Virtual Screening in Lead Discovery: A Viewpoint

EST Lead Informatics, AstraZeneca R&D Mölndal, S-43183 Mölndal, Sweden
Molecules 2002, 7(1), 51-62; https://doi.org/10.3390/70100051
Submission received: 22 October 2001
/
Revised: 9 December 2001
/
Accepted: 12 December 2001
/
Published: 31 January 2002
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Virtual screening (VS) methods have emerged as an adaptive response to massive throughput synthesis and screening technologies. Based on the structure-permeability paradigm, the Lipinski rule of five has become a standard property filtering protocol for VS. Three possible VS scenarios with respect to optimising binding affinity and pharmacokinetic properties are discussed. The parsimony principle for selecting candidate leads for further optimisation is advocated.
The Emergence of Virtual Screening
Massive throughput in synthesis and screening yields, on the average, hundreds of thousands of novel compounds that are synthesised, then screened for various properties, ranging from biological activity and solubility to metabolic stability and cytotoxicity. The economically-driven pressure to deliver the “first-in-class” drug on the market has forced the pharmaceutical industry to embark in a costly, yet untested, drug discovery paradigm: Hunting for the new gene, the new target, the new lead compound, the new drug candidate, finally hunting for the new drug. Just as computational chemistry (computer-aided drug design, molecular modelling, etc) was being hailed as the newest, safest and fastest method to put new chemical entities on the drug market in the late 1980s, so was combinatorial chemistry (applied molecular evolution, multiple parallel synthesis, etc.), combined with high-throughput screening (HTS), hailed as the newest, safest and fastest method in the mid-1990s. These technologies have partially delivered their promise – but not in the manner initially described by enthusiastic supporters [1].
The reality of drug-receptor interactions, at the molecular level, is far too complex to provide a failsafe in silico technology for drug discovery: entropy and the dielectric constant are just two examples of subjects continuously debated among experts. Beyond ligand-receptor interactions, there’s the complexity of multiple binding modes, accessible conformational states for both ligand and receptor, affinity vs. selectivity, plasma protein binding, metabolic stability (site of reactivity and turn-over), absorption, distribution and excretion, as well as in vivo vs. in vitro properties of model compounds. Such difficulties are hardly surmountable by any single computer-aided drug design software. Even if successful, such software could not provide generally applicable models due to the inability of statistically trained models to provide trustworthy forecasts for unknown (previously-not-seen-by-the-model) classes of compounds.
In the combinatorial chemistry field, differences in the reactivity of similar, yet diverse reagents, that often leads to impure, or not at all synthesized, compounds, as well as the reality of inadequate, or missing, synthetic blocks [2], have resulted in moderately successful lead-finding combinatorial libraries [3], focused more on forming carbon-heteroatom [4] bonds, and less on forming carbon-carbon [5] bonds. On the HTS side, it took several years to realise that compound mixtures do not yield reliable results, compared to one compound per well, and that screening potentially reactive species is too often a cause of false hits [6]. Furthermore, single dose/single test procedures require repeat experiments, in order to eliminate the false positives and to confirm the structure and biological activity of the HTS hits. The stability of the compounds in solid form, or in solution, coupled with the compounds’ availability for future HTS, have added enough technical hurdles that might even slow down, not accelerate, the process of drug discovery.
As increased awareness of these technological shortcomings permeates the computational, combinatorial and medicinal chemistry communities, carefully planned strategies emerge. For example, the choice of reagents in synthesis planning is often performed via genetic algorithms [7] or statistical molecular design [8], whereby multifactorial response surfaces are optimised – all in the context of target-related library design, whenever three-dimensional target information is available. Permeability and solubility, key properties required for orally available compounds [9], are increasingly being screened for with computational and experimental methods [10] prior to choosing candidate leads for further optimisation. Due to their impact on the successful progression of drug candidates towards the launching phase [11] it has been recognised that absorption, distribution, metabolism and excretion (ADME) are key properties in the process of drug discovery that need to be optimised early on.
Virtual screening (VS) [12,13] has emerged as an adaptive response to the massive throughput synthesis and screening paradigm. In parallel to developing methods that provide (more) accurate predictions for binding affinity, pharmacokinetic properties, etc. for low-number series of compounds (tens, hundreds), necessity has forced the computational chemistry community to develop tools that screen, against any given target and/or property, millions or perhaps billions of molecules – virtual or not [14]. VS technologies have thus emerged as a response to the pressure from the combinatorial/HTS community [15]. VS methods can be regarded as less accurate, since speed and the possibility to capture most (but not necessarily all) potentially actives are its key attributes. One of the emerging problems in virtual screening is what criteria should one use, other than (predicted) binding affinity [16] and the post-docking filtering [17] protocols, to judge the quality of a (virtual) hit? What strategy should one follow, given the possibility to evaluate, in silico, extremely large virtual libraries? Similar questions arise when evaluating hits during post-HTS data analysis. This paper attempts to address some of these issues, in the context of medicinal chemistry driven lead and drug discovery.
