An Improved Synthesis of 5-(2,6-Dichlorophenyl)-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (a VX-745 analog)

1
Institute of Organic Chemistry, VUT, A-1060 Vienna, Getreidemarkt 9, Austria
2
Iconix Pharmaceuticals, Inc., 830 Maude Avenue, Mountain View, CA 94043, USA
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(12), 959-963; https://doi.org/10.3390/61200959
Submission received: 22 October 2001
/
Revised: 30 October 2001
/
Accepted: 30 October 2001
/
Published: 30 November 2001
Abstract
:An improved procedure for the synthesis of an analog of the p38 inhibitor compound VX-745 is reported.
Introduction
Mitogen-activated protein (MAP) kinases are key enzymes involved in signal transduction and the amplification of cellular responses to stumuli. P38 MAP kinase is a specific member of the MAP kinase family associated with the onset and progression of inflammation, and several groups are working to develop p38 MAP kinase inhibitors as potential treatments for inflammatory and neurological disease. Vertex discovered the 3D structure of p38MAP kinase in 1996 and computer modeling and testing suggested the design of VX-475 [1].
For comparison purposes we needed a sample of a VX-745 analog and followed the synthesis as described in a general procedure for the preparation of p38 inhibitor compounds by Bemis and coworkers [2,3]. As we had difficulties in duplicating some of the experiments and only limited physical data were mentioned by the authors, we herein report our experience with and improvements of the synthesis of this VX-745 analog.
Results and Discussion
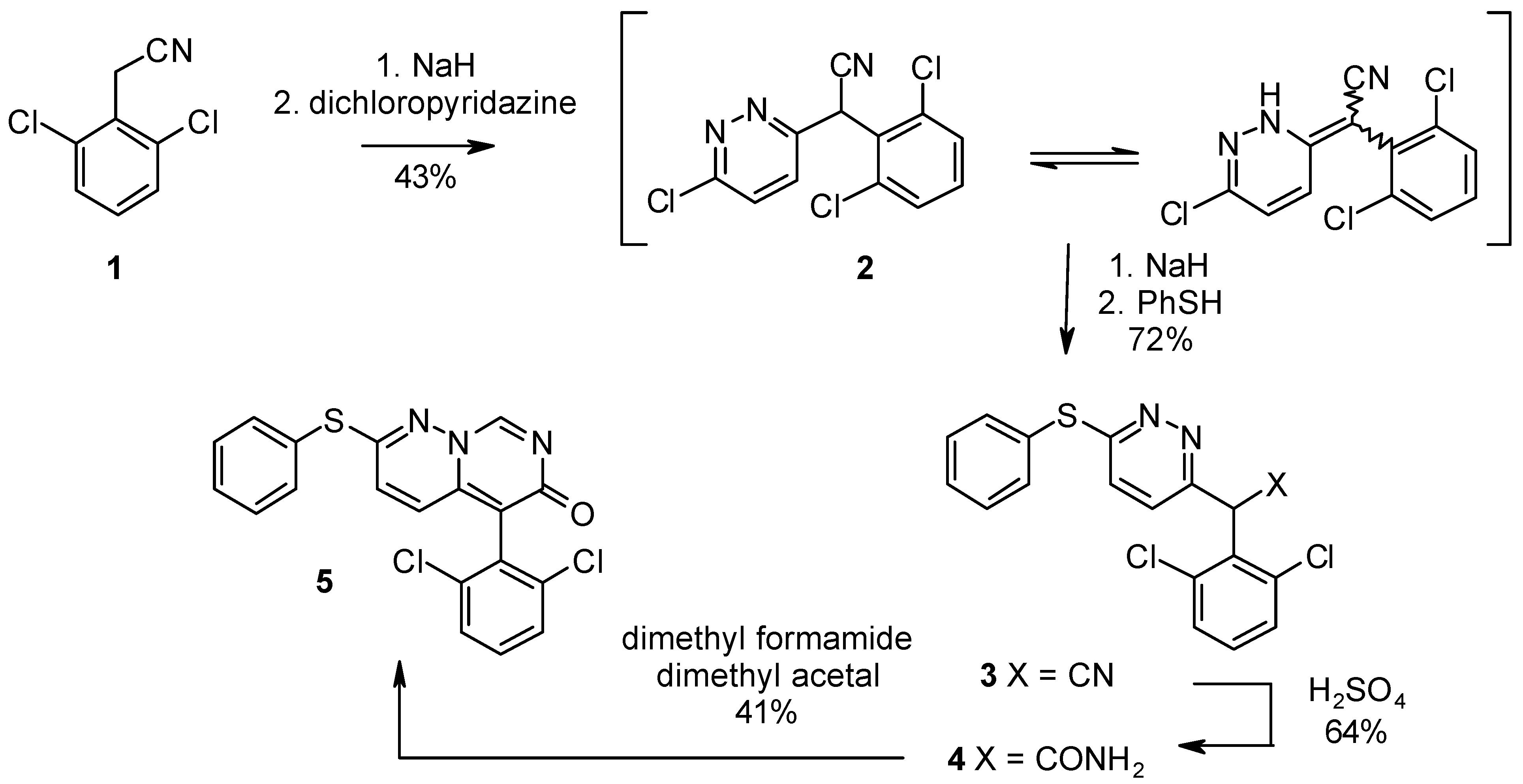
The use of NaH, instead of NaNH2 [2,3], for the deprotonation of dichlorophenylacetonitrile 1 and aqueous workup using satd. NH4Cl (instead of satd. Na2CO3) afforded 2 as a shiny red crystalline product without chromatographic purification. The yield could be improved to 43% on a 9 g scale, whereas Bemis [2,3] reported a yield of 28% without comments on the physical state. HPLC and NMR analysis (no benzylic protons could be detected) revealed the formation of a mixture of tautomers of 2. Since 2 obtained from the modified procedure was quite insoluble in the solvents given by Bemis, DMF was used as solvent for this step. A modified aqueous workup and direct recrystallization of the crude product from EtOH allowed us to avoid column chromatography and gave the desired product 3 as orange crystals in a yield of 72%. The absence of signals of the benzylic proton and the complex peak pattern proved again the formation of at least two tautomeric species. Bemis provides no information on the physical state of 4; while in the general procedure the compounds obtained are described as oils.
