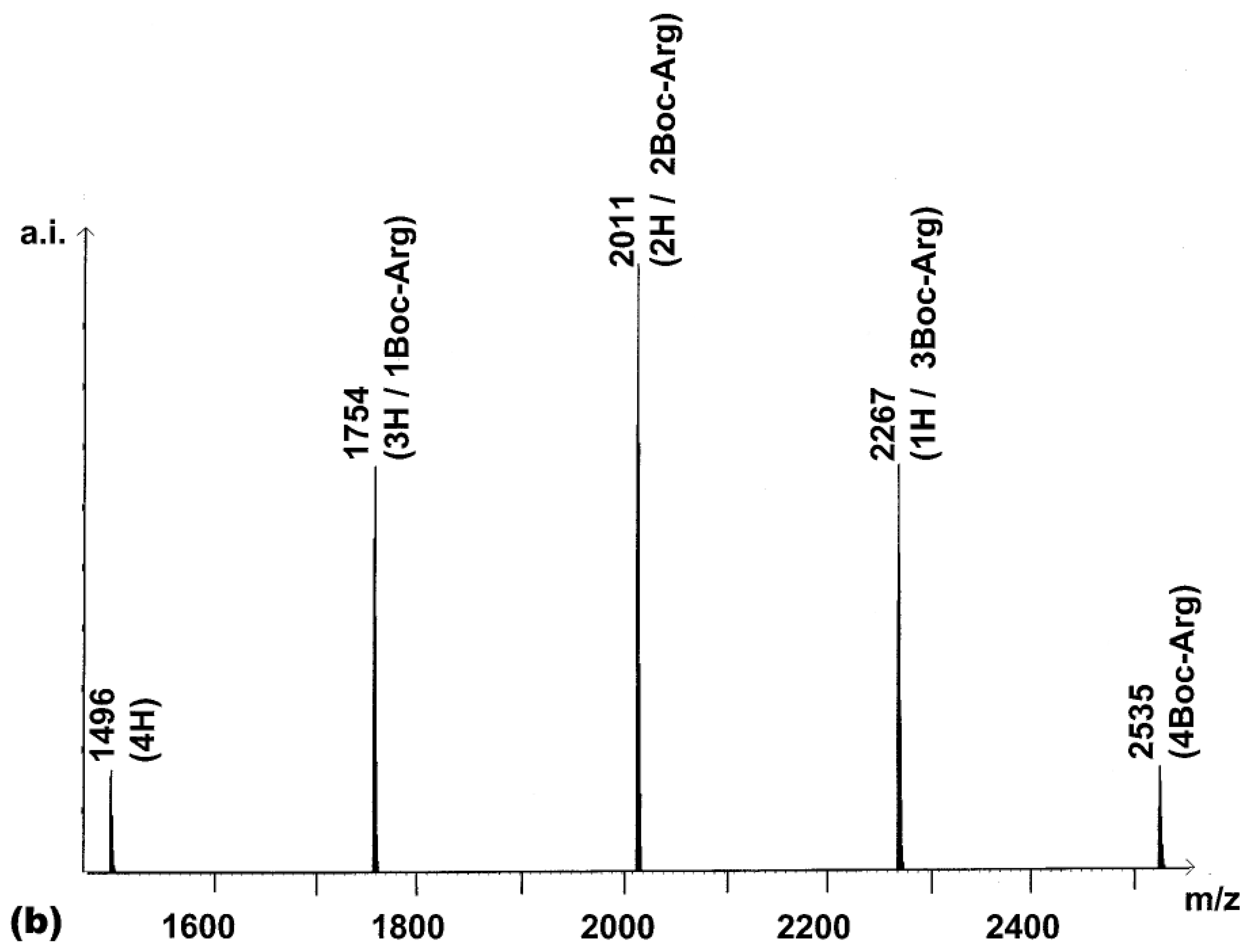
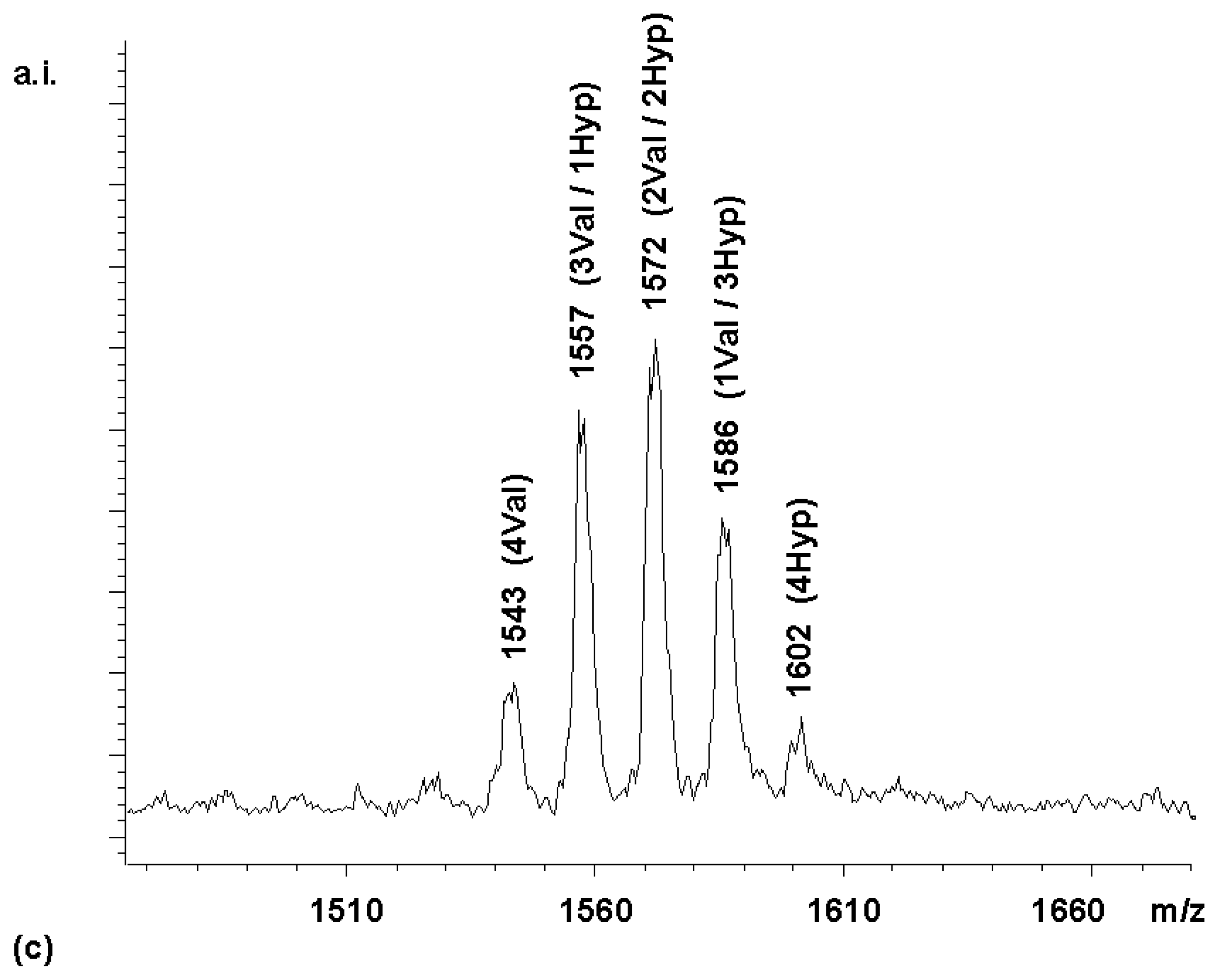
chemset 7a). For this, 2.2 equivalents of activated Boc-Arg-OH were reacted with
2. The MALDI-TOF spectrum of the product of this reaction (
Figure 2a) contained all five peaks expected. In an ideal combinatorial synthesis, these are expected to exist in a ratio of 1:4:6:4:1 (
Figure 2b).