ADME Property Filtering in Virtual Screening
Starting from the comparison of early HTS and combinatorial chemistry hits at Pfizer (up to 1994), Chris Lipinski and coworkers [10] analysed a subset of 2245 drugs from the World Drug Index (WDI), in order to better understand what are the common molecular features of orally available drugs. Using a simplified, yet efficient version of the QSAR paradigm for structure-permeability [18], as suggested [19] by Van de Waterbeemd et al., they have concluded that poor absorption or permeation are more likely to occur when
- The molecular weight (MW) is over 500;
- The calculated [20] octanol/water partition coefficient (CLOGP) is over 5, and
- There are more than 5 H-bond (hydrogen bond) donors (HDO – expressed as the sum of O-H and N-H groups);
- There are more than 10 H-bond acceptors (HAC – expressed as the sum of N and O atoms).
Thus, any pairwise combination of the following conditions: MW > 500, CLOGP > 5, HDO > 5, and HAC > 10, may result in compounds with poor permeability (exceptions are actively transported compounds and peptides). The “rule of 5” (RO5) probability scheme had a major impact in the pharmaceutical industry, as most managers realised that due to its simplicity, it could be implemented early on in drug discovery, and potentially increase the chance of success in drug discovery projects. Chemists use, therefore, the rule of 5 in order to avoid compounds with potential permeability problems. We note here that, while very popular in recent literature [21,22], the polar molecular surface area (PSA) is directly correlated with the sum of HDO and HAC, i.e. it is a RO5-compatible descriptor.
With the exception of CLOGP, all other RO5 criteria are additive, since they can be accurately computed for virtual libraries starting from the reactants – in other words prior to the enumeration step in which products are generated in silico from a list of reagents [24] – provided that those moieties gained or lost during the “virtual reactions” are accounted for. Rapid methods for estimating hydrophobicity exist [25], making it possible to perform RO5 library filtering prior to enumeration. This can save time early on in the process of synthesis planning, and opens further up the possibility of exploring large amounts of reagents – since post-enumeration property estimation is usually much more time-consuming. For example, the time spent on computing the properties on 100 class A, B and C reagents is significantly less than computing the same properties for a fully-enumerated one million compounds {A-B-C} library.
In addition to the RO5 criteria, other reactant-based properties can be used to filter virtual libraries prior to the enumeration stage. With filtering, one can avoid, for example: The presence of large numbers of halogens in the products (even though one or several halogens are allowed in the reactants); The presence of highly flexible unsubstituted, unbranched alkyl chains with e.g., more than 8 CH2 groups; The formation of compounds that exceed the maximum accepted number of rings, or ionisable groups, in the product library. Last, but not least, one should exclude early on those molecules that are reactive toward protein targets: Michael acceptors, ketones, aldehydes, and all manner of suicide inhibitors. Such compounds would not yield leads or hits, but would likely turn to be false HTS positives [26].
In a follow-up paper [27], Lipinski discussed the RO5-compliance from the perspective of putting a drug on the market. While this analysis is focused on solubility and permeability, one can extrapolate to other pharmacokinetic properties such as metabolic stability, excretion and toxicity. Based on screening results from Merck and Pfizer, Lipinski argues that it is much easier to optimise pharmacokinetic properties early on in the process of drug discovery, and attempt to optimise the receptor binding affinity at a later stage. What follows is a more detailed discussion of this viewpoint, with respect to virtual screening.