Scheme 1.

Partial hydrolysis of the nitrile moity of 3 was performed using shorter reaction times than described in the literature and a modified workup, (no neutralization of the H2SO4) to afford 4 as bright orange crystals. Bemis and coworkers again report no details for the final conversion of 4 to 5. When pure 4 was reacted with dimethylformamide dimethyl acetal the VX-745 analog 5 was formed as a yellow precipitate, which crystallized from AcOH/water with a yield of 41% (see Scheme 1).
Conclusions
We have prepared the VX-745 analog 5 in a 4-step sequence and increased the overall yield from 3.5% to 16%. Spectral data, HPLC- and CHN-analysis proved identity as well as high purity (> 99%).
Experimental
General
Melting points were measured on a Kofler hot stage apparatus. 1H-NMR-spectra were recorded on a Bruker AC-200 (200 MHz) pulse Fourier-transform NMR spectrometer in CDCl3, DMSO-d6 or TFA-d1 using tetramethylsilane as an internal standard. Thin layer chromatography (TLC) was performed on precoated plates (Merck TLC aluminum sheets silica 60 F254) with detection by UV light or with phosphomolybdic acid in aqueous EtOH by heating. All reactions were magnetically stirred under an argon atmosphere.
6-Chloro-α-(2,6-dichlorophenyl)pyridazine-3-acetonitrile (2).
Sodium hydride (4.06 g, 84.5 mmol, 55% dispersion in mineral oil) was washed with anhydrous light petroleum (3 x 35 mL) and then suspended in anhydrous THF (30 mL). 2,6-Dichlorophenyl-acetonitrile (1) (13.1 g, 70.4 mmol) in anhydrous THF (50 mL) was added at RT and the resulting mixture stirred for 30 min. at this temperature. 3,6-Dichloropyridazine in anhydrous THF (50 mL) was then added at RT and stirring continued for another 30 min. Saturated NH4Cl solution (350 mL) was added and the layers were separated. The aqueous layer was extracted with EtOAc (3 x 75 mL), the combined organic layers were washed with water (2 x 400 mL) and brine (1 x 400 mL), dried (Na2SO4), filtered and concentrated in vacuo. The residue was dissolved in anhydrous EtOH (150 mL) and crystallized at -20 °C for 48 h. Yield: red crystals (8.70 g, 43%, lit. [2,3] 28%); mp. 124-31 °C.
α-(2,6-Dichlorophenyl)-6-phenylthiopyridazine-3-acetonitrile (3).
Sodium hydride (1.23 g, 28.1 mmol, 55% dispersion in mineral oil) was washed with anhydrous light petroleum (3 x 25 mL) and suspended in anhydrous DMF (10 mL). Thiophenol (3.10 g, 28.1 mmol) in anhydrous DMF (10 mL) was added at RT and stirred for 10 min. 6-Chloro-α-(2,6-dichloro-phenyl)pyridazine-3-acetonitrile (2) (8.00 g, 26.8 mmol) in anhydrous DMF (20 mL) was added at RT and the reaction mixture stirred for 45 min. at 100 °C. The solution was cooled and partitioned between water (1 L) and EtOAc (200 mL). The aqueous layer was extracted with EtOAc (5 x 75 mL), the combined organic layers were washed with water (5 x 400 mL), 1 N NaOH (2 x 250 mL) and brine (1 x 500 mL), dried (Na2SO4), filtered and concentrated in vacuo. The residue was crystallized from anhydrous EtOH (50 mL). Yield: orange crystals (7.15 g, 72%); mp. 188-91 °C (lit. [2,3] oil).
α-(2,6-Dichlorophenyl)-6-phenylthiopyridazine-3-acetamide (4).