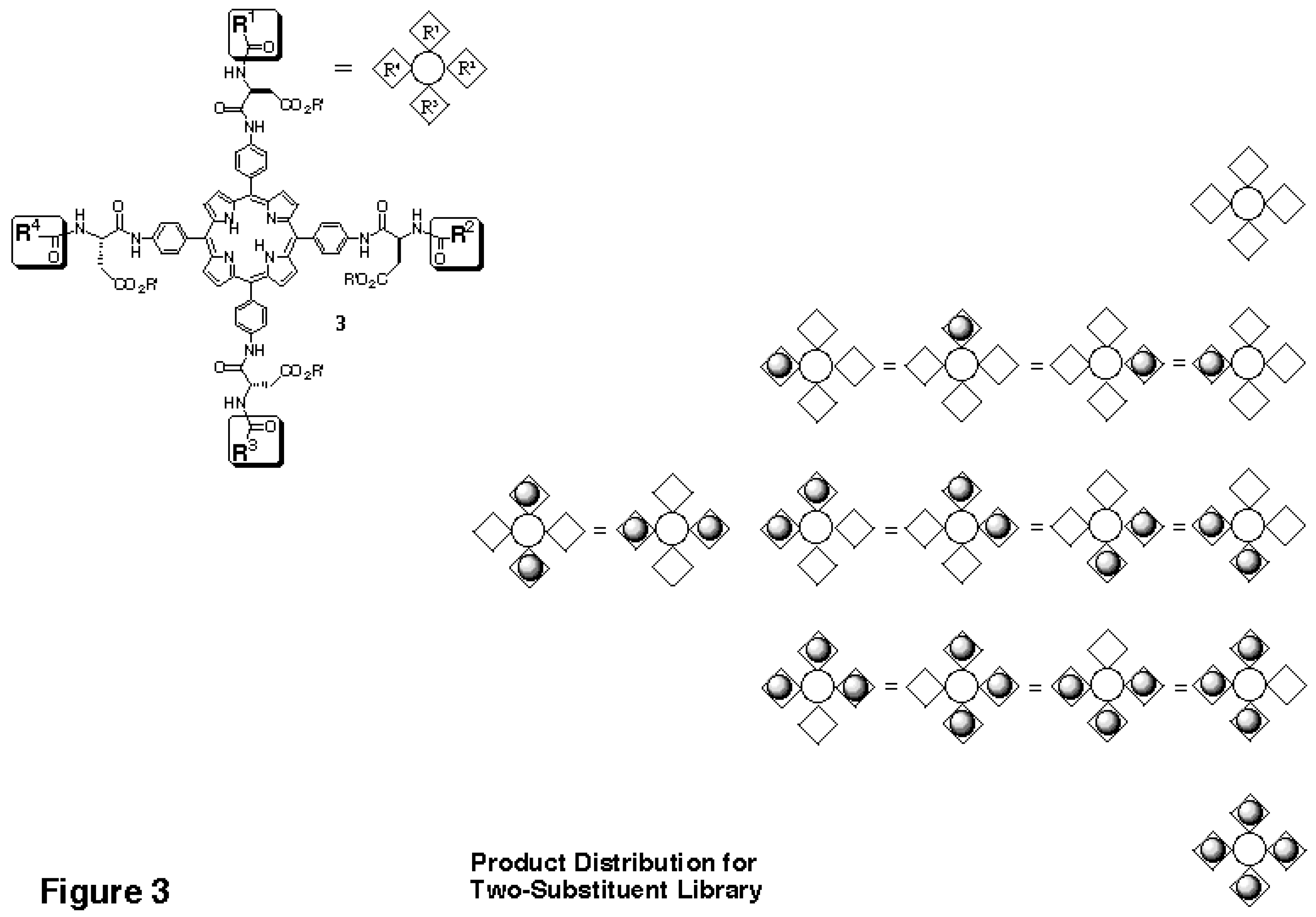
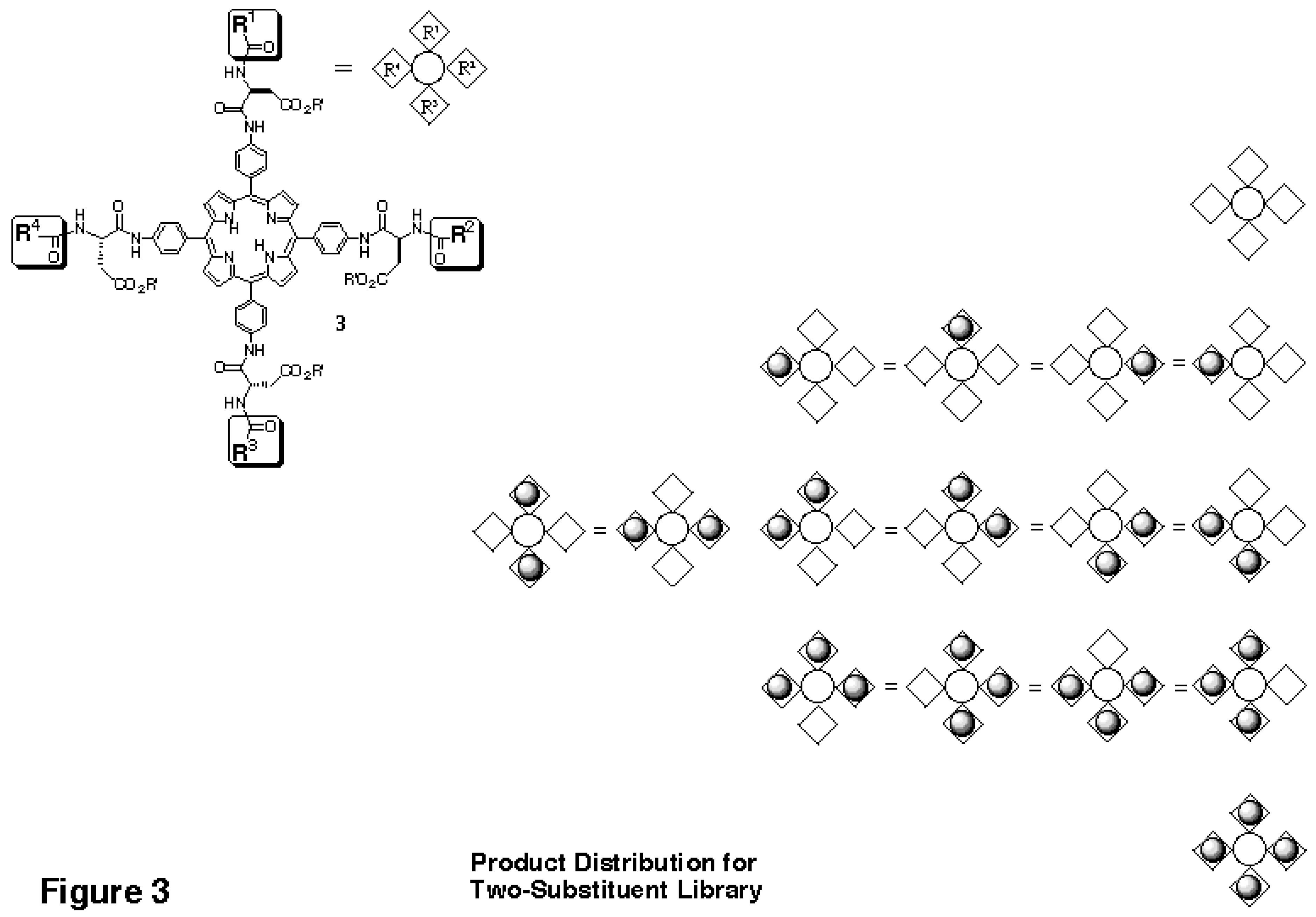
This distribution is the result of the effective symmetry of the porphyrin core, which makes many of the products identical, as shown schematically in
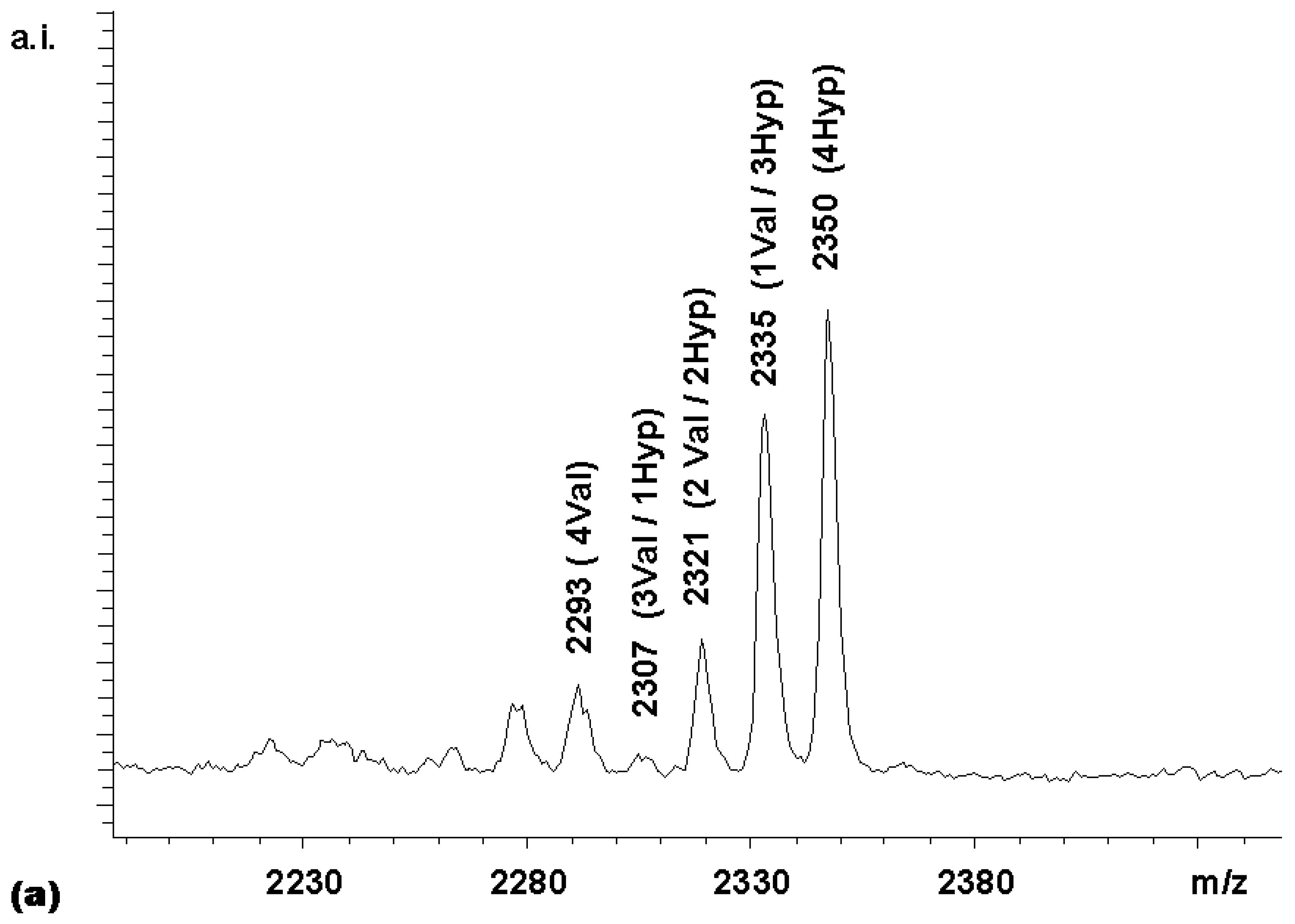
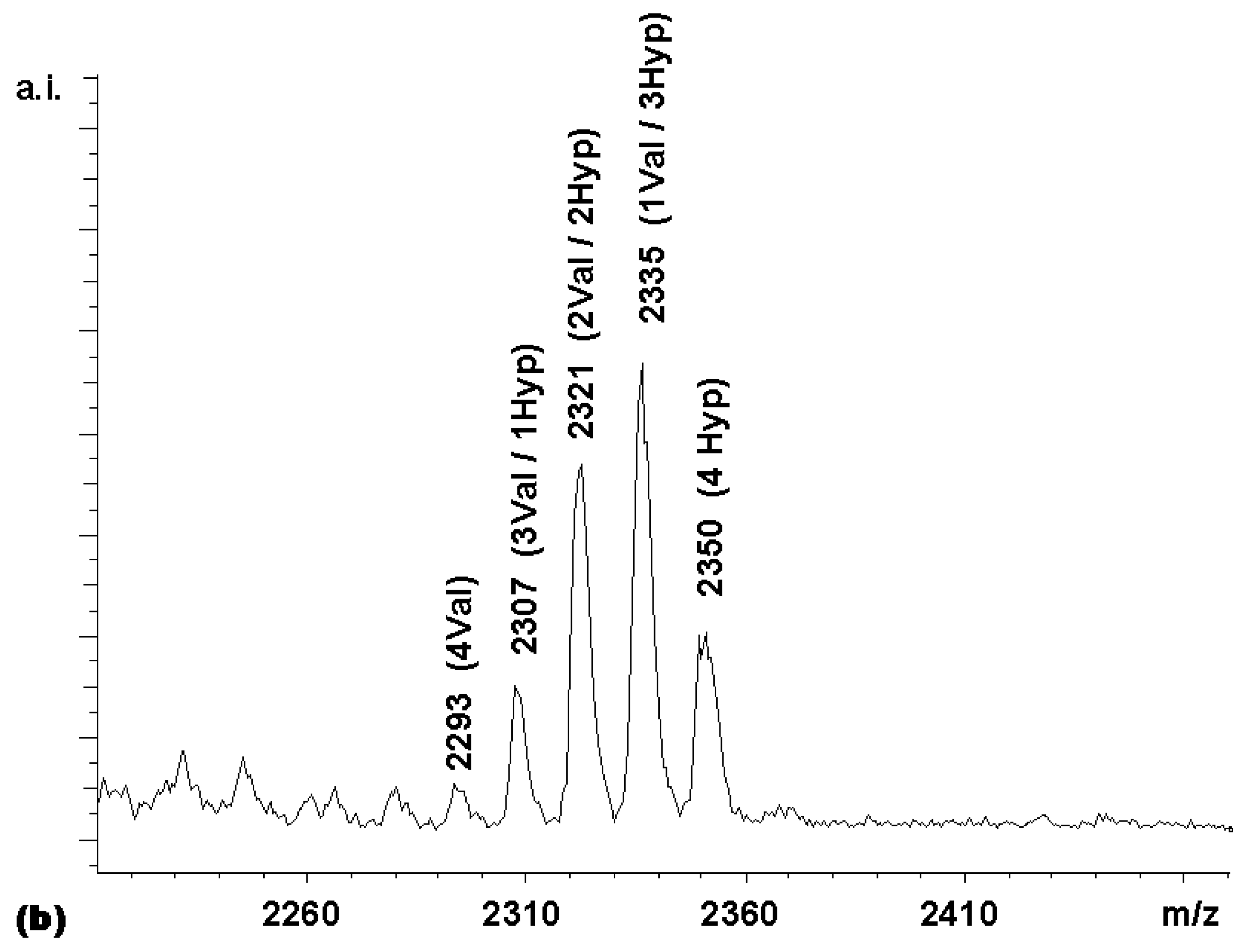
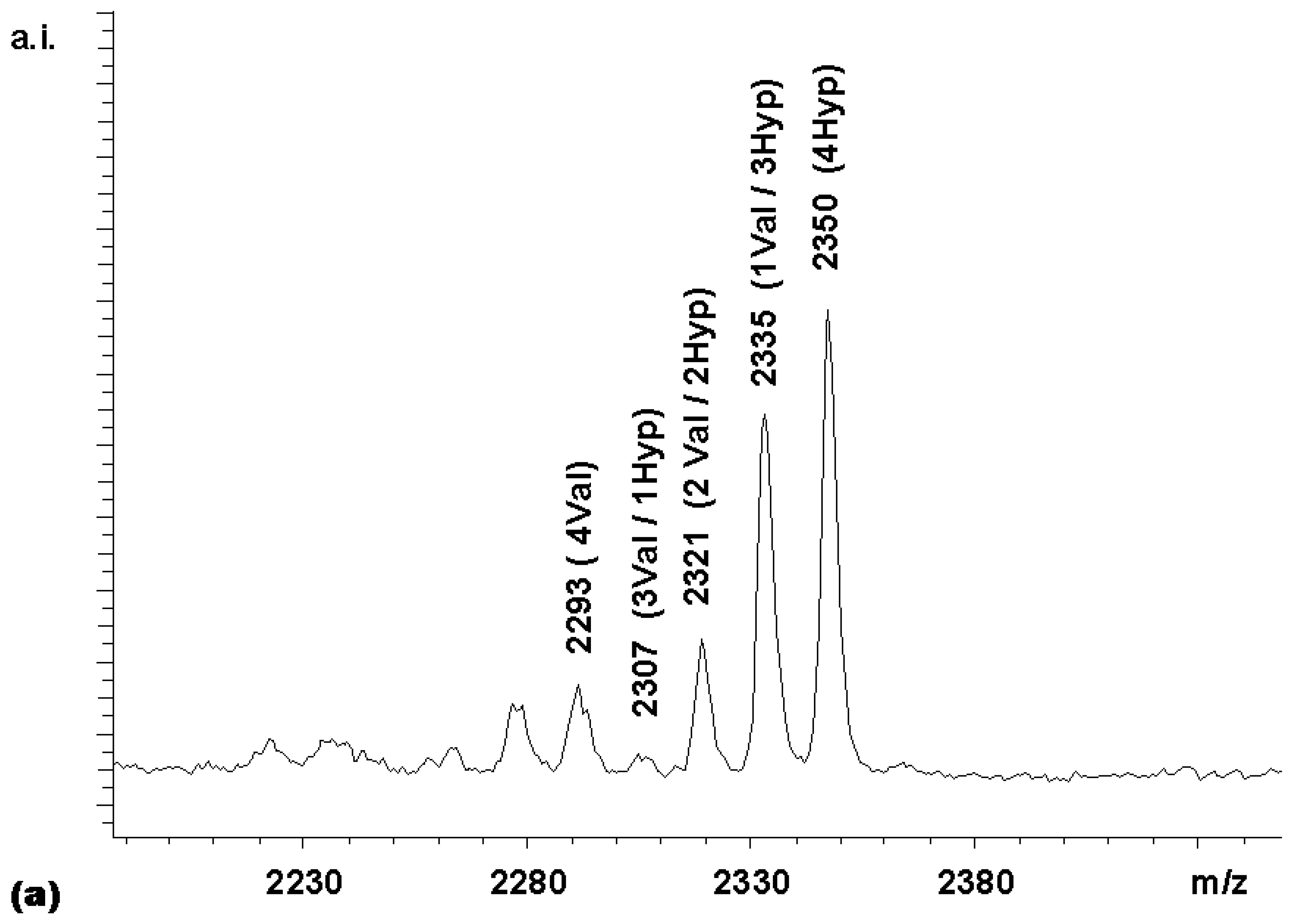
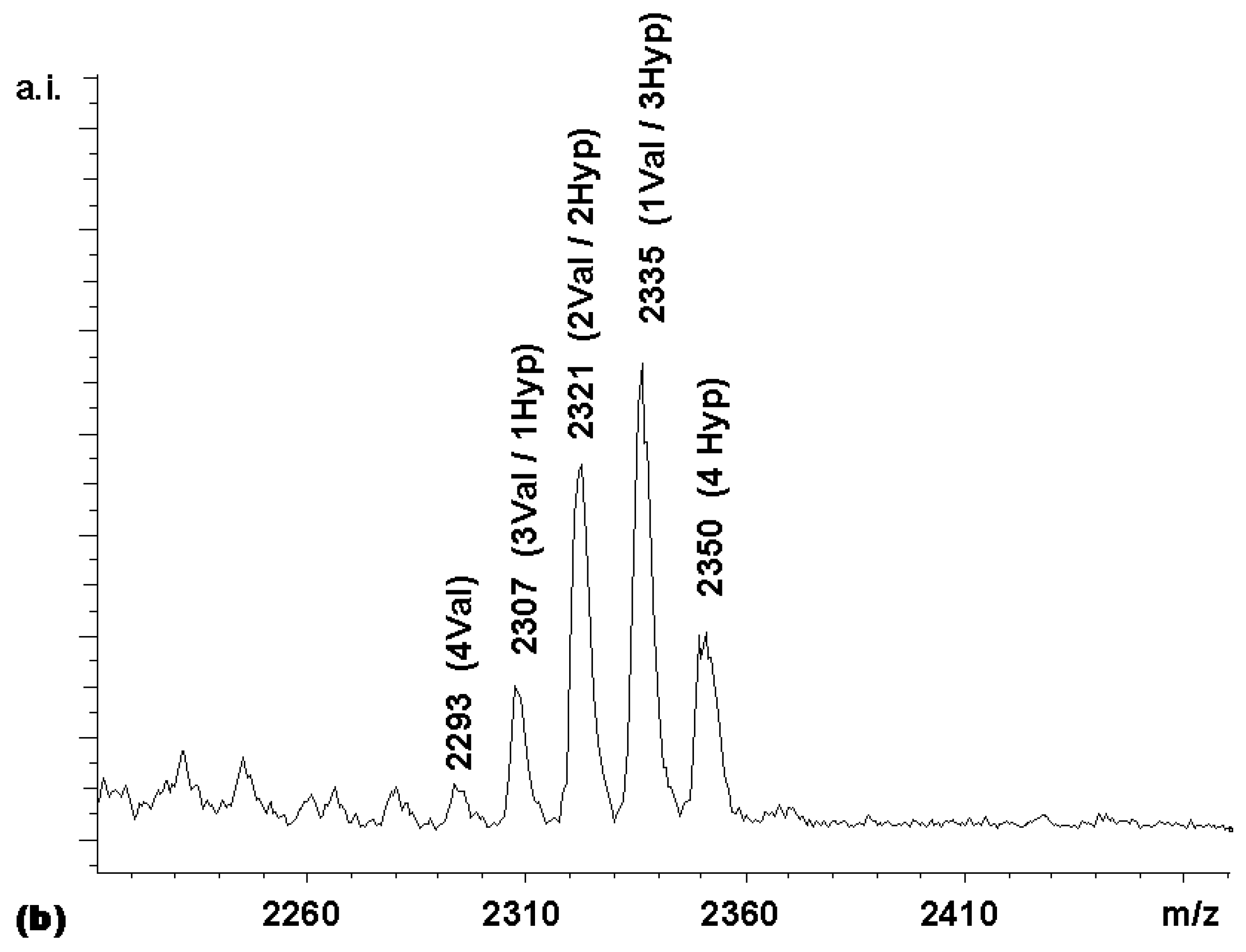
Figure 3. With this expected statistical distribution in mind, a mixed coupling reaction with two carboxylic acid building blocks was performed. The carboxylic acid building blocks chosen were Boc-Val-OH and Boc-Hyp-OH, where Hyp is 4- hydroxyproline. The MALDI-TOF mass spectrum of the library obtained (
Figure 4a) shows a non-statistical distribution of products, with the Boc-Hyp-containing compounds being overrepresented and the compounds bearing the β-branched valine residues being underrepresented. This was an expected result, based on the relative reactivities reported by Houghten and colleagues [
11] for Boc-Pro-OH and Boc-Val-OH. These authors recommended 4.56 equivalents Boc-Pro-OH and 11.9 equivalents Boc- Val-OH in mixtures designed to produce equimolar representation of these two building blocks in peptide libraries synthesized on MBHA resin. Assuming that the reactivity of the 4-hydroxyproline is similar to that of proline, the relative rates in the competitive coupling to the porphyrin core thus seemed to be comparable to those to the amino-groups on the derivatized MBHA solid supports.
