Synthesis of Acridine-based DNA Bis-intercalating Agents

1
The Austin Research Institute, Studley Road, Heidelberg, Victoria 3084, Australia
2
University of Melbourne, Swanston Street Parkville, Victoria 3052, Australia
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(3), 230-243; https://doi.org/10.3390/60300230
Submission received: 12 July 2000
/
Revised: 30 August 2001
/
Accepted: 18 January 2001
/
Published: 28 February 2001
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
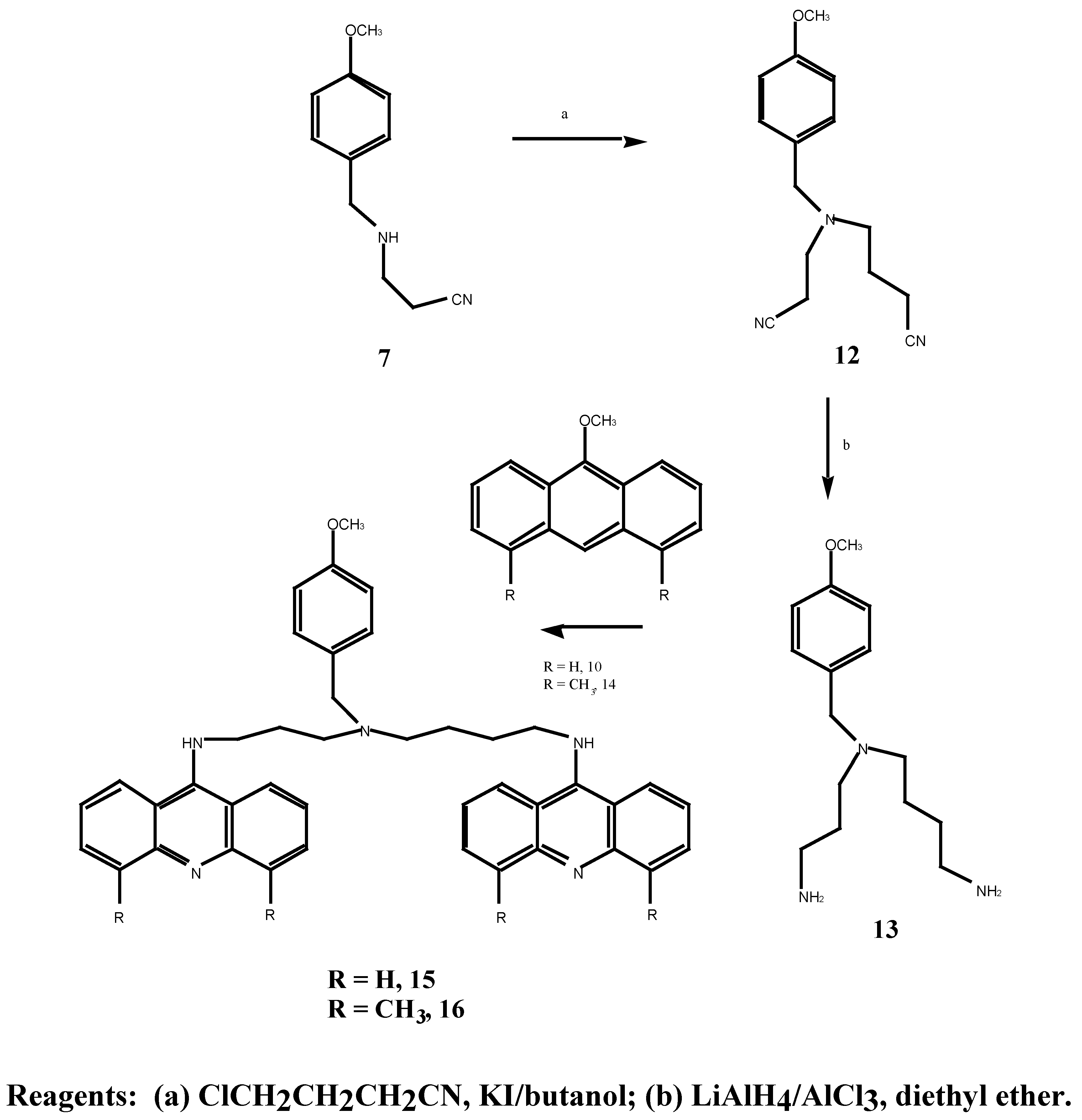
:Methods for the synthesis of N1, N8-bis(9-acridinyl)-N4-(4-hydroxybenzyl)-spermidine and N1, N7-(hydroxybenzyl)-bis-(3-aminopropyl)amine were investigated. Thus monocyanoethylation of 4-methoxybenzylamine followed by treatment with 4-chlorobutyronitrile gave the dinitrile N-(2-cyanoethyl)-N-(3-cyanopropyl)-4-methoxy-benzylamine. Subsequent in situ reduction with lithium aluminium hydride gave the corresponding diamine. Biscyanoethylation of 4-methoxybenzylamine with 2 mole of acrylonitrile followed by reduction yielded the diamine N, N-bis-(3-aminopropyl)-4-methoxybenzylamine. Both diamines reacted smoothly with 9-methoxyacridine to give the bis-(9-acridinyl) compounds 11 and 15 but with 4,5-dimethyl-9-methoxyacridine, the bis compound 16 was produced in only low yields. Demethylation of the dinitriles by a variety of approaches all failed to give the corresponding hydroxybenzyl derivatives. These studies yielded useful methylated tyrosine derivatives which could also be iodinated. This study has been useful for elucidating chemical methods needed for the synthesis of the desired tyrosine-based bis acridine compound and for alerting us to the need to synthesise a more labile protected tyrosine intermediate which will be easily deprotected to afford the desired tyrosine-based bis acridine compound.
Introduction
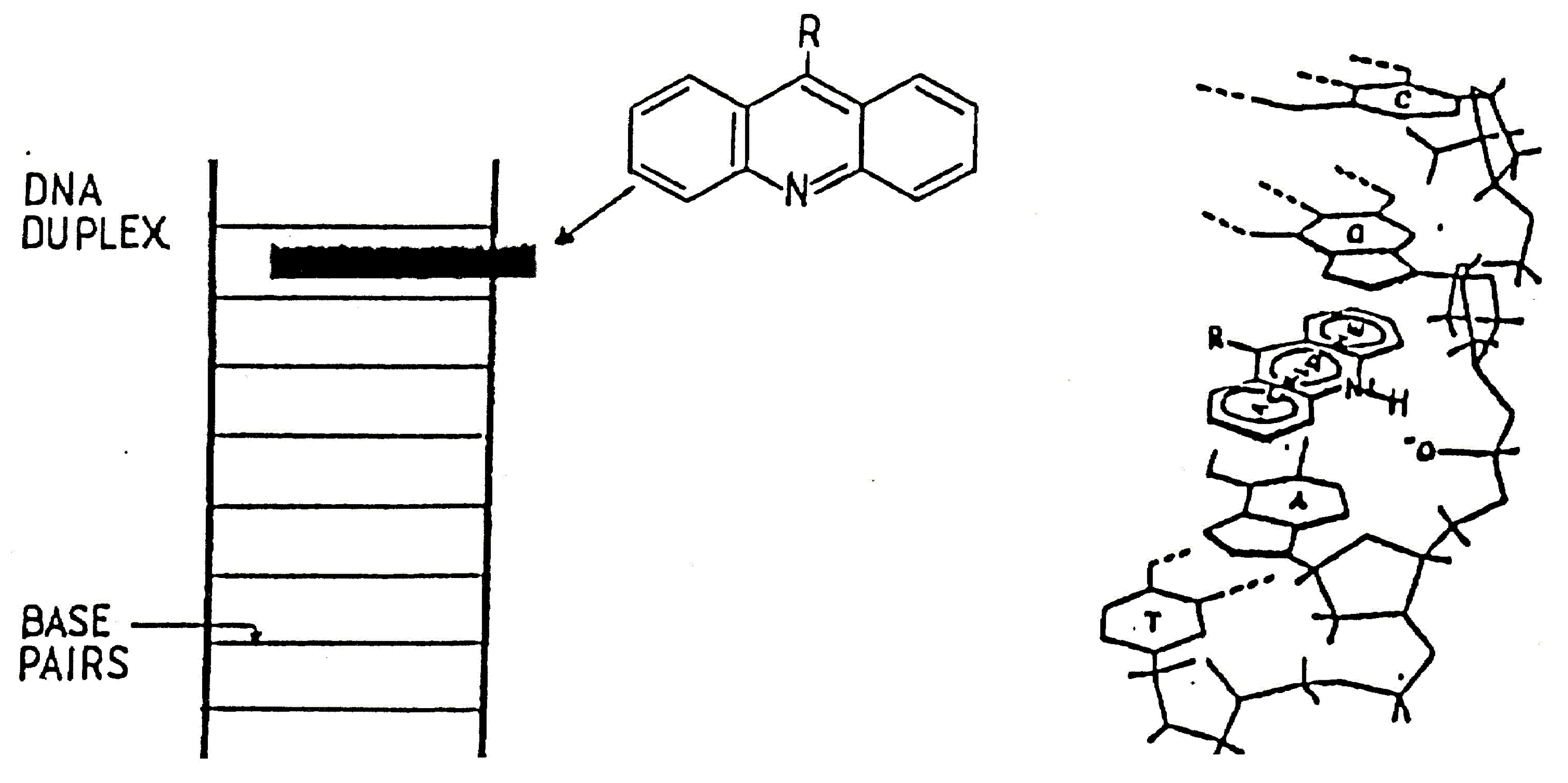
Acridine (1) and its derivatives have long been a well established class of DNA and RNA binding compounds [1].