α-(2,6-Dichlorophenyl)-6-phenylthiopyridazine-3-acetonitrile (3) (6.50 g, 14.8 mmol) was stirred in concentrated H2SO4 (200 mL) at 100 °C for 75 min. The solution was cooled to RT, poured into cold water (4 L) and stirred for 10 min. The precipitate was filtered, washed with water (6 x 100 mL), dissolved in MeOH (25 mL)/EtOAc (100 mL), dried (Na2SO4), filtered, concentrated in vacuo and triturated with iPr2O (2 x 10 mL). Yield: orange crystals (3.70 g, 64%); mp. 120-32 °C (lit. [2,3] oil).
5-(2,6-Dichlorophenyl)-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (5).
α-(2,6-Dichlorophenyl)-6-phenylthiopyridazine-3-acetamide (4) (3.40 g, 8.71 mmol) and N,N-dimethylformamide dimethyl acetal (2.51 g, 18.9 mmol) in anhydrous toluene (60 mL) were stirred at 100 °C for 2 h. The solution was stirred at RT for an additional 2 h, and the precipitate thus formed was collected by suction filtration and washed successively with toluene (2 x 50 mL) and light petroleum (4 x 50 mL). The crude product was dissolved in hot AcOH (10 mL), then water (30 mL) was added dropwise, and the resulting suspension was stirred at RT for 1 h. The precipitate was filtered off and washed with water (5 x 20 mL), iPrOH (3 x 20 mL) and iPr2O (3 x 20 mL) and dried (50 °C/50 mbar) to afford the title compound as yellow crystals. Yield: (1.45 g, 41%, lit. [2,3] ~ 10%); mp.: 262 - 265 °C; Anal. Calcd for C19H11Cl2N3OS: C, 57.01; H, 2.77; N, 10.50. Found: C, 56.73; H, 2.97; N, 10.22; 1H-NMR (CF3COOD) δ 10.00 (s, 1H), 8.33 – 8.09 (m, 8H), 8.04 (s, 2H). The purity of the product was determined to be > 99% by HPLC; HPLC conditions: Injection volume: 10 µL, run time: 20 min, column: Phenomenex Synergi Polar-RP 4, temperature: ambient, solvent A: MeCN : H2O = 97: 3, solvent B: MeCN : 0.1% TFA = 97 : 3, gradient used: see following table.

{kind=link}
| Time | Flow | %A | %B |
|---|---|---|---|
| 0.00 | 1.00 | 30.0 | 70.0 |
| 3.00 | 1.00 | 30.0 | 70.0 |
| 15.00 | 1.00 | 90.0 | 10.0 |
| 20.00 | 1.00 | 30.0 | 70.0 |
| 50.00 | 0.20 | 30.0 | 70.0 |
References
- Dillon, S. B.; Griego, S. D. Use of CSAIDs (cytokine suppressive antiinflammatory drugs) in rhinovirus infection. WO 01/19322, 22 March 2001. [Chem. Abstr. 2001, 134, 242657]. [Google Scholar] Ferraccioli, G. F. VX-745 Vertex Pharmaceuticals. Curr. Opin. Anti-Inflammatory Immunomodulatory Invest. Drugs 2000, 2, 74–77. [Google Scholar]
- Bemis, G. W.; Salituro, F. G.; Duffy, J. P.; Cochran, J. E.; Harrington, E. M.; Murcko, M. A.; Wilson, K. P.; Galullo, V. P. Preparation of annelated pyrimidinones and analogs as p38 kinase inhibitors. WO 98/27098, 25 Jun 1998. [Chem. Abstr. 1998, 129, 81749]. [Google Scholar]
- Bemis, G. W.; Salituro, F. G.; Duffy, J. P.; Harrington, E. M. Preparation of pyrido[1,2-c]-pyrimidin-3-ones or 1,2-dihydro-pyrido[1,2-c]pyrimidin-3-ones as inhibitors of p38. US 6147080, 14 Nov 2000. [Chem. Abstr. 2000, 133, 350242]. [Google Scholar]
- Sample Availability: Available from MDPI.
© 2001 by Molecular Diversity Preservation International (MDPI)
Share and Cite
MDPI and ACS Style
Treu, M.; Jordis, U.; Lee, V.J. An Improved Synthesis of 5-(2,6-Dichlorophenyl)-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (a VX-745 analog). Molecules 2001, 6, 959-963. https://doi.org/10.3390/61200959
AMA Style
Treu M, Jordis U, Lee VJ. An Improved Synthesis of 5-(2,6-Dichlorophenyl)-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (a VX-745 analog). Molecules. 2001; 6(12):959-963. https://doi.org/10.3390/61200959
Chicago/Turabian StyleTreu, Matthias, Ulrich Jordis, and Ving J. Lee. 2001. "An Improved Synthesis of 5-(2,6-Dichlorophenyl)-2-(phenylthio)-6H-pyrimido[1,6-b]pyridazin-6-one (a VX-745 analog)" Molecules 6, no. 12: 959-963. https://doi.org/10.3390/61200959