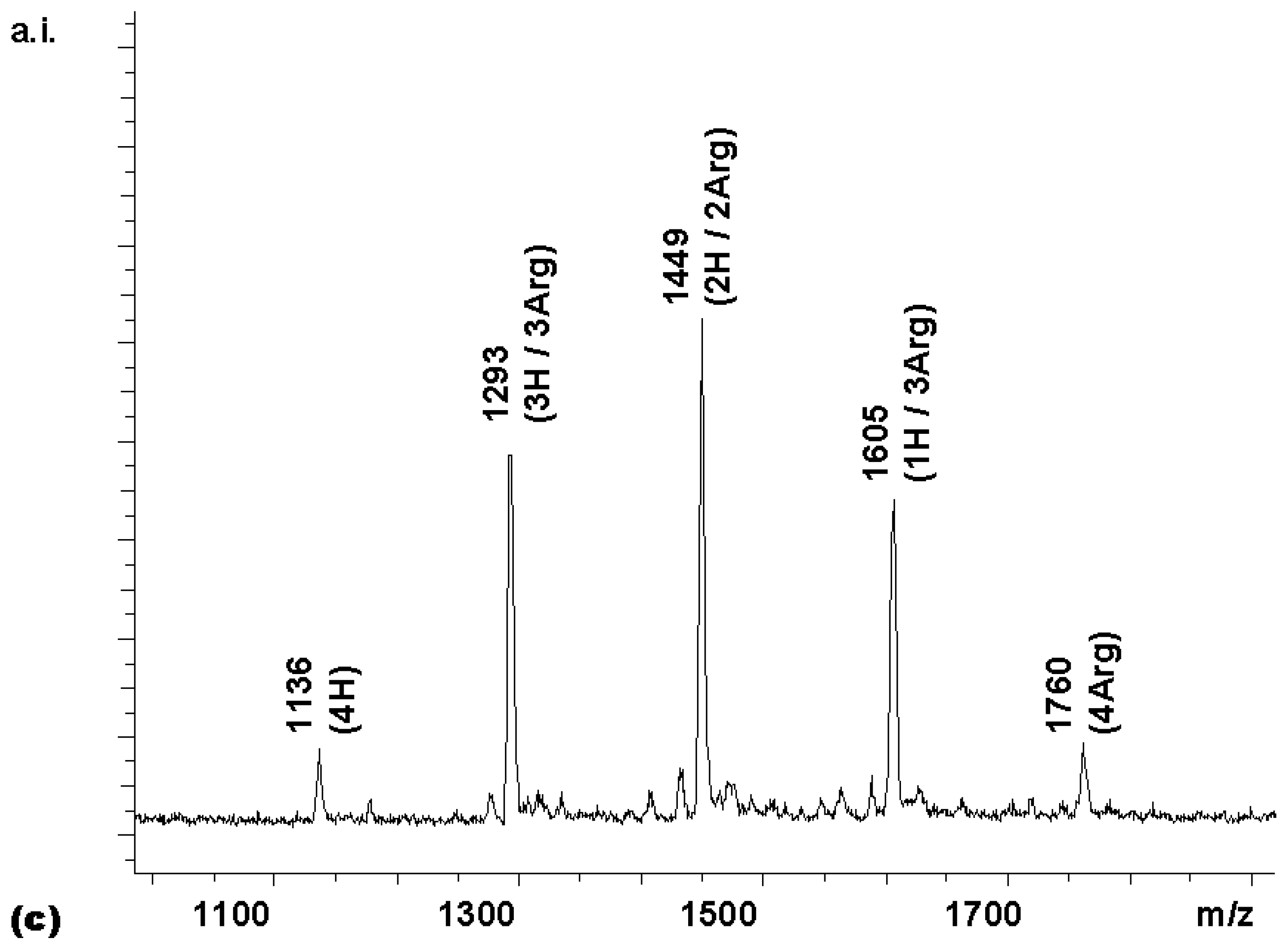
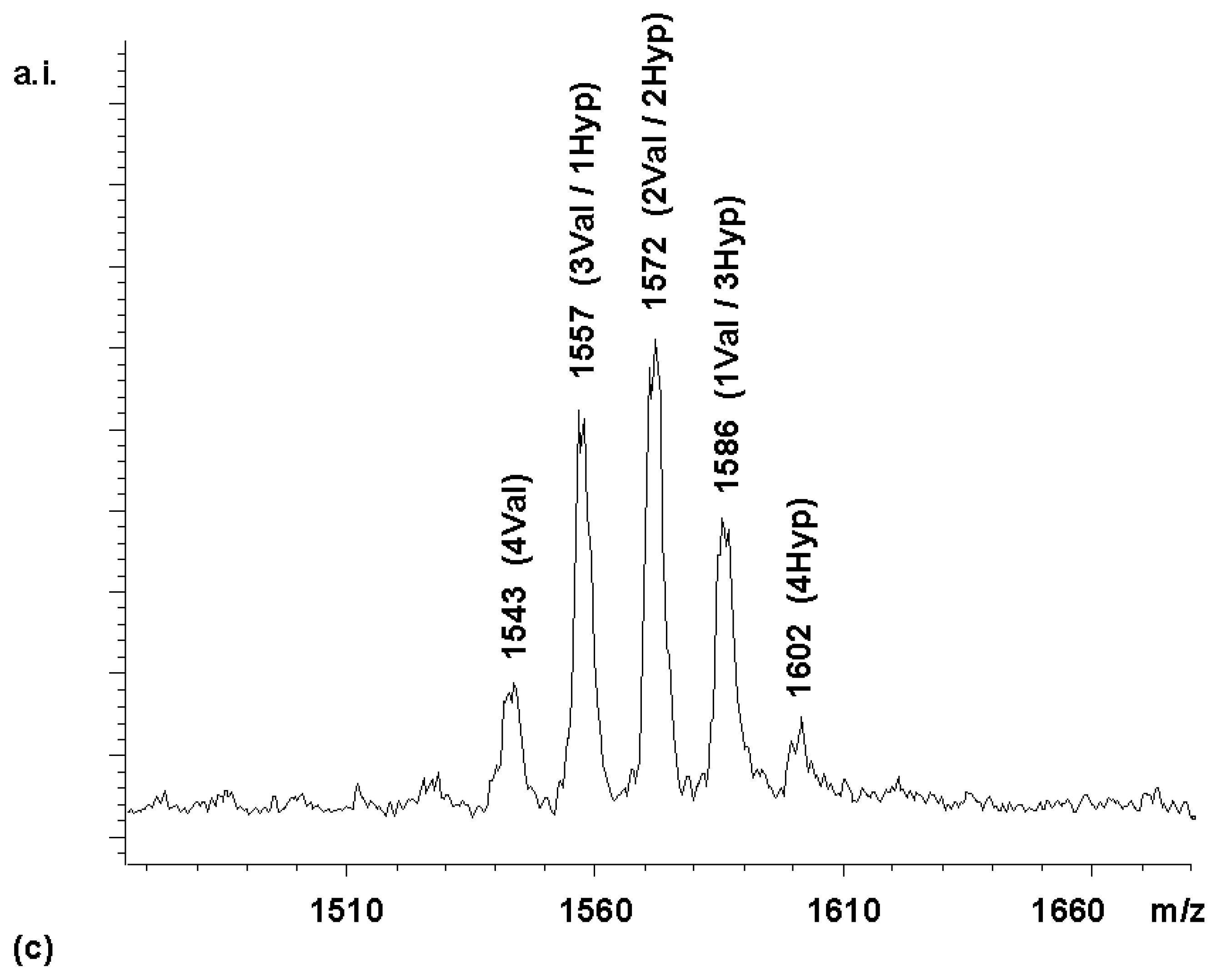
To confirm this, a coupling was performed with a Boc-Val-OH to Boc-Hyp-OH ratio of 3:1. With this reactivity-adjusted mixture, the library whose spectrum is shown in
Figure 4b was obtained. While the peak distribution was still somewhat biased towards the hydroxyproline-bearing compounds in the Boc-protected form, the MALDI spectrum of the fully deprotected library (
chemset 8b) showed a near-statistical distribution.
These differences between peak intensities for chemsets 8a and 8b may either reflect minor losses during the precipitations involved in the deprotection steps or small differences in the desorption/ionization properties of protected and fully deprotected species.