![Molecules 06 00230 i001]()

These compounds have held the interests of biochemists and chemists for some years due to the possibilities of their clinical use. Many types of drugs are known to bind to DNA through inter-calation between consecutive nucleotides in the DNA strand [2]. To this class of compounds belong several anti tumour agents of clinical importance such as actinomycin-D, daunomycin and adriamycin, as well as several drugs used in the treatment of parasitic diseases including quinacrine and ethidium bromide [3]. Thus the synthesis of acridine-derived DNA intercalating molecules that bind preferentially to particular nucleotide sequences appeared an exciting prospect. The mode of binding of acridine molecules involves intercalation of the acridine tricyclic ring between adjacent base pairs in the DNA duplex [4,5]. The acridine moieties are held in place by van der Waals forces supplemented by stronger ionic bonds to the phosphate ions of the DNA backbone (Scheme 1).
Scheme 1.
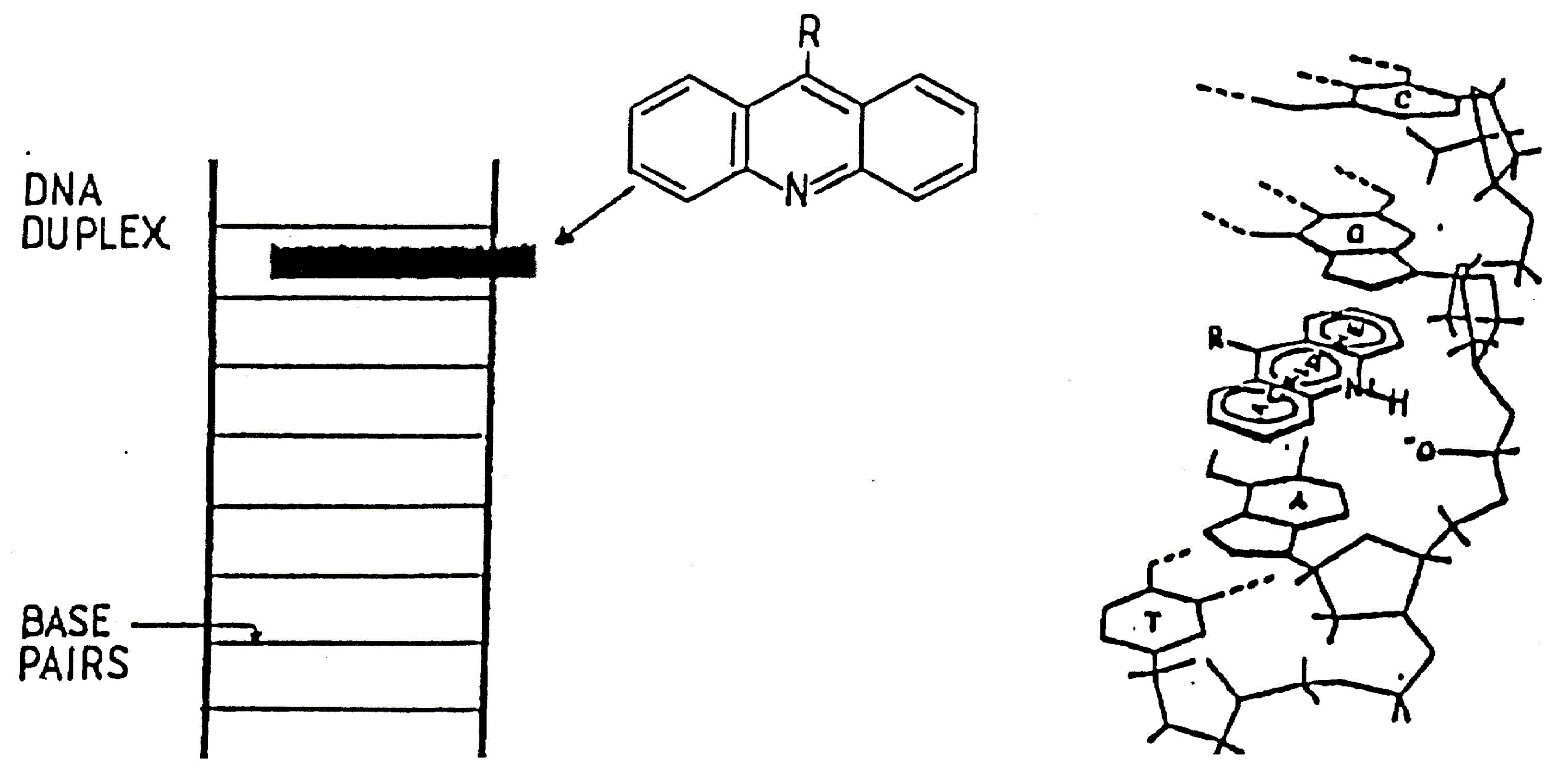
It has been demonstrated by Hurwitz [6] that the binding of acridine molecules interferes with normal DNA function by blocking the DNA starter required by polymerases to synthesise RNA and DNA and hence inhibits protein synthesis. Considerable synthetic attention has been paid to substituted aminoacridines [7]. The incorporation of radioactively labelled iodine provides a method of determining the degree of ligand-DNA interaction, as well as any sequence selectivity. It has been shown the 125I, covalently bound to pyrimidine bases in double stranded DNA or incorporated into a DNA binding compound through and iodine carrier, can cause scission of the DNA helix within three base pairs either side of the decaying atom [8]; the effectiveness of 125I is a consequence of the intense radiochemical damage resulting from the emission of low energy Auger electrons [9].
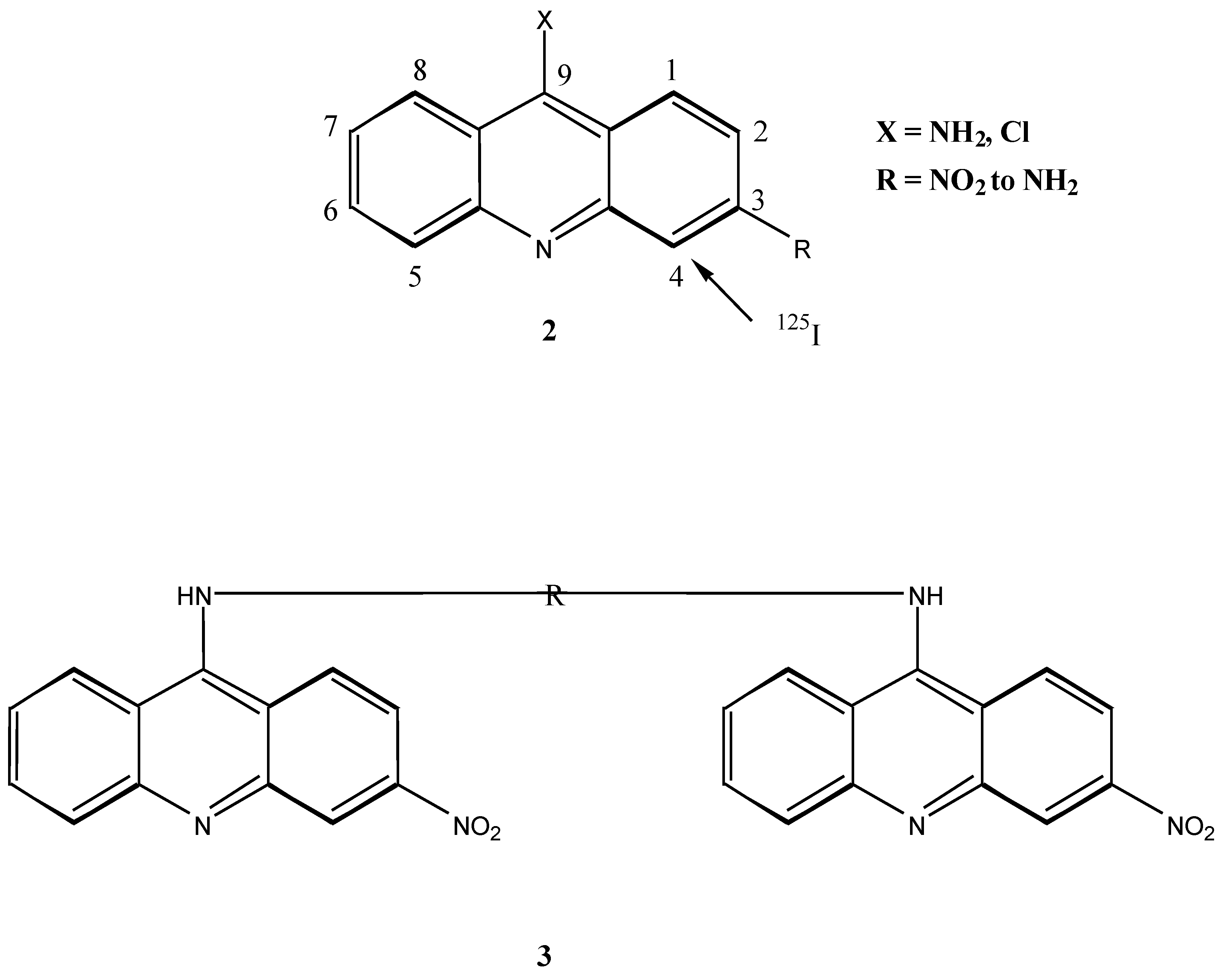
Monoacridines have been shown to bind strongly to DNA. In more recent times however, it has been found that bifunctional intercalaters result in both higher affinity [10] and selectivity [11] of binding. The incorporation of 125I is one method of determining the effectiveness and sequence selectivity of binding. 125I may be introduced into acridines if activating amine groups occupy the C-3 and C-6 positions, directing iodination to the C-4 and C-5 positions respectively. Introduction of the activating amine group has been achieved through the nitro derivative [7], and coupling of the nitroacridines to the linker bis-amino compounds must occur at this stage. However, upon linking of the nitro acridines, reduction of the nitro functions to the corresponding amine functions proved to be difficult [12].
