Synthesis of the Aspidosperma Alkaloid Na-Formyl-16-α-Hydroxyaspidospermidine

Department of Chemistry, Faculty of Science, Constantine University, 25000, Algeria
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(10), 803-814; https://doi.org/10.3390/61000803
Submission received: 1 April 2001
/
Revised: 6 September 2001
/
Accepted: 8 September 2001
/
Published: 30 September 2001
Abstract
:The first total synthesis of Na-formyl-16α-hydroxyaspidospermidine and its isomer via (°)-vincadifformine is described and their structure elucidation using different methods of analysis is reported.
Introduction
The indole alkaloid strictanine (1b) was recently isolated from the fruit of Rhazia stricta Decsne [1] which is an indigenous medicinal plant abundantly found in Pakistan. This medicinal plant has long been used for the treatment of various diseases [2,3]. We report here the first total synthesis of this compound starting from (°)-vincadifformine (2), which was previously prepared from tryptamine hydrochloride by an adaptation of Kuehne’s biomimetic synthesis [4,5]. This prompted us to extend it to more oxygenated alkaloids such as spegazzinine (10a) and spegazzinidine (10b) [6].
Results and Discussion
The existing literature method for the conversion of the readily available (±)-vincadifformine (2) into the key intermediate 16-hydroxyindolenine (3) showed that the latter compound is particularly unstable [7, 8]. Similarly, photo-oxygenation [9] of 2 in the presence of Rose Bengal and sodium thiosulphate as reducing agent to give 16-hydroxyindolenine (3) resulted in low yields of the desired compound. The other product was mainly recovered starting material. This is undoubtely due to the different tungsten lamps used (2x150W). Le Men and co-workers [10] also achieved this convertion in vitro through a multi-step procedure involving the prior oxidation with peroxyacids to afford the 16-hydroxy-1,2-dehydrovincadifformine-Nb-oxide (4) in which the Nb centre was blocked to avoid any spontaneous rearrangement.

Reaction of vincadifformine (2) with m-chloroperbenzoic acid in dry benzene gave, after evaporation of the solvent, a variable yield of 16-hydroxyindolenine-Nb-oxide derivative 4. However, an essentially quantitative yield of 4 was obtained after solvent removal under reduced pressure (t bath < 40°C) when vincadifformine was treated with 3 equivalents of m-chloroperbenzoic acid in the dark for 36h under a nitrogen atmosphere.
The molecular ion was observed at m/z 370, in agreement with the empirical formula C21H26N2O4 which was also supported by microanalysis. The UV spectrum in methanol gave maxima at 223 and 270 nm and the IR spectrum indicated a non conjugated ester at 1738 cm-1 (in contrast with the absorptions at νmax 1670 and 1610 cm-1 in the starting material) and a hydroxy group at 3450 cm-1. The 1H-NMR spectrum showed a multiplet at 8 ppm (OH), a doublet due to the C-9 proton at 7.6 ppm, a multiplet at 7.5-7.1 (3 ArH), the methoxycarbonylmethyl group at 3.95 ppm and the C-18 methyl group as a triplet at 0.5 ppm. Subsequent hydrolysis with 1.25M sodium hydroxide solution, followed by decarboxylation under acidic conditions (pH = 1) at 100°C for 20 min, produced the desired product 5a as a yellow amorphous solid in 98% yield. The presence of the carbonyl function in 5a was confirmed by the long wavelength absorption observed in the UV spectrum [λmax 218, 243 (sh), 300 nm] typical of the indolinic compound and by the absorption in the IR spectrum at 1720 cm-1 (CO). The molecular ion in the mass spectrum was observed at m/z 312 and was consistent with the molecular formula C19H24N2O2.
Scheme 1.

Attempts to reduce the N-oxide group with palladium-hydrogen gave a low yield (32%) of 5b whereas Adam’s catalyst (PtO2) and hydrogen at atmospheric pressure afforded a good yield (80%). However, the major mass spectral fragment of 5b was not observed at m/z 124 as for vincadifformine and its derivatives. Instead, the molecule exhibits a M+-28 peak at m/z 268 and shows its most intense peak at m/z = 138, which is also observed in the 16-dehydrospegazzinidine dimethyl ether 8 [11]. Consequently, the retro Diels-Alder fragmentation is less important in compound such as 5b with a carbonyl group at C-16 which thus alters the typical aspidospermine mass spectral fragmentation pattern. The molecular ion decomposes by expulsion of carbon monoxide to give species b (m /z=268). Subsequent fission of the C-5, C-6 bond therefore produces not the anticipated ion at m/z 124, but rather the ion c (m/z 138, 100%) as the major fragment (Scheme 1).