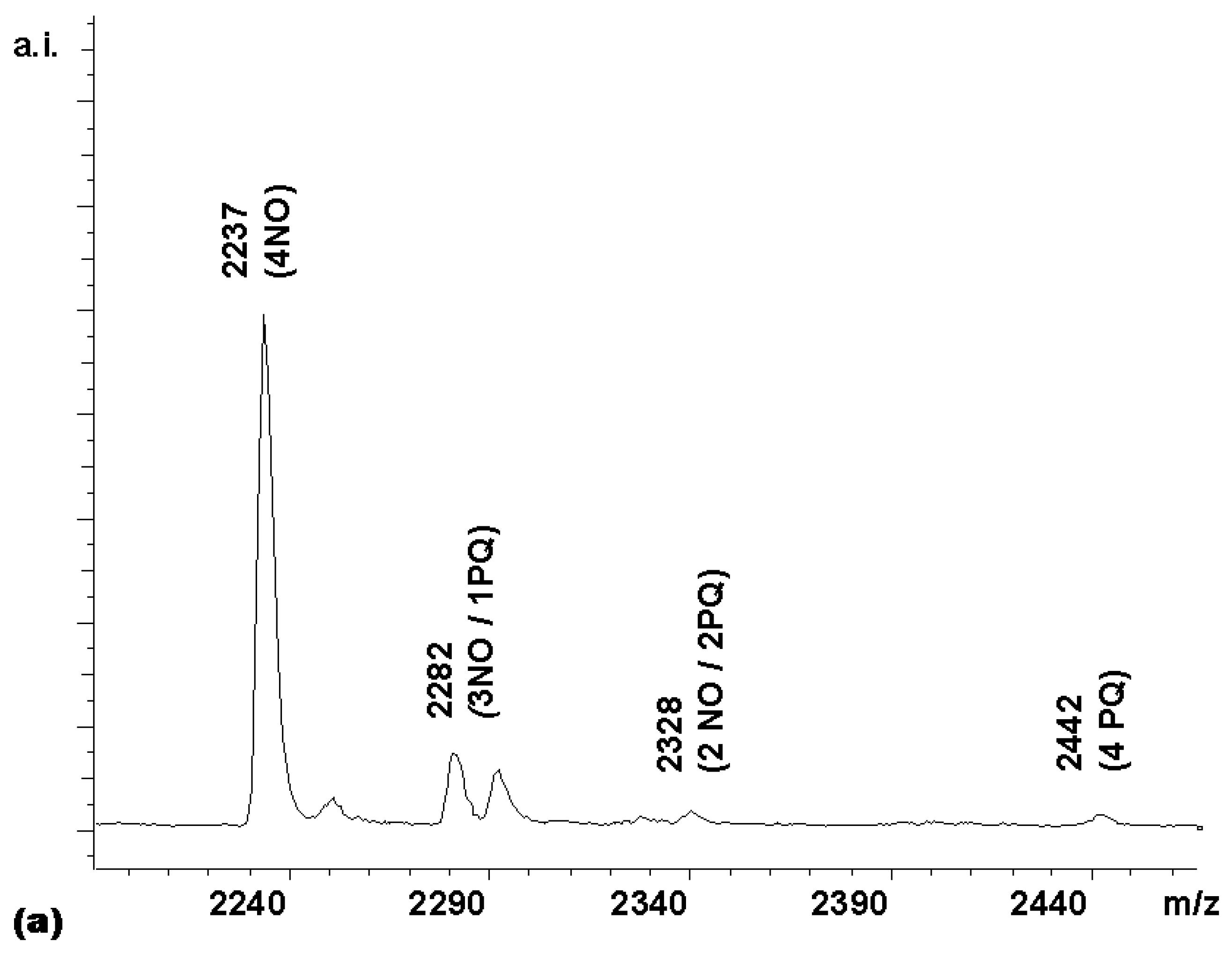
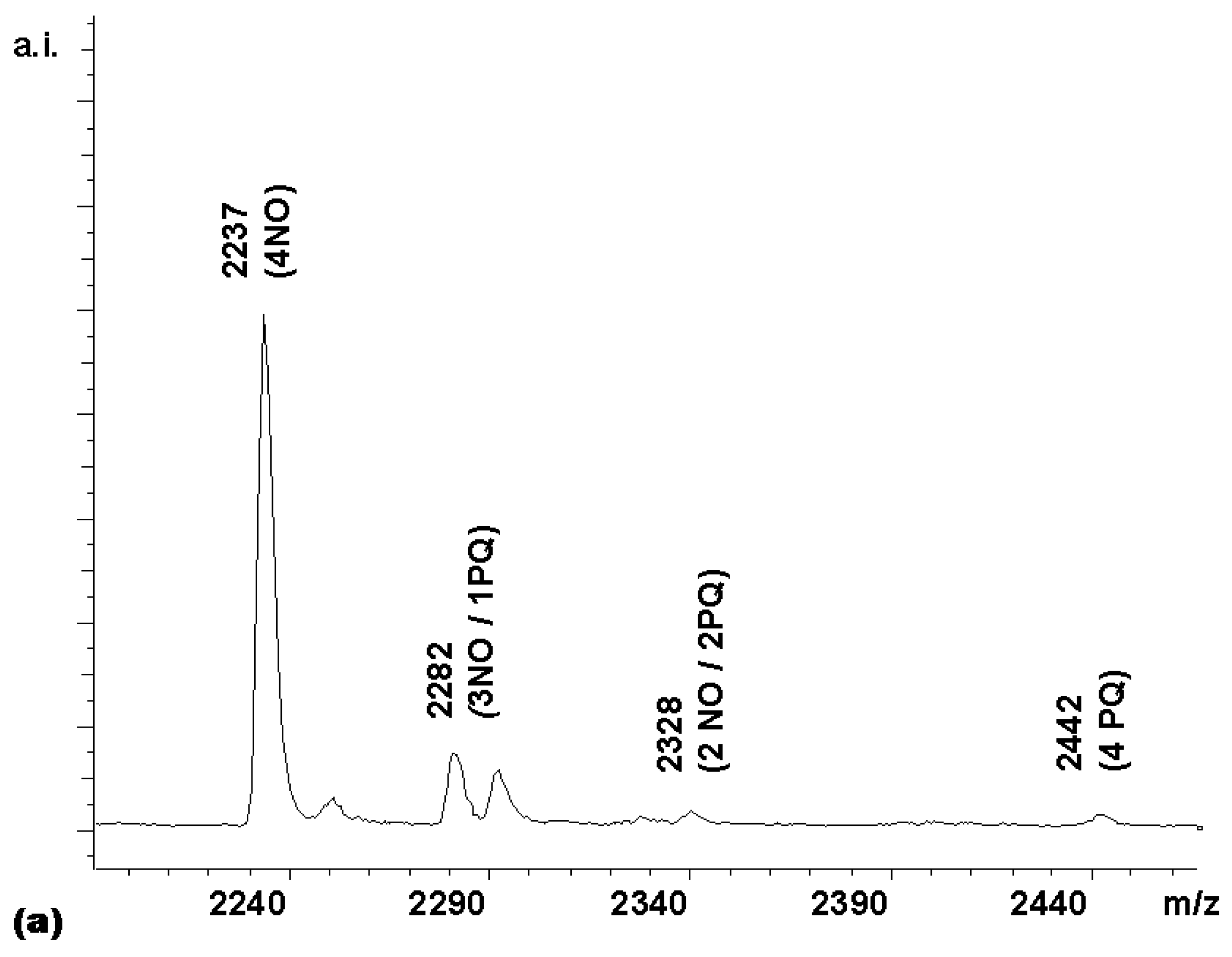
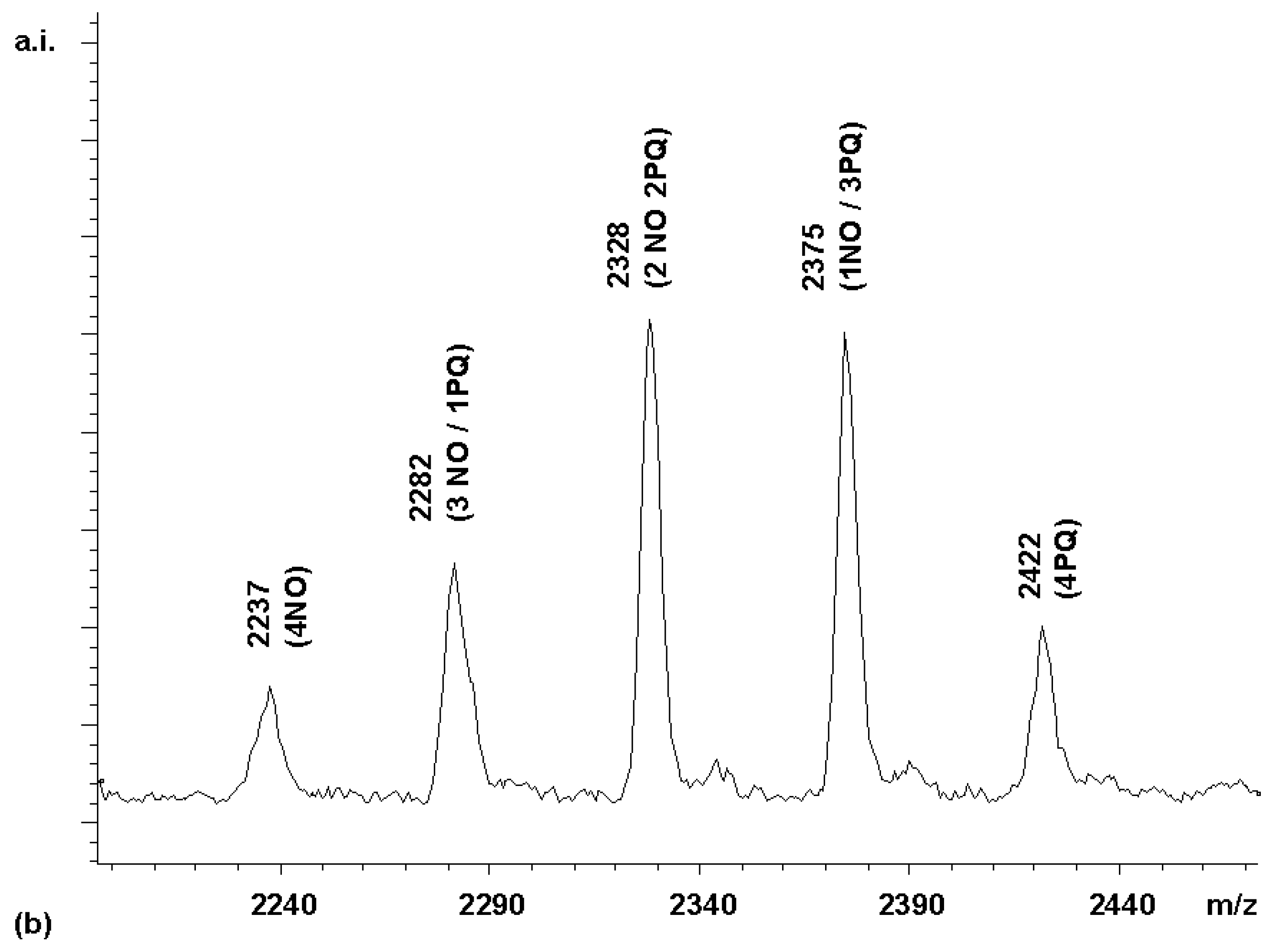
Next, a competitive coupling reaction was performed with the HBTU-activated forms of naphthoxyacetic acid (NO) and phenylquinoline carboxylic acid (PQ). With equimolar quantities of these two carboxylic acids, a product distribution heavily biased towards the less sterically hindered acetic acid derivative, whose α-carbon is a methylene group, was obtained (
Figure 5a).