Scheme 2.
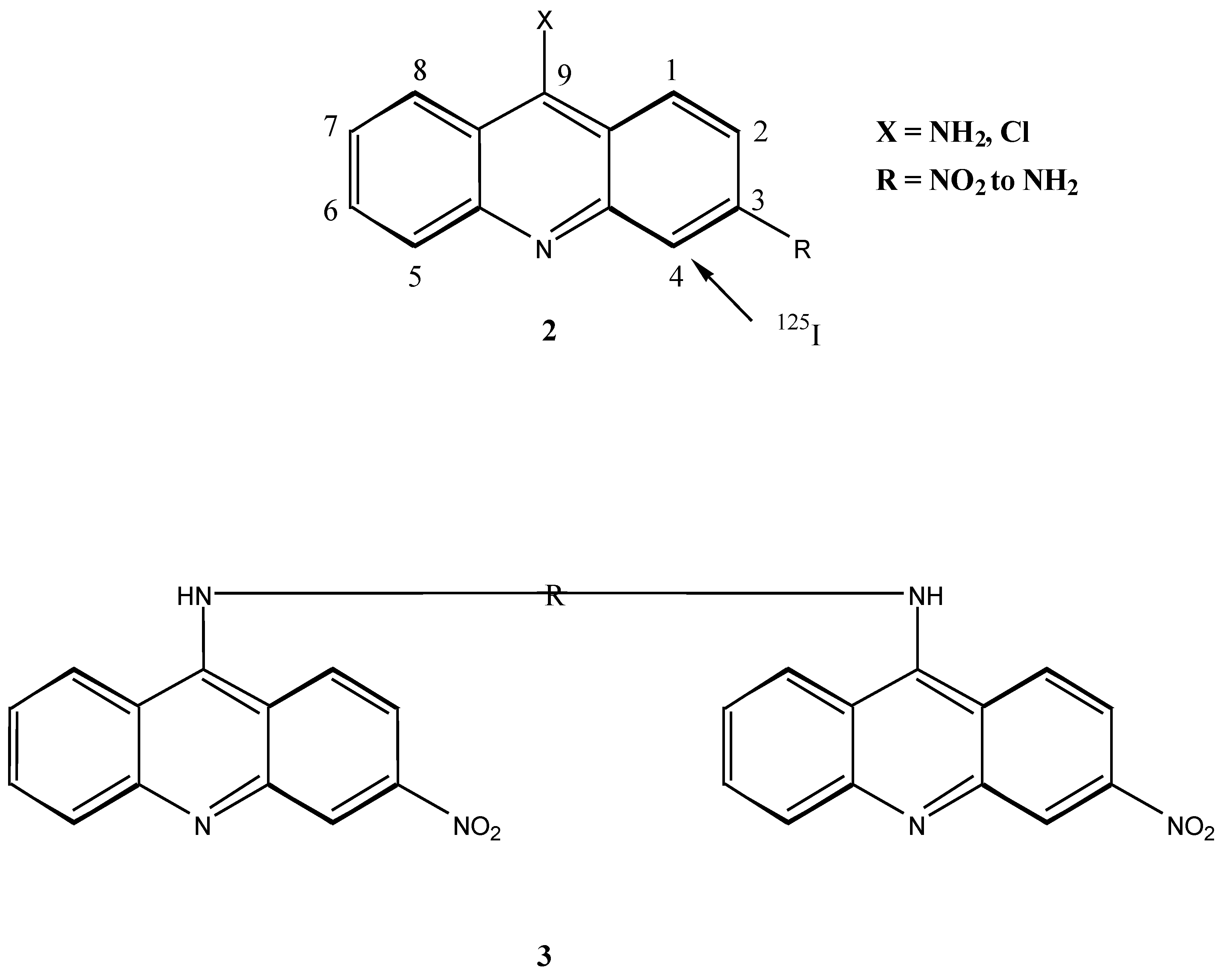
It was therefore proposed that the synthesis of bis-acridinyl DNA binding molecules in which the iodine carrier was incorporated into the linker chain would be an attractive alternative. We report here the synthesis of bis-acridine DNA intercalating compounds 4 with a spermidine type linker chain incorporating an aromatic ring susceptible to iodination. Motivation for the synthesis of a spermidine type linker molecule stems from previous work performed by Le Pecq and co-workers [3] who carried out the first synthetic work on bis-intercalating compounds.
Le Pecq showed that the minimum length required of the acridine linker molecule for effective bis-intercalation into DNA was 10.1 Å, i.e. the length of two base pairs along the DNA axis which is in accordance with the “excluded site model” [3]. In order to conform to this criterion it was decided to use two linker molecules, spermidine (n=4) and bis-(3-aminopropyl)amine (n=3), in the synthesis of the target compound 4.
![Molecules 06 00230 i002]()

The inclusion of a readily iodinated hydroxybenzyl moiety in the linker chain of 4 was proposed in order to take advantage of the use of 125I labelling and DNA sequencing to identify precisely the nucleotide sequence of highest affinity [13]. Two acridine derivatives, acridine and 4,5-dimethylacridine were chosen as the intercalating agents, because both bind strongly to DNA. The 4,5-dimethylacridine derivative bears special significance in that it resembles the active intercalating portion of an effective DNA binding antibiotic antibiotic, actinomycin-D (5) which is known to inhibit transcription. Both 4,5-dimethylacridine and actinomycin-D have methyl substituents at the C-4 and C-5 positions of their respective tricylic rings.
![Molecules 06 00230 i003]()

Results and Discussion
Previous work carried out in our group by Tassigiannakis [12] resulted in the successful synthesis of N,N-bis-(2-cyanoethyl)-4-methoxybenzylamine (8) and N,N-(3-aminopropyl)-4-methoxybenzyl-amine (9) utilizing step (b) of Scheme 3 and step a of Scheme 4. However the attempted preparation of a bis-aminoacridine by treatment of the bis-amine 9 with 9-chloroacridine proved unsuccessful due to the inherent unreactive nature of 9-chloroacridine. It was therefore decided to continue this synthetic work and explore variations of the scheme in the hope of improving the synthesis towards the target bis-aminoacridine 4, with particular attention given to the coupling step with the acridines.