16-Oxoaspidospermidine 5b, when subjected to reduction with sodium borohydride in ethanol, followed by heating, gave a mixture of C-16 epimers, which were separated by column chromatography on kieselgel G using chloroform – methanol (9:1) as eluent. The major product (less polar, 72.5%) was 16α-H,16β-OH aspidospermidine 6a, obtained as colourless plates (m.p. 55 °C), which exhibited UV absorptions at λmax 212, 244 and 300 nm. The exact structure of this compound was deduced from its 1H-NMR spectrum which exhibited signals clearly indicating that a coupling constant (J = 4 Hz) of H - 2 (d, 3.77 ppm) and H – 16 (m, 4.85 ppm) that is compatible with a cis configuration (2α - H, 16α-H). Irradiation of 16-H gave C-2 as a singlet at 3.75 ppm. Therefore the configuration of the hydroxyl group is β. The 13C-NMR spectrum showed nineteen resonances with the C-16 atom giving rise to the resonance at δ 67.65 ppm. The more polar product (6b, 12.5%) was found to be epimeric at C-16 and was compared with that also obtained by Le Men et al. [10] from the indolenine 9. Reaction of 16β-hydroxyaspidospermidine 6a with formic acid and acetic anhydride afforded the N,O-diformyl derivative 7a in 90% yield as colorless plates (m.p.70-72 °C). The infrared spectrum indicated the presence of two characteristic absorptions at 1720 (OCHO) and 1670 cm-1 (NCHO) with no free NH group. In the 1H-NMR spectrum, the two singlets which correspond to the two formyl functions (OCHO) and (NCHO) were observed at 8.9 and 8.6 ppm respectively. Data from the 13C-NMR spectra are reported in Table 1 for comparison.

{kind=link}
{kind=link}
| Carbon N° | 6a | 7a | 1a | 1b |
|---|---|---|---|---|
| 2 | 70.01 | 70.14 | 71.84 | 71.39 |
| 3 | 53.15 | 53.32 | 52.9 | 53.4 |
| 5 | 52.49 | 51.89 | 52.47 | 52.64 |
| 6 | 43 | 44.11 | 41.25 | 42.23 |
| 7 | 54.0 | 66.4 | 65.23 | 64 |
| 8 | 136.01 | 138.46 | 138 | 138.6 |
| 9 | 123 | 123.17 | 124.15 | 123.52 |
| 10 | 118.4 | 116.15 | 116 | 116(w) |
| 11 | 127.61 | 124.79 | 125.11 | 125.43 |
| 12 | 109.26 | 109.05 | 109.66 | 110.54 |
| 13 | 150.35 | 140.97 | 142.23 | 142 |
| 14 | 22.10 | 21.57 | 21.86 | 21.38 |
| 15 | 34.7 | 32.76 | 33.62 | 32.78 |
| 16 | 67.65 | 127.81 | 128.02 | 127.86 |
| 17 | 35.97 | 34.84 | 35.46 | 31.30 |
| 18 | 7.75 | 7.12 | 7.58 | 6.8 |
| 19 | 34.23 | 32.15 | 34.35 | 34.31 |
| 20 | 36.11 | 35.04 | 35.79 | 36.74 |
| 21 | 74.49 | 71.05 | 74.09 | 73.85 |
| NCHO | ….. | 160.18 | 159.82 | 160.2 |
| OCHO | ….. | 160.05 | ….. | ….. |
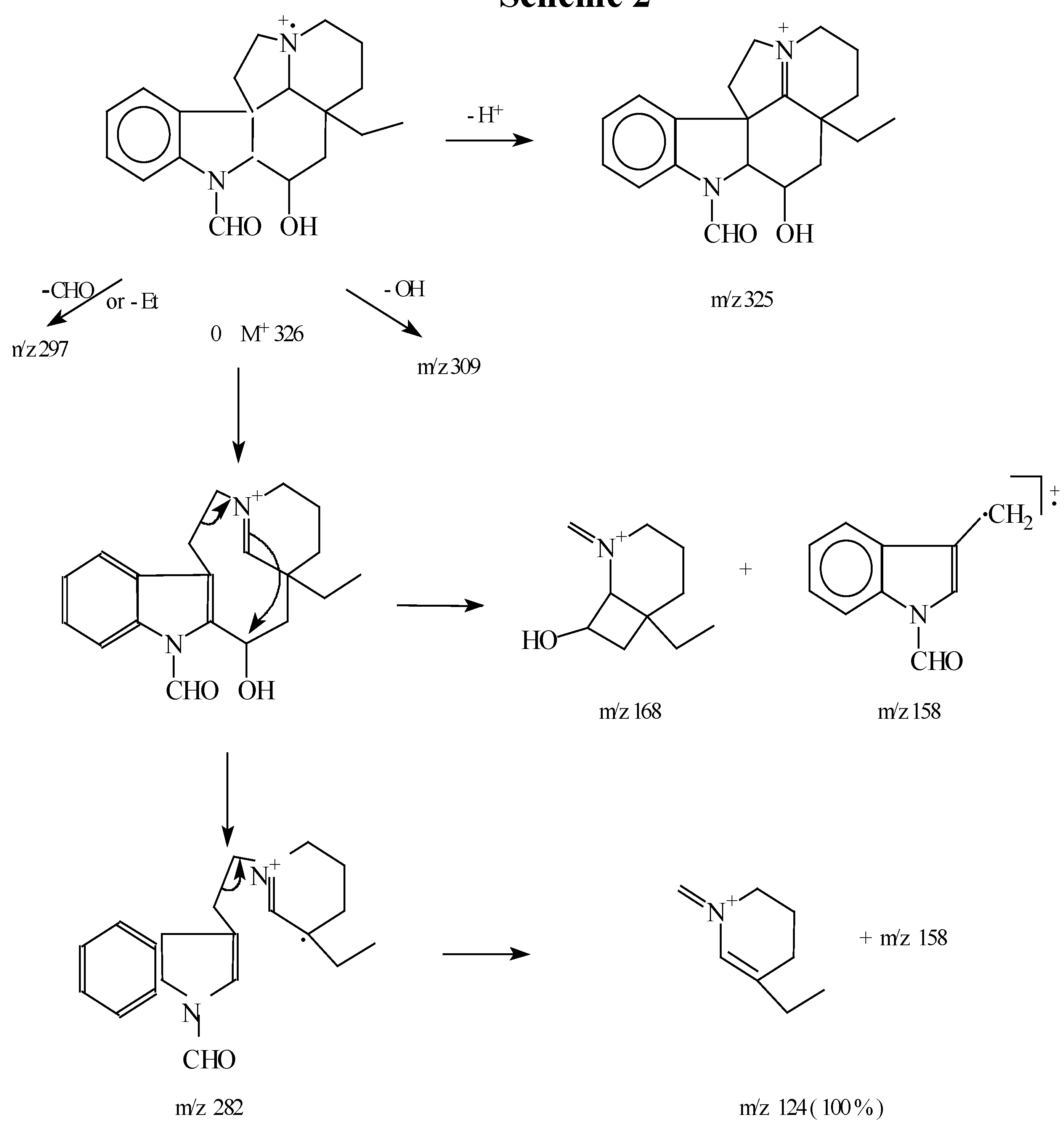
When sodium carbonate was added to a solution of the diformyl 7a compound in methanol the reaction produced Na-formyl-16β-hydroxyaspidospermidine (1a) in 98% yield (m.p. 66-68 °C). The spectroscopic data suggested that the correct relative stereochemistry had been obtained. In the IR spectrum a broad absorption was seen at 3400 cm-1 (OH) and a strong absorption at 1680 cm-1 which was assigned to the Na –formyl group. The UV spectrum in methanol is found to be essentially identical to that reported by Atta –Ur- Rahman [1] for the alkaloid strictanine (1b). The mass spectrum shows the anticipated fragmentation pattern (Scheme 2).