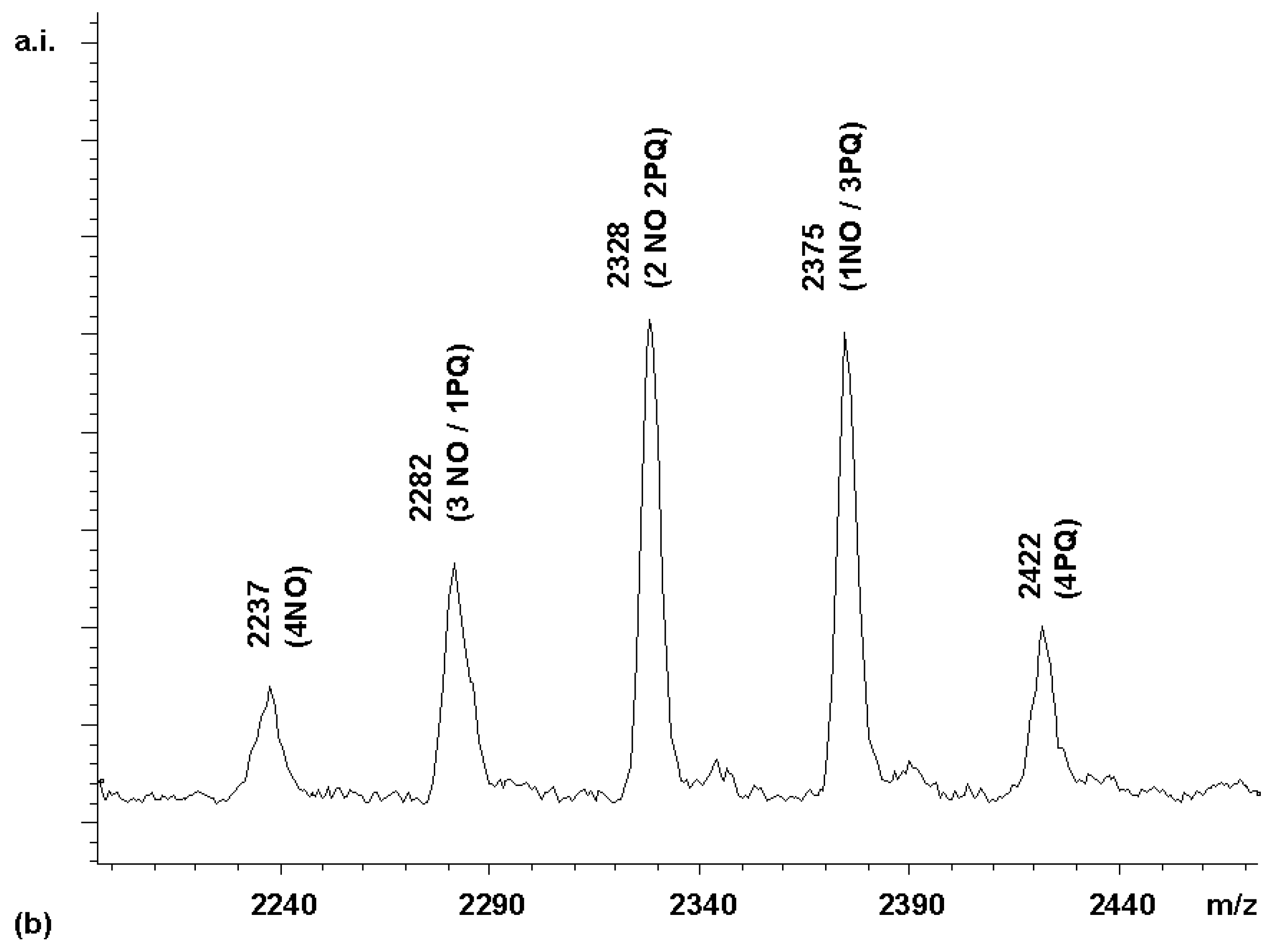
Since the intensity of the (3NO / 1PQ) peak was only 13% of the (4NO) peak, instead of the 400% expected for a statistical distribution, the quinoline acid apparently had only 1/30 of the reactivity of the acetic acid derivative. This is a difference in reactivity difficult to adjust by using an excess of the less reactive component. With a 0.1 M solution of the naphthoxyacetic acid in the coupling mixture, a 3 M solution of the quinoline would be required to make a reactivity-adjusted mixture. To obtain a near-statistically distributed library, a stepwise coupling procedure was therefore devised instead. First, a coupling mixture (4.5 equivalents) of the less reactive of the two acids, i.e. the phenylquinoline carboxylic acid, was added to a solution of porphyrin
2. This led, after 25 min, to a roughly statistically distributed library of PQ-bearing and unreacted amino groups, as evidenced by MALDITOF MS. To this was then added a coupling mixture of the napthoxyacetic acid to react the remaining free amino groups. The MALDI spectrum of the resulting library (
chemset 9) was gratifying, in that it contained a close-to-ideal product distribution (
Figure 5b).
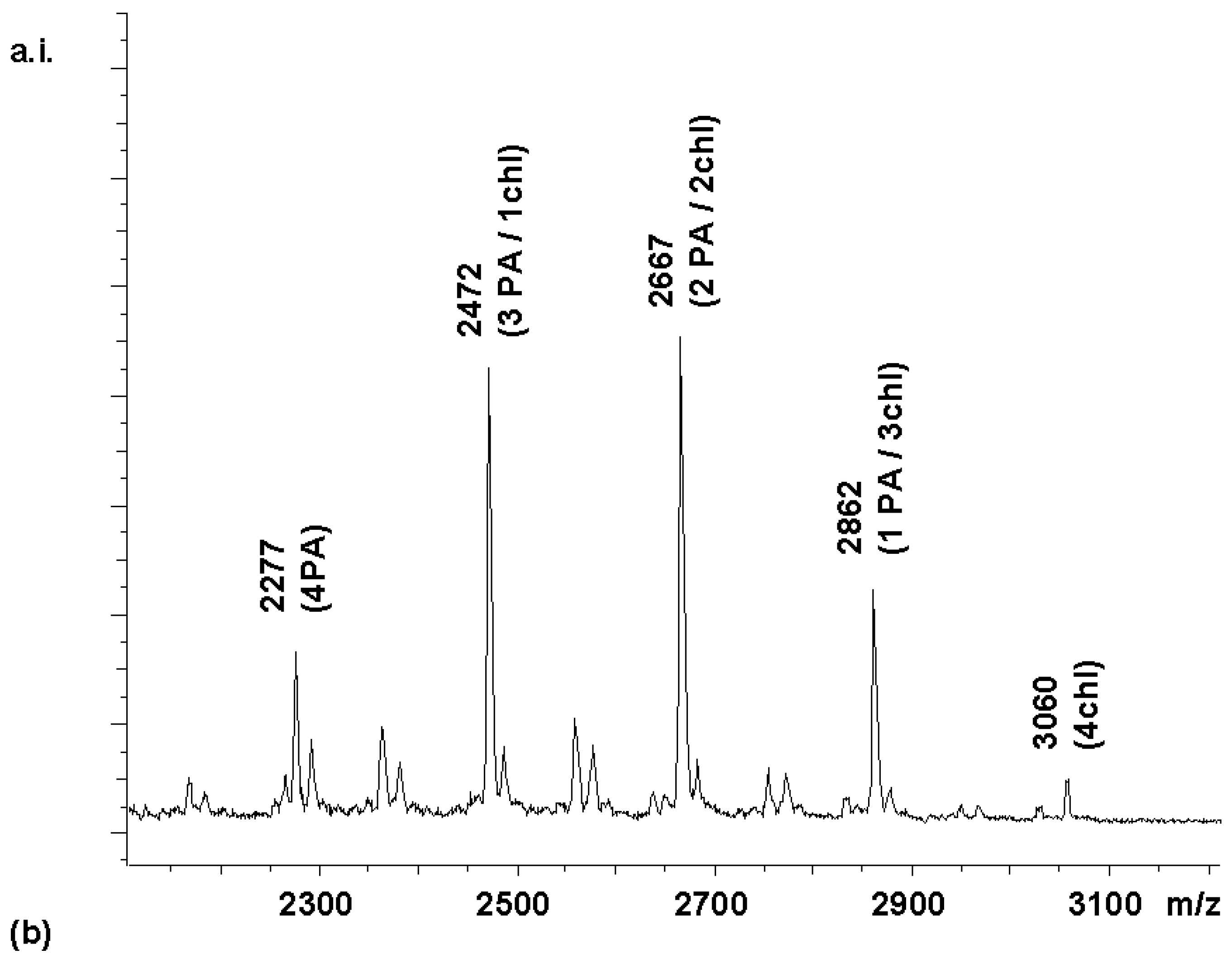
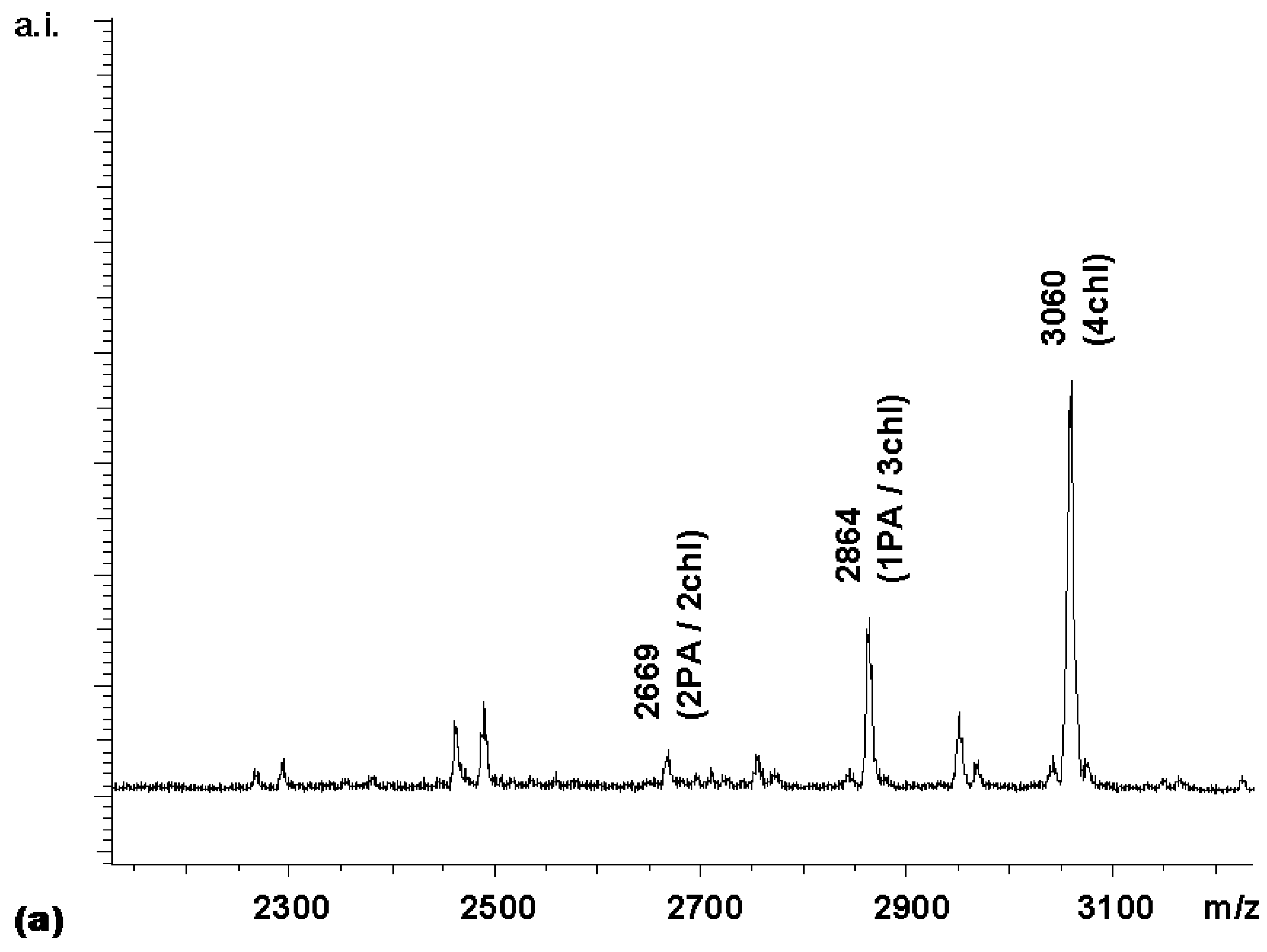
To extend this approach of stepwise coupling to a pair of even more diverse carboxylic acids,
chemset 10 (
Scheme 1) was prepared. The outcome of the competitive coupling with these building blocks was non-trivial to predict. Both reactants contain functional groups that can, in principle, react with the activated ester formed from their carboxylic acid functionalities. The cholic acid contains three hydroxyl groups that can form esters, and the phenylanthranilic acid contains an aromatic secondary amine that may react to give an amide. Ester formation between primary hydroxyl groups and HBTU-activated acids has previously been used in the synthesis of triester solid supports [
12].
Figure 6a shows the MALDI spectrum of the library obtained when coupling
2 with an equimolar mixture of the two activated carboxylic acid building blocks. The less sterically hindered (and electronically less deactivated) bile acid gained the upper hand in the coupling reaction. When the same reaction was performed in a step-wise manner by adding the activated anthranilic acid derivative first (4 equivalents, 20 min), followed by addition of another 4 equivalents of activated cholic acid, a near-statistical distribution of products was obtained (
Figure 6b).
Discussion
The combinatorial syntheses presented here can be monitored directly via MALDI-TOF mass spectrometry, without a need for cleavage from a solid support or even work-up. Further, the detection technique is at least three orders of magnitude more sensitive than NMR spectroscopy, suitable for automation, and produces trivial-to-predict signals for each non-isobaric library component. This allows for a convenient monitoring of coupling reactions. The disadvantage of the technique is that quantitation via MALDI is much less reliable than integration of NMR signals. An equimolar mixture of three tetrasubstituted porphyrins of general structure