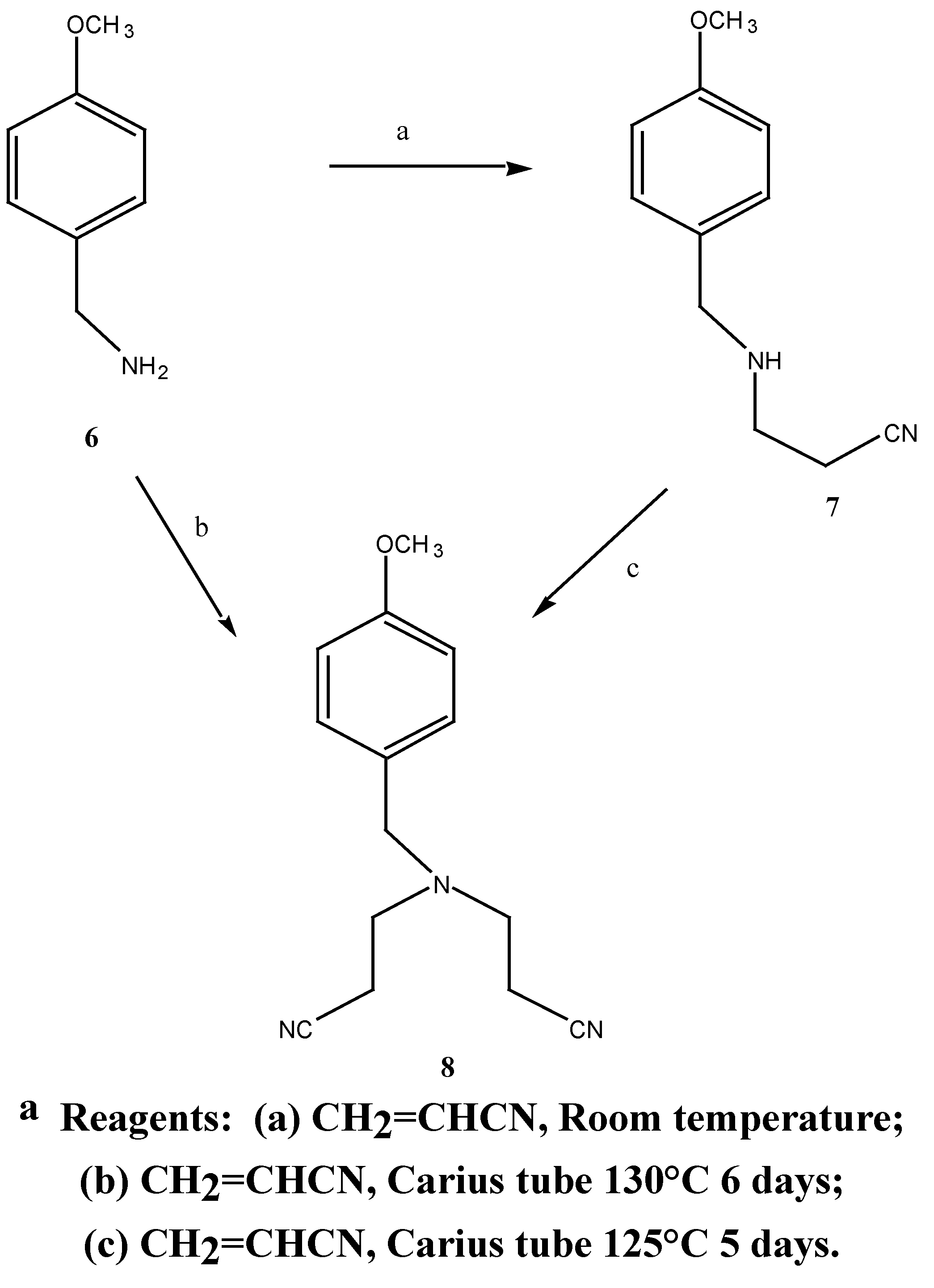
The synthetic approach to N,N-bis-(3-aminopropyl)-4-methoxybenzylamine (9) was patterned on the synthesis of the non-substituted benzylamine compounds by Bergeron and co-workers [14]. Two routes were employed in the synthesis of N,N-bis-(2-cyanoethyl)-4-methoxybenzylamine (8), Scheme 3.
Scheme 3a.
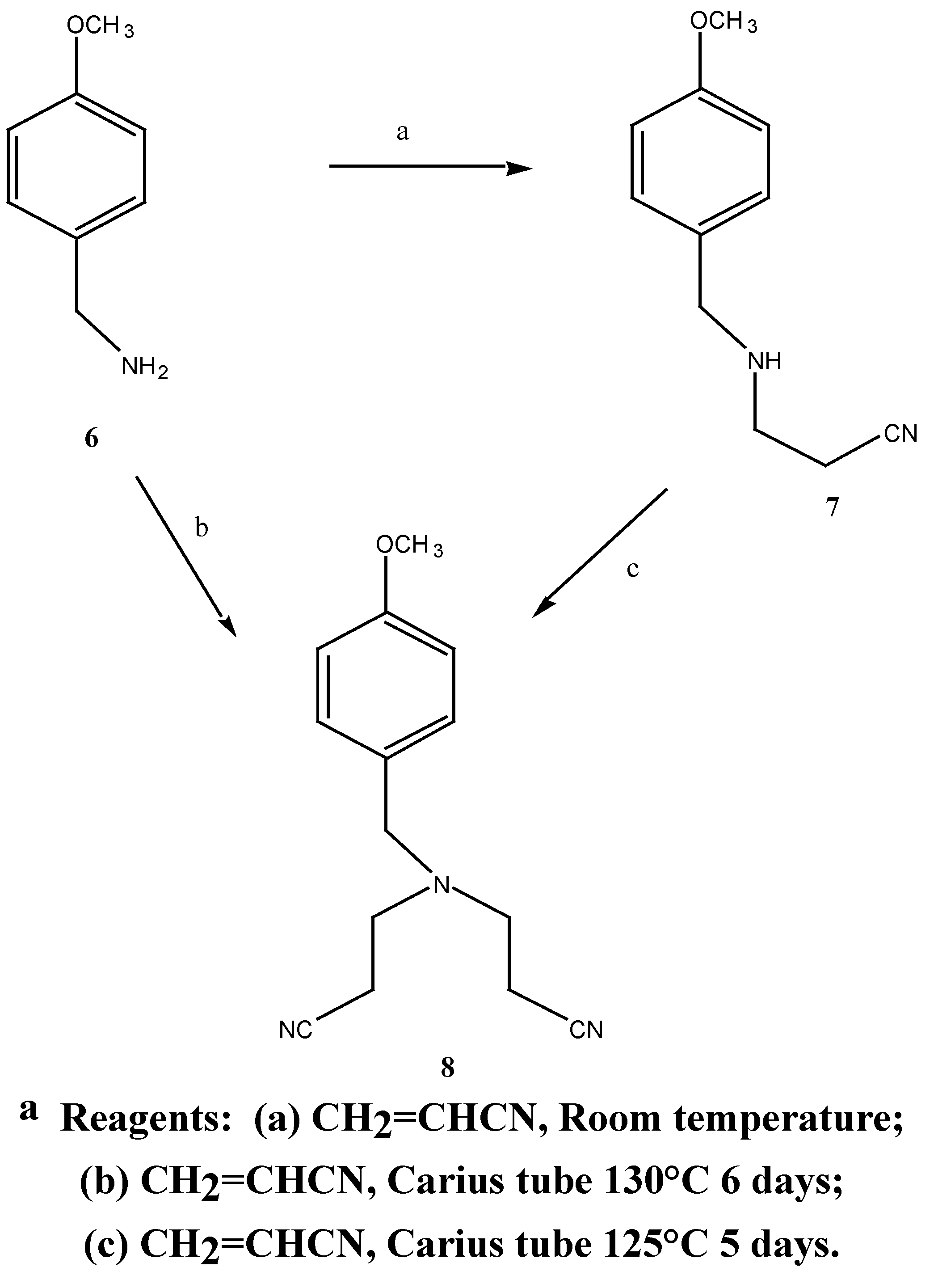
Treatment of methoxybenzylamine 6 with acrylonitrile at room temperature afforded the mono-nitrile (7) in good yield (81%). The appearance of two triplets at δ 2.44 and 2.88 ppm in the 1H-NMR spectrum was attributed to the C-9 and C-10 methylene protons. Treatment of the mononitrile 7 with excess acrylonitrile and a trace of hydroquinone in a Carius tube [15] (sealed under vacuum) at 125°C for five days afforded the dinitrile 8 in 92% yield. The 1H-NMR spectrum showed the correct integration for the methylene protons of the identical cyanoethyl chains resonating as triplets at δ σ2.41 and 2.84 ppm. Hydroquinone, previously omitted from the reaction procedure [14], proved essential in preventing polymerisation of the reaction mixture at such high temperatures.
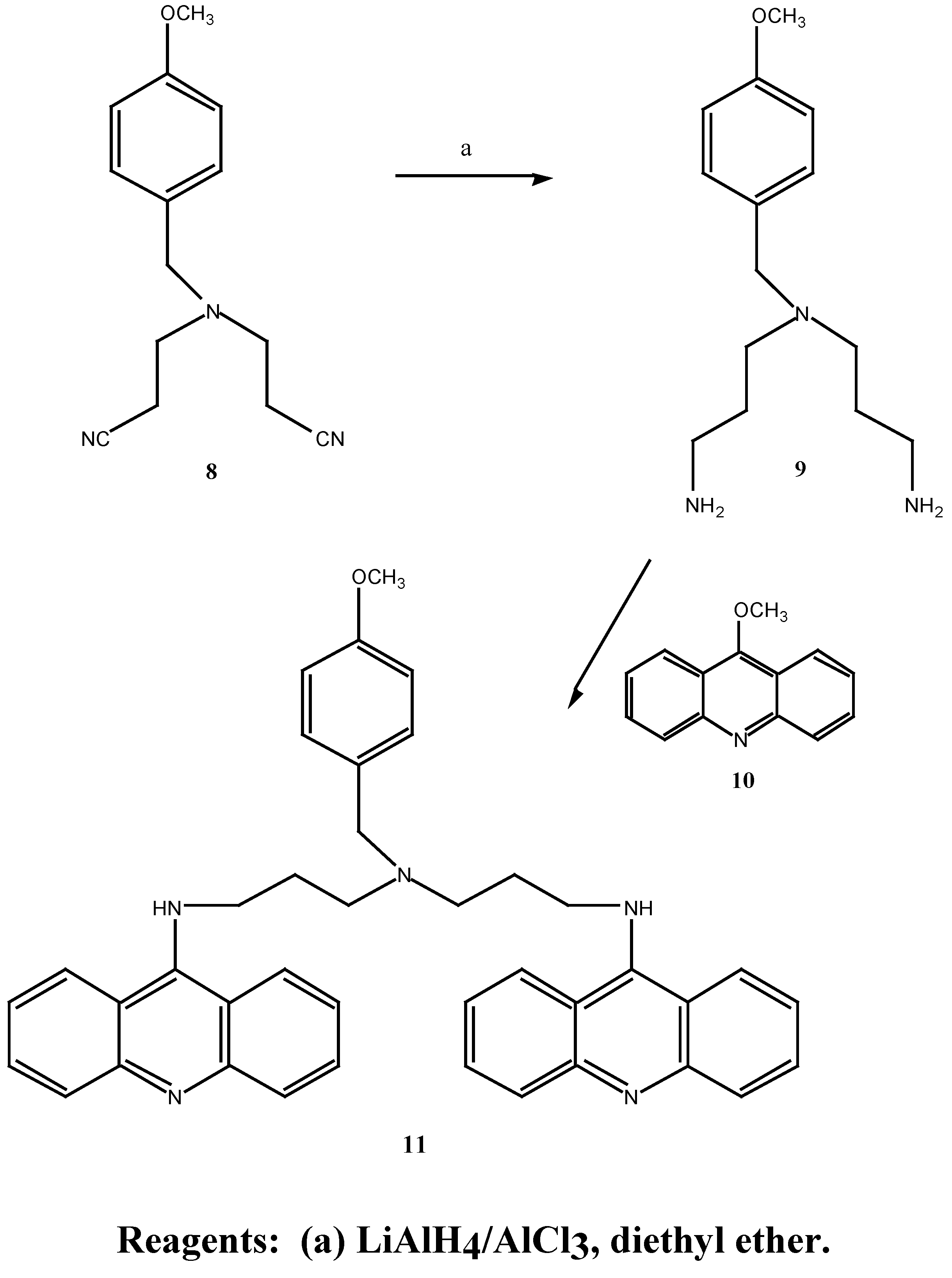
Synthesis of 8 was also achieved in one step by treating 4-methoxybenzylamine with excess acrylonitrile in the presence of hydroquinone in 72% yield (Scheme 5), comparable to the overall yield above. The dintrile 8 previously reported [12] as a liquid, was obtained as a low melting solid. Subsequent reduction of 8 with lithium aluminium hydride and aluminium chloride in diethyl ether gave the bis-amine 9. The 1H-NMR. spectrum revealed a singlet at δ 1.57 ppm corresponding to the
amine protons.