Scheme 2.

In the 1H-NMR spectrum the N-formyl proton (NCHO) was seen as two singlets (relative intensity 7:1) at 9 and 8.72 ppm respectively. Saturation of one peak caused the disappearance of the other one. These are the two s-cis/s-trans geometrical isomers resulting from restricted rotation, well-known for amides. The H-12 proton was observed as a doublet at 8.03 ppm due to the ortho-coupling to H-11
Experimental
General
Melting points were determined on a Kofler hot-stage apparatus and are uncorrected. IR spectra were recorded on either a Perkin–Elmer 1420 or a 1310 spectrophotometer. UV absorption spectra were obtained on a Unicam PU 8800 spectrometer. NMR spectra were recorded on either a JEOL FX90QFT (90 MHz for 1H and 13C), a GE QE 300 (300 MHz for 1H and 13C) or a Bruker 400 MHz spectrometer (400 MHz for 1H and 13C). Solutions in deuteriochloroform, with tetramethylsilane as internal standard were used, unless otherwise stated. J values are given in Hz. Mass spectra were recorded on a Kratos MS 25 instrument; accurate mass measurements were carried out on a AEI/Kratos MS 902/50 spectrometer.
Preparation of aldimine
Cyclohexylamine (49.59 g, 0.5 mmol) was placed in 250mL three-necked, round bottomed flask fitted with a thermometer, mechanical stirrer, and an addition funnel, then cooled to –5 °C in an ice- salt bath. Butyraldehyde (36.05 g, 0.5 mol) was carefully added dropwise at such a rate that the temperature remained below 5° C. Stirring was continued for 30 minutes. The solution was then allowed to warm to room temperature, poured into a separatory funnel, and the water produced during the reaction was removed. The reaction was allowed to go to completion by leaving it standing for 24h in the presence of K2CO3 (16 g) and KOH pellets (16 g). Filtration followed by distillation gave the desired aldimine (65 g, 68%) as a colorless liquid, b.p. 80-84 °C / 20 mm Hg; 1H-NMR: δH (CDCl3, 90 Mz) 7.7 (1H, t, J=5.14 Hz, =CH-), 3 - 1.2 (14 H, m), and 1 ppm (3 H, t, J: 7.4 Hz, -CH3).
Preparation of lithium diethylamide
A solution of diethylamine (7.31 g, 0.1 mol), dry hexamethylphosphoramide (17.92 g, 0.1 mol), dry benzene (20 mL), and lithium (0.69 g, 0.1 atmg) was stirred vigorously between 20-25 ° C for 3 h.
Lithium salt of the imine
Lithium diethylamide solution in dry tetrahydrofuran (15 mL) was cooled to –60 °C and the imine (15.3 g , 0.1 mol) was added via a syringe at such a rate that the temperature remained between –60°C and –65 °C during the addition. After 30 min stirring the reaction mixture was allowed to warm to –10 °C for 2 h.