3 with four thymine acetic acid residues, four nalidixic acid residues, or four cholic acid residues, for example, gives relative peak intensities of 2.2 :5.7 : 2.2, i.e. depending on the structure of the terminal para substituents the signal intensity varies by up to 2.6-fold. Therefore, unless a calibration experiment is performed first, one has to treat the relative peak intensities obtained for this library with an error factor of approximately 3. Though this is a serious drawback, obtaining a library whose composition is statistical within a factor of 3 is often acceptable, particularly when SMOSE experiments [
5] are performed to identify hits.
In the reactivity tests performed, the aminoacylated tetraanilinoporphyrin behaves like a micro-solid support reacting at four sites. The "microsupport" is small enough to be detected directly via MALDITOF mass spectrometry. In fact, the medium size of the scaffold facilitates MALDI analysis, since it shifts the peaks of interest into a mass range above 1500 Daltons, where no interference from matrix peaks occurs. Further, the peak distribution for a two-component tetraphenylporphyrin library (
Figure 3) is well suited to measure relative reaction rates of building blocks with substantially different reactivities. The ratio between the A3B and the 4B peaks of a library bearing building blocks "A" and "B" is 4:1 in a statistically distributed library. As a result, even if one of the two building blocks has only 1/30 of the reactivity of the other, a readily measurable signal intensity (13% of the dominating 4B peak) is still obtained for the A3B peak, if B is the less reactive component (compare
Figure 5a). This peak would be only 3.3% of the predominating one in the case of the corresponding peptide library with its "peptide-A" and "peptide-B" peaks. This may make this technique useful for investigators who want to test the reactivity of new building blocks prior to employing them in large scale library syntheses. Perhaps even Hammett-Parameters for reactions relevant for combinatorial syntheses [
13] could be measured in MALDI-monitored reactions employing porphyrin cores.
Substituted tetraphenylporphyrins have recently been shown to be high affinity ligands for protein surfaces [
14] and short single-stranded DNA [
15]. Since the libraries reported here are efficiently prepared in solution, i.e. without the unpredictability of solid support-based reactions [
16], porphyrin-based chemsets may become attractive for the search for other functional porphyrins. With two-component libraries, obtained step-wise with structurally diverse and non-equireactive building blocks, an interesting structure space may be searched.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}