Scheme 4.
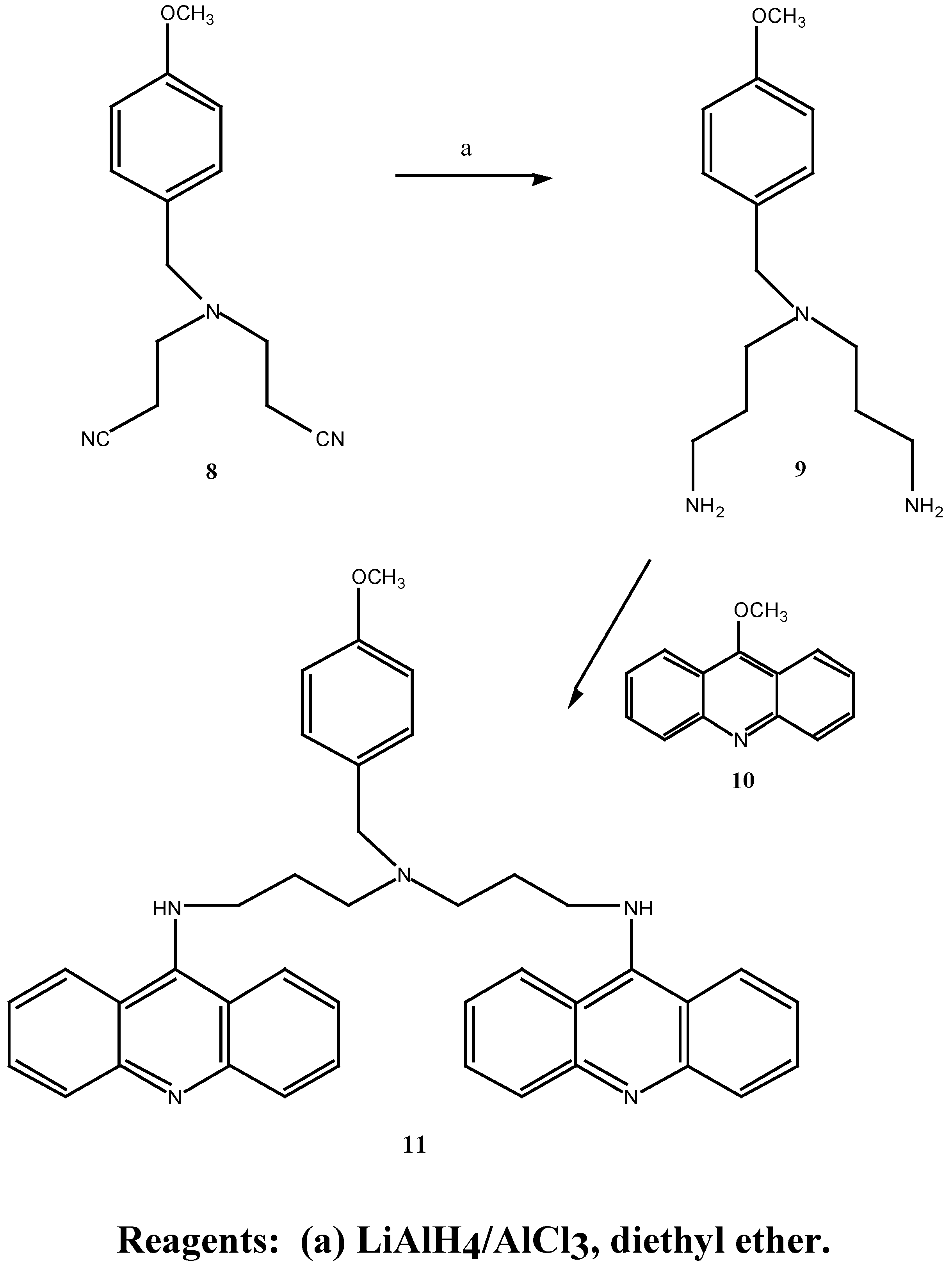
N1, N7-bis-(9-acridinyl)-N-(4-methoxybenzyl)-bis-(3-aminopropyl)amine (11) was prepared in good yield by treatment of the bis-amine 9 with two equivalents of 9-methoxyacridine (10) in methanol at room temperature, Scheme 4. 13C-NMR spectral analysis revealed that bis coupling of the acridine derivative had occurred, with seven acridine carbon resonances present (δ 112.0, 119.7, 122.7, 129.3, 130.7, 133.4, 129.9 ppm) at approximately twice the intensity of the bis-amine methylene carbons. An improvement on previous work [12] was the use of 9-methoxyacridine in the coupling reactions rather than 9-chloroacridine. Linking of the acridine moiety by displacement of the methoxy function required only stirring at room temperature whereas high temperatures were required and long reaction times were required for any coupling of the bis-amine to 9-chloroacridine.
The synthesis of N-(3-aminopropyl)-N-aminobutyl)-4-methoxybenzylamine (13) outlined in Scheme 5, was based on the synthesis of the non-substituted benzylamine compounds by Bergeron and co-workers [16].
Scheme 5.