Synthesis of chloroaldehyde
To a stirred solution of 1-bromo-3-chloropropane (15.74 g, 0.1 mol) in anhydrous tetrahydrofuran (200 mL), cooled to –70°C, the solution of the lithium salt of the aldimine was added dropwise via syringe. After the addition was complete the resulting reaction mixture was allowed to warm to –10°C and kept there for 2h. Stirring was then continued overnight at room temperature. The reaction mixture was treated with 3N hydrochloric acid at –20°C, then stirred at room temperature for 5h. The mixture was extracted with ether (3 x 150 mL). The combined ethereal layers were washed with Na2CO3 (40mL), dried (Na2SO4), and then concentrated under reduced pressure. The residue was purified by chromatography on Kieselgel G (180 g) using ethyl acetate as eluent to give 5-chloro-2-ethylpentanal (11.49 g, 77.4-89%) as a colouress oil, b.p. 100°C/0.5 mmHg; UV: υmax (CHCl3) 1720 (CHO), 650 cm-1; 1H-NMR: δH (CDCl3, 90MHz) 9.5 (1H, d, J=2.5 Hz, -CHO), 3.5 (2H, m, -CH2Cl), 2.4 (2H, m), 1,8 (4H, m), and 0.9 (3H, t, J=7,2 Hz, -CH3).
Preparation of (°)-vincadifformine (2)
A mixture of 1-carbomethoxy-1-methyl-1,2,3,4-tetrahydro-β-carboline (5 g, 20.4 mmol), p‑toluenesulphonic acid and 5-chloro-2-ethylpentanal (3.66 mL, 24.9 mmmol) in toluene (420 mL) was refluxed for 100 h under a nitrogen atmosphere in a Dean-Stark apparatus. To the hot solution, diazabicycloundecene (DBU) (6.33 mL, 49.9 mmol) was added and heating continued for 18 h. The reaction mixture was allowed to cool to room temperature, then concentrated under reduced pressure. The brown residue was purified by chromatography on Kieselgel G (180 g), using dichloromethane - ether (7:1) as eluent; this gave (°) vincadifformine (5.83 g, 81.2 %), which was recrystallised from acetonitrile and obtained as colouress prisms; m. p. 125 °C (lit.[4], 124-25 °C); Anal. Found: C, 74.65; H, 7.75; N, 8.3; calc. for C21H26N2O2: C, 74.55; H, 7.7; N, 8.3 %; IR υmax (CHCl3) 3380 (NH), 1720 (CO2Me), 1670, 1610, 1360, 1050, 750 and 610 cm –1; UV λmax (MeOH) 222, 296, 324 nm, and λ min256, 304 nm; 1H-NMR δ H (CDCl3, 300 Mhz) 8.9 (1 H, br s, -NH), 6.65-7.2 (4 H, m, Ar-H), 3.75 (3 H, s, -CO2CH3), 3.2 - 0.7 (15 H, m), and 0.6 ppm (3 H, t, J=7 Hz, -CH3); 13C-NMR: δC 169.05 (-CO2Me), 167.61 (C-2), 143.17 (C-13), 137.77 (C-8), 127.36 (C-11), 120.29 (C-9), 120.43 (C-10), 109.23 (C-12), 92.52 (C-16), 72.5 (C-21), 55.42 (C-7), 51.69 (C-3), 50.9 (-OCH3 ), 50.56 (C-5), 45.1 (C-6 ), 38.08 (C-20), 32.8 (C-5), 25.57 (C-17), 29.31 (C-19) and 7.09 ppm (C-18); MS m/z (%) 338 (M+, 5.6), 144 (0.4), 143 (0.5), 130 (0.6), 125 (9.4), 124 (100) and 96 (1.9).
16-Hydroxy-1,2-dehydrovincadifformine (3)
(°) Vincadifformine (0.5 g, 1.47 mmol) in a solution of Rose Bengal (1.5 x 10-4M) in methanol - water (6:1, 37.5 mL) containing sodium thiosulphate (0.35g, 2.2 mmol) and 2N sodium hydroxide solution (0.5 mL) was irradiated for 1 h between 20-30 °C using two 150W tungsten lamps in the presence of oxygen. Sodium tetrathionate was filtered off and the solvent removed under reduced pressure (t bath < 40°C), then the crude product was filtered through a short column of neutral alumina to remove the sensitiser using chloroform - methanol (19:1) as eluent. After removal of the solvent, the crude products were chromatographed on silica gel (15 g), eluting with chloroform - ethyl acetate (2:1) to give two main fractions: (a) starting material, verified by TLC comparison with an authentic sample, IR and NMR; (b) the second fraction contained 16-hydroxy-1,2-dehydrovinca-difformine (3) (151 mg, 30%) as an orange solid, m.p. 109-110°C (lit.[9] m.p. 112°C); IR: νmax (CHCl3) 3450 (br), 1738 (unconjugated ester cabonyl) and 1450 cm -1; UV: λmax (MeOH) 210, 224 (sh), 264 nm and λmin 240 nm ; 1H-NMR: δH (CDCl3, 90 MHz) NH was absent, 7.6 (1 H, s, 9-H), 7.1‑7.4 (3 H, m, Ar-H), 3.96 (3 H, s, -CO2Me) 3.8-1.2 (15 H, complex) and 0.5 (3 H, t, J=6.6 Hz, ‑CH3); MS: m/z (%) 354 (M+., 72.2), 336 (25.9) [M+-H2O], 295 (26.9) [M+ -CO2Me], 266 (Et, 95), 138 (9.6) and 124 (22.5).