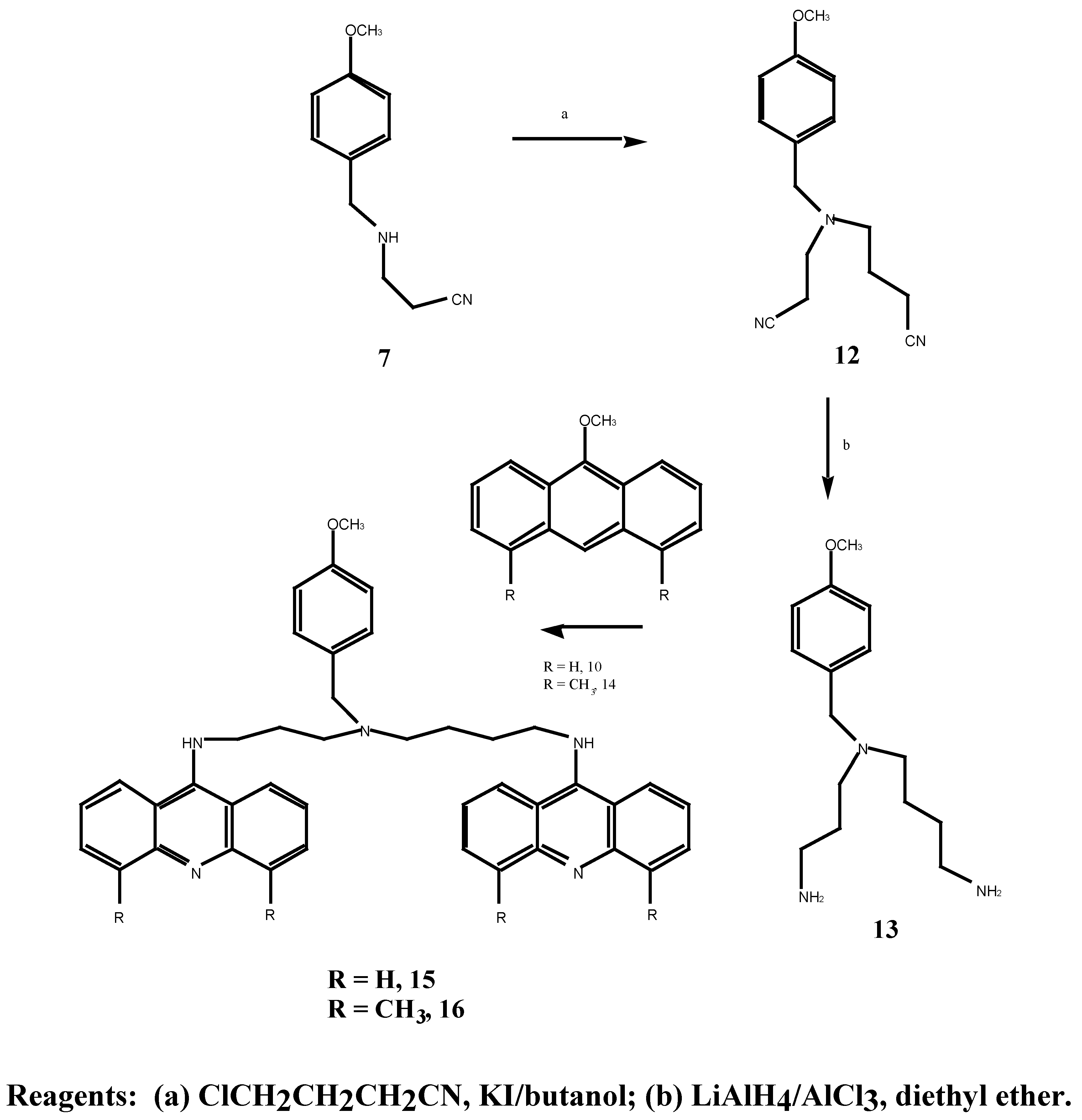
The mononitrile 7, as prepared in Scheme 3, was alkylated with 4-chlorobutyronitrile and potassium iodide in 1-butanol at 117°C with potassium carbonate as the base [17], affording the dinitrile 12 in 50% yield. In addition to the disappearance of the singlet (δ 1.62) produced by the secondary amine proton, the 1H-NMR spectrum contained a doublet of doublets (δ 1.79 ppm) attributed to the C-10 methylene protons, and a complex multiplet (δ 2.35-2.80) corresponding to the other eight methylene protons at C-9, C-11, C-13 and C-14. The dinitrile 12 was reduced in the same manner as 8 to yield the bis-amine 13 in 34% yield. The disappearance of the nitrile 13C-NMR signals at δ 40.4 and 42.0 ppm provided positive evidence of the reduction.
Treatment of 13 with 9-methoxyacridine 10 in methanol, afforded the previously unreported bis-aminoacridine 15 in 82% yield. The 13C-NMR spectrum was consistent with bis-acridine coupling to the diamine 13 with relatively intense resonances at δ 112.9, 13.3, 113.7, 121.6, 122.6, 124.4, 130.6 and 155.1 ppm. In an analogous manner, it was expected that treatment of the bis-amine 13 with 4,5-dimethyl-9-methoxyacridine (14) would result in bis coupling of the acridine derivative to yield the bis-aminoacridine 16. However, coupling proved ineffective even after applying higher temperatures and longer reaction times. Due to low yields of the bis-aminoacridine obtained, conclusive characterisation of the coupled compound was not achieved. The 4,5-dimethylacridine thus appears less reactive towards amines than the unsubstituted acridine 10.
9-Methoxyacridine (10) used in the coupling reactions described earlier, is readily synthesised by standard procedures [18,19]. The preparation of 4,5-dimethyl-9-methoxyacridine (14) however has not been previously reported. Based on Albert’s acridine synthesis [19], this acridine derivative could be obtained via the corresponding 4,5-dimethyl-9-chloroacridine (18). Preparation of 18 was attempted according to the original method Neuman and Powell [20] through an Ullmann condensation of o-bromotoluene and 3-methylanthranilic acid. Isolation of the intermediate condensation product was not successful, however, with the availability of 4,5-dimethylacridone (17) [21] we proceeded to synthesise the desired 9-methoxy compound 14 by the method outlined in Scheme 6. Thus treatment of 17 with phosphorus oxychloride gave 4,5-dimethyl-9-chloroacridine (18) in 84% yield and subsequent treatment with metallic sodium in methanol afforded the 9-methoxy derivative 14. The appearance of a singlet at δ 4.19 ppm in the 1H-NMR spectrum indicated the presence of a methoxy group.
Scheme 6.

Although the 4-methoxybenzylmoiety may be iodinated under a variety of conditions, ultimately bis-aminoacridine with a hydroxybenzyl moiety would be more desirable targets since they are more susceptible to iodination. In addition, iodination with 125I has to be performed at the final step of the synthesis so the method used must be mild enough so as not to damage the rest of the molecule. Thus a number of demethylation experiments were carried out on N-2-(2-cyanoethyl)-N-(3-cyanopropyl)-4-methoxybenzylamine (12) with intentions of deprotecting the hydroxy function and reprotecting with a more acid labile protecting group such as tertiary butyl or methoxymethyl. After reduction of the dinitrile 12 to the diamine 13 and coupling to the acridines, the protecting group could then be easily removed in trifluoroacetic acid to give and analogue of tyrosine, a naturally occurring amino acid.
Demethylation using chlorotrimethylsilane and sodium iodide by Olah’s method [22] failed to produce the desired tyrosine analogue. Trimethylsilane appeared to attack the tertiaryamine in preference to the methoxy function. Integration of the multiplet at δ 1.61-2.82 ppm in the 1H-NMR spectrum indicated that the four-carbon nitrile chain had been cleaved. The appearance of a small singlet at δ 1.30 ppm corresponds to the proton of the resulting secondary amine. Deprotection of the hydroxybenzyl moiety of 12 was then attempted using sodium borohydride and iodine according to the method of Long [23]. This procedure also proved unsatisfactory, resulting in cleavage of the long nitrile chain, in addition to the recovery of a small percentage of starting material.
Treatment of 12 with boron tribromide in dichloromethane as outlined by McOmie and co-workers [24] also failed to produce the tyrosine analogue, resulting in significant decomposition of the dinitrile. Use of a less powerful ether-cleaving agent such as boron trichloride or boron trifluoride has not been attempted.
Future work
Our future chemistry will involve the synthesis of a more labile protected tyrosine derivative and the subsequent alkylation of the amino group followed by the attachment of the acridine moieties and iodination of the tyrosine moiety to afford the desired iodinated molecule.
Experimental
General
Melting points were determined on a Kofler hot stage microscope and are uncorrected. Micro-analysis were performed by the AMDEL Australian Microanalytical Service, Melbourne. Infrared spectra were recorded as thin films on sodium chloride discs using a Perkin-Elmer 457 grating spectrophotometer, unless otherwise stated. Electronic absorption spectra were recorded in ethanol using a Varian Super Scan 3 spectrophotometer. Mass spectra were recorded using a VG Micromass 7070F instrument at 70 eV. Proton NMR spectra were recorded at 100MHz using a Jeol FX-100 spectrometer. Sample solutions were prepared in deuterochloroform containing tetramethylsilane as an internal reference. Chemical shifts have been quoted on the δ scale in p.p.m. relative to tetra-methylsilane. Carbon-13 NMR spectra were recorded at 25 MHz using a Jeol FX-100 spectrometer. Deuterochloroform was used as solvent with tetramethylsilane as an internal reference. Chemical shifts were quoted on the δ scale in p.p.m. relative to tetramethylsilane. All reactions were performed under a dry nitrogen atmosphere. Tetrahydrofuran was distilled from sodium benzophenone ketyl under dry nitrogen immediately before use. Methanol was dried over type 3Å molecular sieves and distilled from iodine and magnesium turnings immediately before use. 1-Butanol was dried over type 4Å molecular sieves while all benzene and diethyl ether was sodium dried. Gel filtration was performed on Sephadex LH-20 using 0.005N acetic acid in 4:1 methanol-chloroform as eluent, unless otherwise stated. Preparative thin layer chromatography was performed on Merck Kieselgel 60 GF254 (200-300 mesh). Analytical thin layer chromatography (t.l.c) was performed on Merck Kieselgel 60 GF254 precoated plates.
Synthetic methods
N-(2-cyanoethyl)-4-methoxybenzylamine (7). A mixture of 4-methoxybenzylamine (6) (11.02 g,
80.0 mmol) and acrylonitrile (4.97g, 93.0 mmol) was stirred for 45 hours at room temperature. Fractional distillation of the reaction mixture gave N-(2-cyanoethyl)-4-methoxybenzylamine (7) (12.2 g, 81 %) as a clear oil b.p. = 245°C at 0.5 mmHg. MS m/z (M+) 190 1H-NMR δ 1.6, s, NH; 2.44, t, J = 5.6 Hz, CH2CN; 2.88, t, J = 6.5 Hz, NHCH2; 3.70, s, PhCH2; 3.78, s, PhCH2; 3.78, s, OCH3; 7.04, d, J3,5 9.0 Hz, H-3, H-5; 7.26, d, J2,6 9.0 Hz, H-2, H-6. 13C-NMR δ 131.5 (C-1), 129.2 (C-2, C-6), 113.8 (C-3, C-5), 158.0 (C-4), 55.2 (C-7), 52.5 (C-8), 44.2 (C-9), 18.7 (C-10), 118.7 (C-11). Anal. found C 69.3, H 7.5; C11H14N2O requires C 69.5, H 7.4 %.