16-Hydroxy-1,2-dehydrovincadifformine-Nb-oxide (4)
A stirred solution of (°)-vincadifformine (0.2 g, 0.58 mmol) in anhydrous benzene (6 mL) was treated in small portions with m-chloroperbenzoic acid (224 mg, 1.3 mmol, 2.2 equiv.) at ambient temperature, in the dark. Stirring was continued for 36 h under a nitrogen atmosphere. The solvent was removed under reduced pressure (t bath < 40°C) and the residue was taken up in dichloromethane. The mixture was washed with 10 % sodium sulphite solution (30 mL), 5% sodium bicarbonate solution, and water, then dried (MgSO4) and concentred under reduced pressure (t bath < 40°C). Chromatography on silicagel G (30 g) using a 5:3.9:1:0.1 mixture of benzene - methanol - ether and ammonia as eluent afforded 16-hydroxy-1, 2-dehydrovincadifformine Nb-oxide (4) (170 mg, 77.9%), which was recrystalised from dichloromethane-ether to give colourless prisms, m.p. 176-78 °C (dec.); Anal. Found: C, 67.9; H, 7.1; N, 7.55; Calc. for C21H26N2O4: C, 68.1; H, 7.0; N, 7.6 %); IR: νmax (CHCl3) 3450 (OH), 3100-2900, 2809, 1738 (unconjugated ester carbonyl), 1570, 1450, 1280-1230 and 663 cm -1; UV: λmax (MeOH) 223, 270 nm; 1H-NMR: δH (CDCl3, 90 MHz), 8.0 (1 H, m, -OH) 7.6 (1 H, dd, 9-H), 7.5-7.1 (3 H, m, Ar-H), 3.95 (3 H, s, -CO2Me), 2.64 (1H, s, 21-H), 3.4-1.2 (14 H, complex) and 0.5 ppm (3H, t, J=7 Hz, 18-H); 13C-NMR: δC 180 (C-2), 171.87 (C-22), 152 (C-13), 146 (C-8), 127.98 (C-11), 127.79 (C-10), 124.6 (C-9), 120.05 (C-12 ), 83 (C-21), 76.6 (C-16), 67 (C-3), 65 (C-5), 58 (C-7), 53.11 (- CO2CH3), 40.62 (C-17), 36.43 (C-20), 35.11 (C-19), 31.46 (C-15), 18.8 (C-14), 7.4 (C-18); MS: m/z (%) 370 (M+, 10.2), 253 (15.4), 252 (3.3), 267 (5.1), 144 (9.1), 130 (18.2) and 124 (30.9).
The less polar fraction contained vincadifformine-Nb-oxide (36 mg, 18 %), m.p. 160°C (dec.), IR: νmax (CHCl3) 1690 and 1615 cm-1; UV: λmax (MeOH) 227, 297 and 330 nm; 1H-NMR: δH (CDCl3, 90 MHz) 7.65-7.2 (4 H, m, Ar-H), 3.9 (3 H, s, - CO2Me), 3.8 (1 H, s, 21-H), 3.45-1.2 (15 H, m) and 0.5 (3H, t, -CH3); MS: m/z 354 (M+, 86), 353 (39.7), [M+-17], 295 (34) [M+-CO2Me], 294 (6.4), 252 (100), 265 (4.5), 144 (6.3), 143 (12.9) and 124 (21.3).
16-Oxoaspidospermidine-Nb-oxide (5a)
m-Chloroperbenzoic acid (1.128 g, 6.5 mmol) was added in small portions to a stirred solution of vincadifformine (1 g, 2.9 mmol) in dry benzene (150 mL) under a nitrogen atmosphere. The reaction mixture was stirred continuously for 24 h at room tempurature in the dark, after which the solvent was removed under reduced pressure (t bath < 40°C). The residue was taken up in 1.25 M sodium hydroxide solution (100 mL) and the resulting solution was stirred at ambient temperature under a nitrogen atmosphere in the dark for 2.5 h. Then the solution was acidified with 1.25 M hydrochloric acid solution to pH 1, and stirred vigorously at 100°C for 20 minutes under a nitrogen atmosphere. After cooling the mixture was extracted with ether (5 x 60 mL). The aqueous phase was then made alkaline with sodium hydroxide solution, saturated with NaCl and extracted with dichloromethane (5 x 60 mL). The combined organic fraction were dried (Na2SO4) and evaporated under reduced pressure to give 16-oxoaspidospermine-Nb-oxide (5a) (0.915g, 98 %) as a yellow amorphous solid; IR νmax (CHCl3) 3040-2880 cm-1, 1700 (-CO), 1610, 1470, 1275, 1110, 920, 715 and 670 cm-1; UV: λmax (MeOH) 218, 243 (sh) and 300 nm; 1H-NMR: δH (CDCl3, 90 MHz) 8.25 (1 H, br, NH), 7.5-6.5 (4H, m, Ar-H), 5 (1H, s, H-21) 4-1.1 (16 H, complex) and 0.7 ppm (3H, t, J=6.5 Hz, -CH3); MS: m/z (%) 312 (M+, 0.9), 296 (6.9) [M+-16], 295 (19.2), 294 (100), 293 (59.6), 268 (4), 144 (16.8), 138 (32.3), 130 (34.4) and 124 (20.1).