N-(2-Cyanoethyl)-N-(3-cyanopropyl)-4-methoxybenzylamined (12) A solution of 4-chloro-butyronitrile (2.46 g, 25.0 mmol) in anhydrous n-butanol (9.1 mL) was added over 2 hours to a mixture of N-(2-cyanoethyl)-4-methoxybenzylamine (5.07 g, 40.0 mmol), anhydrous sodium carbonate (4.22g, 40.0 mmol) and potassium iodide (0.64 g, 4.0 mmol) at 117°C. The mixture was stirred for an additional 20.5 hours at 117°C. After cooling to room temperature, the mixture was filtered and the solid washed with diethyl ether. The combined filtrate and washing were extracted with 3N HCl (3 x 40 mL) and the resulting acid solution was washed with ether (2 x 40 mL), made basic with potassium carbonate and extracted with ether (3 x 70 mL). The final ethereal extracts were dried over sodium sulphate, filtered and evaporated to give an oil (4.45 g, 50.0 %). Subsequent distillation afforded 3.24 g (43.3 %) of the desired N-(2-cyanoethyl)-N-(3-cyanopropyl)-4-methoxybenzylamine (12) as a light yellow oil b.p. = 235° at 0.06 mmHg. MS m/z 257 (M+); 1H-NMR δ 1.79, q, , J 6.0 Hz, CH2CH2CH2, 2.46, m, 2 x NCH2, 2 x CH2CN, 3.54, s, PhCH2, 3.79, s, OCH3, 6.87, d, , J 9.0 Hz, H-3, H-5, 7.23, d, , J 9.0 Hz, H-2, H-6. 13C-NMR δ 113.7 (C-1), 129.7 (C-2, C-6), 113.9 (C-3, C-5), 158.9 (C-4), 57.8 (C-7), 55.2 (C-8), 49.2 (C-9), 14.6 (C-10), 119.7 (C-11), 51.8 (C-12), 23.4 (C-13), 118.9 (C-15). Anal. found C 69.7, H 7.3, N 16.2; C15H19N3O requires C 70.0, H 7.3, N 16.3 %.
N-(3-Aminopropyl)-N-(4-aminobutyl)-4-methoxybenzylamine (13). A solution of anhydrous aluminium chloride (2.04 g, 15.3 mmol) in anhydrous diethyl ether (21 mL) was rapidly added to a suspension of lithium aluminium hydride (0.6 g, 15.8 mmol) in anhydrous ether (30 mL). After vigorous stirring of the mixture, a solution of N-(cyanoethyl)-N-(cyanopropyl)-4-methoxy benzylamine (12) (1.47 g, 6.0 mmol) in anhydrous diethyl ether (21.0 mL) was added over 2 hours and the reaction was stirred at room temperature for 29 hours. The solution was cooled to 0°C, quenched with 30 % aqueous potassium hydroxide (w/v; 30 mL) and the ethereal layer decanted. The combined ethereal extracts were washed with cold saturated aqueous sodium chloride (2 x 25 mL). Evaporation of the diethyl ether gave a light brown oil which was taken up in chloroform (60 mL), dried over anhydrous magnesium sulphate, filtered and the chloroform evaporated under reduced pressure. A crude oil was obtained (0.73 g) which upon fractional distillation afforded 0.52 g (34 %) of the desired N-(3-aminopropyl)-N-(4-aminobutyl)-4-methoxybenzylamine (13) as a light yellow oil. MS m/z 265 (M+); 1H-NMR δ 1.12, s, 2 x NH2, 1.49, m, 2 x (H-10, H-11, H-14), 2.50, m, 2 x (H-9, H-12, H-13, H-15), 3.47, s, PhCH2, 3.79, s, OCH3, 7.20, d, , J 10 Hz, H-2, H-6. Anal. found C 67.9, H 10.0, N 16.2; C15H27N3O requires C 67.9, H 10.2, N 15.8 %.
N1,N8-Bis-(9-acridinyl)-N4-(4-methoxybenzyl)spermidine (15). N-(3-aminopropyl)-N-(4-amino-butyl)-4-methoxybenzylamine 13 (0.34 g, 128 mmol) was dissolved in methanol (2.0 mL) and added to 9-methoxyacridine 10 (0.54 g, 2.39 mmol). The mixture was stirred for 70 hours at 30°C, the methanol was evaporated under reduced pressure followed by gel filtration to afford 250 mg (30 %) of the desired bis-aminoacridine 15 as an orange gum. 1H-NMR δ 1.5-3.3, m, 7 x CH2, 2.22, s, 2 x NH, 3.55, s, PhCH2, 3.71, s, OCH3), 7.02-7.96, m, H-2, H-6, H-3, H-5, and acridine protons, 20H. Anal. found C 70.1, H 6.7; C41H41N5O requires C 69.5, H 5.9 %.
N1,N8-Bis-(4,5-dimethyl-9-acridinyl)-N4-(4-methoxy benzyl)spermidine (16). N-(3-aminopropyl)-N-(4-aminobutyl)-4-methoxybenzylamine 13 (60 mg, 0.23 mmol) was dissolved in tetrahydrofuran (1.0 mL) and added to 4,5-dimethyl-9-methoxyacridine (14) (85 mg, 0.18 mmol). The mixture was stirred for 48 hours at room temperature, then stirred at 35°C for 8 hours. Evaporation of the tetra-hydrofuran under reduced pressure followed by gel filtration afforded the desired N1,N8-bis-(4,5-dimethyl-9-acridinyl)-N4-(4-methoxy benzyl)spermidine (16) as an orange gum. 1H-NMR δ 1.1-3.2, m, 8 x CH2, 2.21, s, 2 x PhCH3, 3.42, s, OCH3, 7.02-7.81, m, H-2, H-6, H-3, H-5 and acridine protons. UV (log E), 226, 266, 398 (4.02, 3.86, 3.0). vmax 3400, 2950, 2150, 1570, 1410, 1250, 1180, 1025 cm-1.
N,N-Bis-(2-cyanoethyl)-4-methoxybenzylamine (8): Method 1:-A mixture of 4-methoxybenzyl-amine (0.406 G, 2.9 mmol), an excess of acrylonitrile (0.85 g, 16.0 mmol) and a trace of hydro-quinone was added to a Carius tube [15]. The tube was sealed under vacuum (0.1 mmHg) and the tube was heated at 45° for 8 hours, then 130°C for 140 hours. The resulting amber brown liquid was distilled under vacuum to afford 0.53 g (72 %) of the desired N,N-bis-(2-cyanoethyl)-4-methoxy-benzylamine (8) as a colourless oil. b.p. = 225°C at 0.21 mmHg, which formed a white solid, m.p. = 48°C on cooling to room temperature.
Method 2:- A mixture of N-(2-cyanoethyl)-4-methoxybenzylamine 7 (1.63 g, 8.6 mmol), an excess of acrylonitrile (1.69 g, 31.8 mmol) and a trace of hydroquinone was added to a Carius tube. The tube was sealed under vacuum (0.3 mmHg) and heated at 125° for 144 hours. The resulting brown oil was distilled under vacuum to afford 1.93 g (92.4 %) of the desired N,N-bis-(2-cyanoethyl)-4-methoxybenzylamine (8) as a light yellow oil. b.p. = 220°C at 0.2 mmHg. MS m/z 243 (M+); 1H-NMR δ 2.41, t, 2 x (H-10, H-13), J 6.6 Hz, 2.84, t, 2 x (H-9, H-12), J 6.0 Hz, 3.6, s, PhCH2), 3.77, s, OCH3, 6.85, d, , J 9.0 Hz, H-2, H-6, 6.89, d, , J 8.6 Hz, H-3, H-5, 7.20, d, , J 10 Hz, H-2, H-6. 13C-NMR δ 116.0 (C-1), 129.8 (C-2, C-6), 113.9 (C-3, C-5), 159.1 (C-4), 57.4 (C-7), 55.2 (C-8), 49.2 (C-9), 16.7 (C-10), 118.8 (C-11), 49.2 (C-12), 16.7 (C-13). Found M+. = 243.135; C14H17N3O requires M+. = 243.137.
N,N-Bis-(3-aminopropyl)-4-methoxybenzylamine (9):- A solution of aluminium chloride (2.66 g, 19.8 mmol) in anhydrous diethyl ether (25 mL) was rapidly added to a suspension of lithium aluminium hydride (0.75 g, 19.8 mmol) in anhydrous diethyl ether (35 mL). The mixture was vigorously stirred and a solution of N,N-bis-(2-cyanoethyl)-4-methoxybenzylamine (1.52 g, 6.27 mmol) in anhydrous diethyl ether (25 mL) was added over 1.5 hours. After stirring at room temperature for 50 hours the mixture was cooled to 0°, quenched with aqueous 30 % potassium hydroxide (w/v; 30 mL) and the ethereal layer decanted. The remaining emulsion was extracted with diethyl ether (4 x 100 mL) and the combined diethyl ether extracts washed with saturated aqueous sodium chloride (2 x 25 mL). Evaporation of the diethyl ether under reduced pressure gave a brown oil which was taken up in chloroform (50 mL), dried over anhydrous magnesium sulphate, filtered and the chloroform evaporated under reduced pressure. Fractional distillation of the resulting oil afford 0.391 g (25 %) of the desired N,N-bis-(3-aminopropyl)-4-methoxybenzylamine (9) as a light yellow oil. b.p. = 190° at 0.4 mmHg. MS m/z 251 (M+); 1H-NMR δ 1.57, s, 2 x NH2, 2.37-2.68, m, 6 x CH2, 3.72, s, PhCH2, 3.79, s, OCH3, 6.85, d, , J 9.0 Hz, H-2, H-6, 7.22, d, , J 9.0 Hz, H-3, H-5. UV (log E) 271 nm (3.20). Found M+. = 251.19975; C14H25N3O requires M+. = 251.19975.