16−Oxoaspidospermidine (5b)
A suspension of 16-oxoaspidospermidine-Nb-oxide (5a) (0.9 g, 2.8 mmol) and platinium (IV) oxide (122 mg, 0.53 mmol) in methanol (20 mL) was hydrogenated at room temperature and atmospheric pressure for 3 h. The catalyst was removed by filtration. Concentration under reduced pressure gave 16-oxoaspidospermidine (0.75g, 88 %), which was recrystallised from methanol to afford colouress prisms; m.p. 108-112 °C; Anal. Found: C, 68.3; H, 7.65; N, 7.5%; Calc. for C19H24N2O: C, 68.5; H, 7.5; N, 8%); IR: νmax (nujol) 3345, 1720 (CO), 1610, 1475, 1385 and 730 cm –1; UV: λmax (MeOH) 210, 240 and 292, λmin 262 nm; 1H-NMR: δH (MeOD, 300 MHz) 7.22-6.81 (4H, m, Ar-H), 4.9 (1H, s, 21-H), 4-0.98 (16H, complex), and 0.79 ppm (3H, t, J=7 Hz, -CH3); MS: m/z (%) 296 (M+, 8.8), 268 (10) [M+-CO], 144 (18.6), 138 (100), 130 (6.4), and 124 (18.1).
16–Hydroxyaspidospermidine (6)
To a solution of 16–oxoaspidospermidine (5b) (1.71 g, 5.7 mmol) in ethanol (400 mL) sodium borohydride (5.97 g, 0.15 mol) was added in portions with stirring at ambient temperature. After the addition was complete, the resulting mixture was refluxed for 4h, then allowed to cool before being diluted with water. The mixture was concentrated under reduced pressure and the residue was taken up in water (100 mL), then extracted with dichloromethane (3 x 200 mL). The combined organic phases were dried (MgSO4) and then concentrated under reduced pressure. The residue was purified by chromatography on Kieselgel G (40 g) using chloroform - methanol (9:1) as eluent, which gave two main fractions: (a) the less polar fraction contained colouress plates of 16β-hydroxyaspidospermidine (6a) (1.24 g, 72.5%), m.p. 55°C; Anal. Found: C, 76.75; H, 9.05; N, 9.0; Calc. for C19H26N2O: C, 76.5; H, 8.70; N, 9.0%; IR: νmax (CHCl3) 3420 (-OH), 3000-2705, 1600, 1480, 1460, 1312, 1250, 1030, 900, and 630 cm -1; λ max (MeOH) 212, 244, 300; 1H-NMR: δ H (CDCl3, 400 MHz) 7.05 (2H, m, Ar-H), 6.7 (1 H, t, J=7 Hz), 6.55 (1H, d, J=7 Hz, Ar-H), 4.85 (1H, m, 16-H), 3.77 (1H, d, J=4 Hz, 2-H), 3-0.8 (17H, m), and 0.55 ppm (3H, t, J=7.5 Hz, -CH3); 13C-NMR: δC 150.37 (C-13), 136.01 (C-8), 127 (C‑11), 123 (C-9), 118.4 (C- 10), 109.26 (C-12), 74.49 (C-21), 71.01 (C-2), 67.65 (C-16), 54.1 (C-7), 53.15 (C-3), 52.49 (C-5), 43.0 (C-6), 36.11 (C -20), 35.97 (C-17), 34.7 (C-15), 34.23 (C-19), 22.10 (C-14) and 7.75 ppm (CH3) ; MS: m/z (%) 298 (M+, 5.7), 254 (8.7), 144 (5.3), 138 (1.5), 130 (5.5) and 124 (100); (b) The more polar fraction corresponds to the second epimer, 16α-hydroxyaspido-spermidine (6b) (212 mg, 12.3 % ), which was obtained as an amorphous solid; IR: ν max (CHCl3) 3400 cm-1(OH), 3000, 2940, 2860, 2700, 1605, 1480, 1460, and 670 cm -1; UV: λmax 212, 242 and 296 nm; 1H-NMR: δH (CDCl3, 300 MHz) 7.25-6.6 (4H, m, Ar-H ), 3.6 (1H, m, 16-H), 4 (1H, s, -OH), 3.5 (1H, m, H-2), 3.8-0.8 (16 H, m), 0.6 (3H, t, J=7 Hz, -CH3); MS: m/z (%) 298 (M+, 5.8), 249 (6.2), 168 (18.3), 144 (13.5), 138 (3.1), 130 (16.1) and 124 (100).
Na-Formyl-16β- formyloxyaspidospermidine (7a).
To a stirred solution of 16β-hydroxyaspidospermidine (6a) (121 mg, 0.4 mmol) in formic acid (5.47 mL, 6.6 mg, 0.14 mol) at 0°C, acetic anhydride (0.54 mL, 59 mmol) was added dropwise over 10 minutes. The resulting reaction mixture was stirred at room temperature for 18 h and then concentrated under reduced pressure. The residue was purified by chromatography on kieselgel G (20 g) with dichloromethane/methanol (2%) as the eluent, to give the Na-formyl-16β-formyloxyaspidospermidine 7a (130 mg, 90.1 %) as colourless plates, m.p. 70-2 °C; Anal. Found: C, 71.4; H, 7.6; N, 8; HRMS M+, m/z 354.19138; C21H26N2O3 requires C, 71.18; H, 7.34; M+, m/z 354.1913; IR:νmax (CHCl3) 3019, 2943, 2900, 2800, 2740, 1720, 1670, 1600, 1480, 1460, 1400, 1361 and 1180 cm-1; UV: λmax (MeOH) 210, 250, 282, 290, λ min 226, 270 nm; 1H-NMR: δH (CDCl3, 300 MHz), 8.9 (1H, s, -CHO), 8.6 (1H, s, -CHO), 7.9 (dd, J=9.3 and 18 Hz, 12-H), 7.15-7 (3H, m, Ar-H), 5.75 (1H, m, 16-H), 4.6 (1H, d, J=5Hz, 2-H), 3 (2H, m), 2.4-0.8 (13H, m), and 0.7 (3H, t, J=6Hz, -CH3); 13C-NMR: δC 160.187, 160.35 (‑NCHO), 160.05, 157.89, (-OCHO), 140.97 (C-13), 138.46 (C-8), 127.81, 127.62 (C-16), 124.79, 124.64 (C-11), 123.17, 122.28 (C -9), 116.153 (C- 10) 109.05 (C-12), 71.05, 70.29 (C-21), 70.14, 68.8 (C-2), 66.4,63.7 (C - 7), 53.32, 52.96 (C- 3), 51.89, 51.76 (C - 5), 41.11, 40.67 (C–6), 35.04, 34.94 (C-20), 34.84, 34.64 (C-17), 32.76, 32.15 (C-15), 27.9, 26.61 (C-19), 21.57, 21.46 (C-14) and 7.12, 6.95 (-CH3); MS: m/z (%) 354 (M+, 1.9), 326 (0.9), 325 (0.9), 282 (3.9), 144 (4.5), 130 (5.2), 124 (100) and 29 (CHO, 15.9).
Na-Formyl -16β - hydroxyaspidospermidine (1a)
To a stirred solution of Na-formyl-16β-formyloxyaspidosperminidine (7a) (76 mg, 0.2 mmol) in methanol (1 mL) sodium carbonate solution (1 mL) was added. The resulting reaction mixture was stirred at room temperature for 15 minutes. Concentration under reduced pressure gave a residue which was taken up in ethyl acetate, washed with water (15 mL), and dried over magnesium sulphate. Removal of the solvent under reduced pressure gave a yellow solid, which was purified by chromatography on Kieselgel G (25 g) using dichloromethane/ether (1:1) as eluent, which gave Na- formyl-16β-hydroxyaspidospermidine (1a) (68.5 mg, 98 %) as colouress plates, m.p. 66-68 °C; Anal. Found: C, 73.40; H, 8.15; N, 8.25%; C20H26N2O2 requires C, 73.6; H, 8.0; N, 8.5%); IR νmax (CHCl3) 3360 (br, OH), 3010, 2945, 2860, 2798, 2738, 1680, 1600, 1490, 1460, 1365, 1335, 1180, 1080, 908 and 675 cm -1; UV: λmax (MeOH) 210, 253, 260 (sh), 280, 290 nm and λ min 266, 270 nm; 1H-NMR: δH (CDCl3, 300 Mhz) 9-8.72 (1 H, 2s, -NCHO), 8.03 (1H, d, J=9Hz, 12-H), 7.2-7 (3 H, m, Ar-H), 5 (1H, br s, 16-H), 4.15 (1H, d, J=4.6 Hz, 2-H), 3.2-0.6 (16 H, m) and 0.55 ppm (3 H, t, J =7.4 Hz, ‑CH3); 13C-NMR: δC 159.82 (NCHO), 142.233 (C-13), 138 (C-8), 128.02 (C-6), 125.11 (C-11), 124.15 (C-9), 116 (C–10), 109.6 (C-12), 74.1 (C-21), 71.84 (C-2), 65.23 (C-7), 52.9 (C-3), 52.47 (C-5), 41.26 (C-6), 35.79 (C-20), 35.46 (C-17), 34.35 (C-19), 33.62 (C-15), 21.86 (C-14), 7.58 (C-18); MS: m/z (%) 326 (M+, 5.7), 325 (0.9), 309 (0.2), 298 (0.6), 297 (0.4), 282 (5.4), 149 (6.1), 144 (2.8), 143 (1.5), 138 (2.5), 130 (1.9), 124 (100), 122 (1), 110 (1.7), 109 (2.6), 96 (2.3).