N1,N7-Bis-(9-acrinyl)-N4-(4-methoxybenzyl)-bis-(3-aminopropyl) amine (11). N,N-Bis-(3-amino-propyl)-4-methoxybenzylamine (9) (40.0 mg, 0.16 mmol) was dissolved in methanol (1.5 mL), added to 9-methoxyacridine (10) (66.0 mg, 0.32 mmol) and the mixture was stirred at room temperature for 63 hours. Evaporation of the methanol under reduced pressure, followed by gel filtration afforded 88.0 mg (91 %) of the desired N1,N7-bis-(9-acrinyl)-N4-(4-methoxybenzyl)-bis-(3-aminopropyl) amine (11) as an orange-red gum. 1H-NMR δ 2.17, s, 2 x NH, 2.31-3.65, m, 6 x CH2), 3.74, s, PhCH2, 3.80, s, OCH3, 6.82-7.61, m, H-3, H-5, H-2, H-6 and acridine protons, 20 H; UV (log E) 225, 267, 414 nm. 4.3, 4.45, 4.36. Found M+. = 605.31543; C40H39N5O requires M+. = 605.31543.
9-Chloroacridine (21). Prepared from 2-carboxydiphenylamine (1.0 g, 4.6 mmol) and freshly distilled phosphorus oxychloride 3.0 mL, 32.7 mmol) according to Albert [19] and obtained as pale yellow crystals (0.37 g, 37 %). M.p. = 120°C (lit [27] = 120°C). MS m/z 215 (M+).
9-Methoxyacridine (10). Prepared from 9-chloroacridine (0.5 g, 2.4 mmol) and a sodium methoxide solution generated from sodium (0.1 g, 4.4 mmol) in methanol (5.8 mL) by the method of Albert [19] and obtained as dark yellow crystals. Recrystallisation from light petroleum (60-80°C) afforded 0.31 g (63 %) of the desired 9-methoxyacridine (10) as pale yellow crystals, m.p. = 99-101°C (lit [19] = 103°C). 1H-NMR δ4.26, s, OCH3, 8.0, m, acridine protons.
4,5-dimethyl-9-chloroacridine (18). 4,5-dimethylacridone (20) (1.0 g, 4.5 mmol) and freshly distilled phosphorus oxychloride (7.0 mL) were refluxed at 130°C for 1.5 hours. Unused phosphorus oxychloride was then distilled off under reduced pressure. The residue was allowed to cool and then thinned with chloroform (10.0 mL). The solution was then poured into a mixture of ice (40.0 g) and concentrated aqueous ammonia (33 %) (10.0 mL) with heat being liberated. The solution was stirred, the chloroform layer separated and the aqueous phase extracted with chloroform (2 x 30 mL). The combined chloroform extracts were dried over anhydrous magnesium sulphate, filtered and the chloroform evaporated under reduced pressure to give a yellow powder which upon recrystallisation from petroleum ether (b.p. 60-80°C) afforded 0.83 g (84 %) of the desired 4,5-dimethyl-9-chloro-acridine (21) as yellow-green needles m.p. = 150-151°C (lit [29] 151-152°C). MS m/z 241 (M+); 1H-NMR δ 2.94, s, 2 x CH3, 7.42-8.31, m, acridine protons.
4,5-Dimethyl-9-methoxyacridine (14). 4,5-Dimethyl-9-chloroacridine (220 mg, 0.91 mmol) was added to a methoxide solution generated from sodium (160 mg, 6.95 mmol) in methanol (15.0 mL) and tetrahydrofuran (0.5 mL). The solution was gently refluxed for 40 hours and allowed to cool and the methanol reduced under vacuum. The residue was taken up in benzene, filtered from the salt and the benzene evaporated under reduced pressure to give a yellow powder which was recrystallised from heptane to afford 145 mg (67 %) of the desired 4,5-dimethyl-9-methoxyacridine (14) as yellow crystals m.p. = 90-91°C. MS m/z 237 (M+); 1H-NMR δ2.93 (6H, s, 2 x CH3), 4.19 (3H, s, OCH3), 7.32-8.17 (6H, m, acridine protons). Anal. Found C 80.0, H 6.4, N 5.9%; C16H15NO requires C 80.6, H 6.4, N 5.9 %. UV (log E) 225, 357, 373, 392 nm. (4.63, 3.81, 3.80, 3.66).
References
- Blake, A.; Peacocke, A.R. Biopolymers 1961, 6, 1225.
- Lerman, L.S. J. Mol. Biol. 1961, 3, 18–20.
- Le Pecq, J.B.; Le Bret, M.; Barbet, J.; Roques, B. Proc. Natl. Acad. Sci. U.S.A. 1977, 72, 2915–2199. [CrossRef]
- Sakore, T.D.; Jain, S.; Tsai, C.; Sobell, H.M. Proc. Natl. Acad. Sci. U.S.A. 1962, 74, 188. [CrossRef]
- Waring, M.J. J. Mol. Biol. 1970, 54, 247. [CrossRef]
- Hurwitz, J.; Furth, J.; Malamy, M.; Alexander, M. Proc. Natl. Acad. Sci. U.S.A. 1962, 48, 1222. [CrossRef] [PubMed]
- Martin, R.F.; Kelly, D.P. Aust. J. Chem. 1979, 32, 2637–2646. [CrossRef]
- Martin, R.F. Int. J. Radiat. Biol. 1977, 32, 553.
- Krish, R.E.; Sauri, C.J. Int. J. Radiat. Biol. 1975, 27, 553–560.
- Wakelin, L.P.G.; Creasy, T.S.; Waring, M.J. FEBS Lett 1979, 104, 261–265.
- Shultz, P.G.; Dervan, P.B. J. Am. Chem. Soc. 1983, 105, 7748–7750.
- Tassigiannakis, W. B.Sc.(Hons.) Thesis, University of Melbourne, 1982.
- Martin, R.F.; Holmes, N. Nature 1983, 302, 452–454. [CrossRef]
- Bergeron, R.J.; Burton, P.S.; McGovern, K.A.; Kilne, S.J. Synthesis (Comm). 1981, 731–733.
- Cannellakis, E.S.; Shaw, Y.H.; Hanmers, W.E.; Sahartz, R.A. Biochem. et Biophys. Acta. 1976, 418, 277–289.
- Bergeron, R.J.; McGovern, K.A.; Channing, M.A.; Burton, P.S. Synthesis (Comm). 1980, 45, 1589–1592.
- Bruson, H.A. Org React. 1949, 5, 79.
- Acheson, R.M. Acridines, 2nd Ed ed; J. Wiley and Sons: New York, 1973. [Google Scholar]
- Albert, A. The Acridines, 2nd Ed ed; Edward Arnold Publishers: London, 1966. [Google Scholar]
- Newman, M.S.; Powell, W.H. J. Org. Chem. 1961, 26, 812–815.
- Cain, B.F.; Atwell, G.J. J. Med. Chem. 1976, 19, 1124–1129.
- Olah, G.A.; Narang, S.C.; Gupta, G.B.; Malhotra, R. Tetrahedron 1969, 44, 1247.
- Long, L.H.; Freeguard, G.F. Chem and Ind. (London) 1965, 223.
- McOmie, J.F.W.; Watts, M.L.; West, D.E. Tetrahedron 1969, 24, 2289.
- Kluge, A.F.; Untch, K.G.; Fried, J.H. J. Am. Chem. Soc. 1972, 94, 7827. [CrossRef]
- Gould, F.E.; Johnson, G.S.; Ferris, A.F. J. Org. Chem. 1960, 25, 1658–1660. [CrossRef]
- Brown, H.C.; Choi, Y.M.; Narasimhan, S. J. Org. Chem. 1982, 47, 3153–3163. [CrossRef]
- Allen, C.F.H.; McKee, G.H.W. Org. Synth. 1939, 19, 6–9.
- Albert, A.; Gledhill, W.J. J. Soc. Chem. Indust. 1942, 61, 159–160.
- Sample Availability: Samples are available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
MDPI and ACS Style
Moloney, G.P.; Kelly, D.P.; Mack, P. Synthesis of Acridine-based DNA Bis-intercalating Agents. Molecules 2001, 6, 230-243. https://doi.org/10.3390/60300230
AMA Style
Moloney GP, Kelly DP, Mack P. Synthesis of Acridine-based DNA Bis-intercalating Agents. Molecules. 2001; 6(3):230-243. https://doi.org/10.3390/60300230
Chicago/Turabian StyleMoloney, Gerard P., David P. Kelly, and P. Mack. 2001. "Synthesis of Acridine-based DNA Bis-intercalating Agents" Molecules 6, no. 3: 230-243. https://doi.org/10.3390/60300230