Na-Formyl-16α-hydroxyaspidospermidine (1b)
To a solution of 16 α-hydroxyaspidospermidine (90 mg, 0.3 mmol) in formic acid (4 mL, 4.87g, 0.1 mol) at 0 °C, acetic anhydride (0.4 mL, 17.6 mmol) was added dropwise. The mixture was stirred at room temperature for 18 h, then concentrated under reduced pressure. The residue was taken up in methanol (1 mL), and sodium carbonate solution (1 mL) was added. The reaction mixture was then stirred for 20 minutes. Removal of the solvent under reduced pressure gave a yellow oily residue which was purified by chromatography on silica gel G (15 g) with dichloromethane/5% methanol as the eluent, to afford the Na-formyl-16α-hydroxy-aspidospermidine (1b) (76 mg, 77 %) as a clear, yellow oil; HRMS: Found: M+, m/z 326.200, C20H26N2O2 requires M+, m/z 326.1994); IR: νmax (CHCl3) 3450 (br, OH), 3010, 2940, 2860, 1660, 1405, 1125 cm -1; UV: λmax (MeOH) 212, 253, 260 (sh), 280, 290 nm; 1H-NMR: δH (CDCl3, 400 MHz) 9-8.86 (1H, 2s, NCHO), 8.08 (1H, d, J=9.1 Hz, 12-H), 7.3-7 (3H, m, Ar-H), 4.37 (1H, d, J=7.5 Hz, 2-H) and 0.7 ppm (3H, t, J=7 Hz, CH3); 13C-NMR: δC 160.2 (NCHO), 142 (C-13), 138.62 (C-8), 127.86 (C-16), 125.43 (C-11), 123.52 (C-9), 110.54 (C‑2), 73.85 (C-21), 71.39 (C-2), 64 (C-7), 53.40 (C-3), 52.64 (C-5), 42.23 (C-6), 36.74 (C-20), 34.31 (C-19), 32.78 (C-15), 31.30 (C-17), 21.38 (C-14) and 6.8 (C-18); MS: m/z (%) 326 (M+, 123), 325 (4.3), 309 (6.3), 298 (4.4), 297 (1.8), 282 (8.1), 152 (6.7), 149 (9.4), 144 (36.2), 143 (42.8), 138 (15.8), 130 (51.7), 124 (92.5), 122 (5.1), 110 (188), 109 (6.6), 96 (16.4).
Acknowledgements
We wish to thank the Ministry of Education, Algeria, for financial support and we are indebted to Dr. J. Fisher (Leeds) and Dr. R. T. Brown (Manchester) for both recording and their useful discussions of the NMR spectra.
References
- Atta –Ur –Rahman; Malik, S. Phytochemistry 1987, 26, 589.
- Khan, A. A.; Ashfaq, M.; Ali, M. N. Pharmacognostic Studies of Selected Indigenous Plant of Pakistan; Pakistan Forest Institute, Peshawar; 1979; p. 75. [Google Scholar]
- Chopra, R. N.; Nayar, S. L.; Chopra, I.C. Glossary of Indian Medicinal Plants; C. S. I. R.: New Delhi, 1956; p. 212. [Google Scholar]
- Kuehne, M. E.; Hafter, R.; Roland, D. M. J. Org. Chem. 1978, 43, 3705.
- Kuehne, M. E.; Matsko, T. H.; Bohner, J. C.; Kirkemo, C. L. J. Org. Chem. 1979, 44, 1063.
- Belferdi, F.; Bayoud, S.; Belattar, A. Magister Thesis, Constantine, 1999.
- Hugel, G.; Lévy, J.; Le Men, J. C.R. Seances Acad. Sci. Ser. C 1972, 274, 1350.
- Hugel, G.; Gourdier, B.; J. Lévy, J.; Le Men, J. Tetrahedron 1980, 36, 511.
- Calabi, L.; Danieli, B.; Lesma, G.; Palmisano, G. J. Chem. Soc. Perkin Trans. 1 1982, 1371.
- Hugel, G.; Gourdier, B.; Lévy, J.; Le Men, J. Tetrahedron Lett. 1974, 1597.
- Orazi, O. O.; Cornal, R. A.; Holker, J. S. E.; Djerassi, C. J. Org. Chem. 1956, 21, 979.
- Sample Availability: Samples are available from the authors.
2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Belferdi, F.; Belattar, A. Synthesis of the Aspidosperma Alkaloid Na-Formyl-16-α-Hydroxyaspidospermidine. Molecules 2001, 6, 803-814. https://doi.org/10.3390/61000803
AMA Style
Belferdi F, Belattar A. Synthesis of the Aspidosperma Alkaloid Na-Formyl-16-α-Hydroxyaspidospermidine. Molecules. 2001; 6(10):803-814. https://doi.org/10.3390/61000803
Chicago/Turabian StyleBelferdi, Fatiha, and Abdelhamid Belattar. 2001. "Synthesis of the Aspidosperma Alkaloid Na-Formyl-16-α-Hydroxyaspidospermidine" Molecules 6, no. 10: 803-814. https://doi.org/10.3390/61000803