Three Possible Scenarios
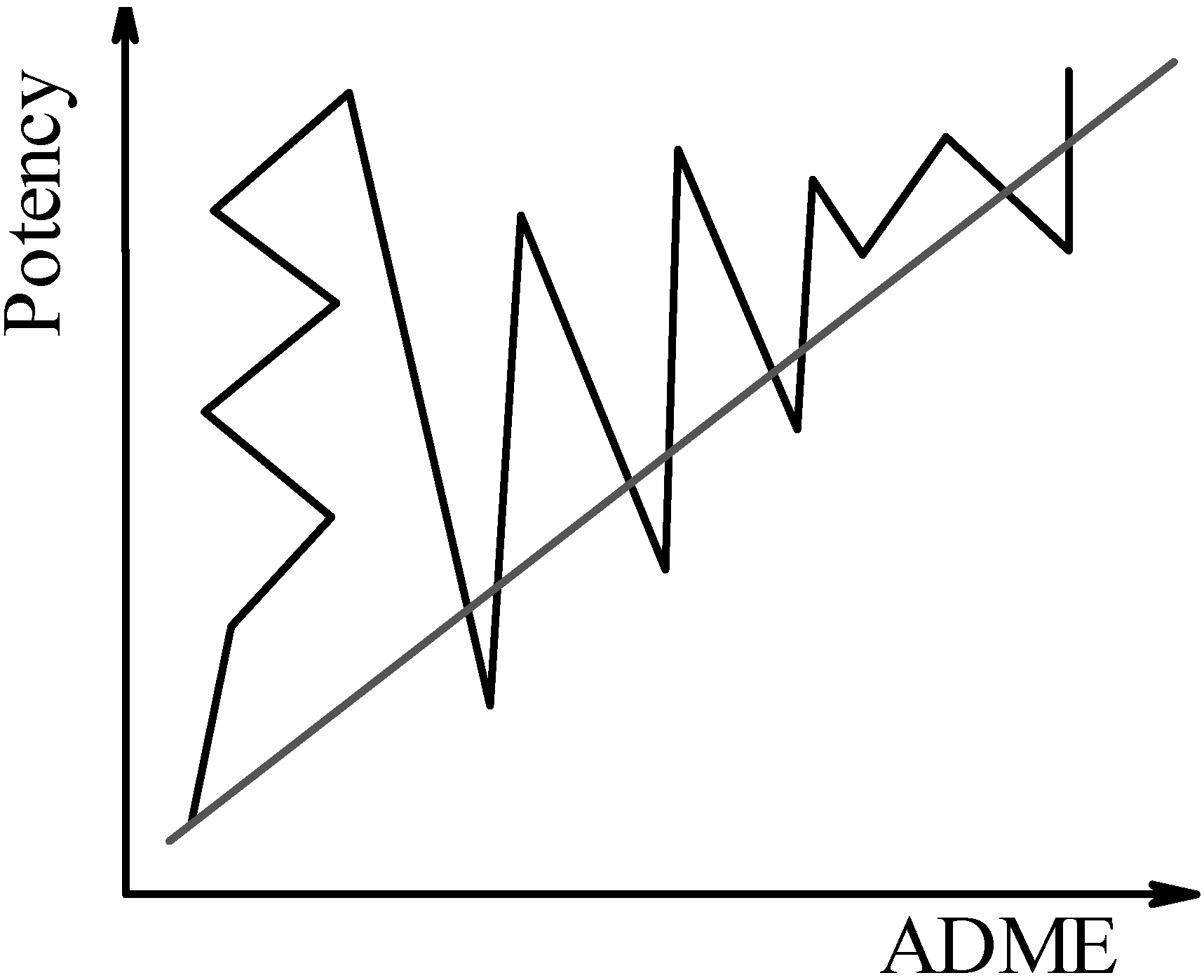
The three possible scenarios for VS in drug discovery are graphically illustrated below. In all three figures, the continuous, jagged line illustrates the number of steps (e.g., the progression of leads in drug discovery, or the progression of hits in virtual screening) needed to reach the “good potency, good ADME properties” stage (upper right corner). The dashed line illustrates the “convergent” drug discovery process.
In the first scenario, one ignores ADME properties until later in the process, while focusing on the results of the docking first. Since docking software relies on scoring functions [28] to rank the various solutions, one should rely on target-trained (more accurate), or consensus [16] scoring schemes, in order to evaluate the results. As previously discussed [29], Figure 1 illustrates the drug discovery paradigm, as implemented in the pharmaceutical industry some decades ago. Receptor binding assays, often in vitro, were the sole criterion for progressing a lead structure to in vivo pharmacokinetic testing, other than the intellectual property position. This strategy was not entirely successful, since only one in ten candidate drugs succeeds through clinical trials to reach the marketed drug status – causing the top 50 pharmaceutical companies to spend, on the average, 750 millions USD for each of the 21 truly novel drugs launched during the past decade [30]. Pharmacokinetic property optimisation is a rather complex undertaking that is likely to require changes in those molecular determinants that are responsible for binding affinity and specificity, e.g., hydrogen bonds (that are directional). It was recently argued that even hydrophobic interactions have some directionality [31].
Figure 1.
First scenario: Binding affinity (potency) is optimised first; ADME properties are optimised at a later stage.
Figure 1.
First scenario: Binding affinity (potency) is optimised first; ADME properties are optimised at a later stage.

Changes in the structures of high-affinity compounds lead, more often than not, to significant drops in potency. The trial-and-error paradigm involves, in this scenario, alterations in the receptor-interacting moieties in order to achieve good ADME properties – a process that may be demanding in terms of time and resources. In terms of virtual screening, the docking and scoring solutions would then be filtered using pharmacokinetic-based prediction schemes [19,32,33,34] at the virtual hit level, i.e., by choosing those compounds that are predicted to have the highest affinity by the scoring functions. This way, a small percentage of the virtually screened compounds are evaluated with ADME prediction schemes, and compounds with potentially good pharmacokinetic properties are likely to be discarded due to more modest performance in terms of predicted potency.
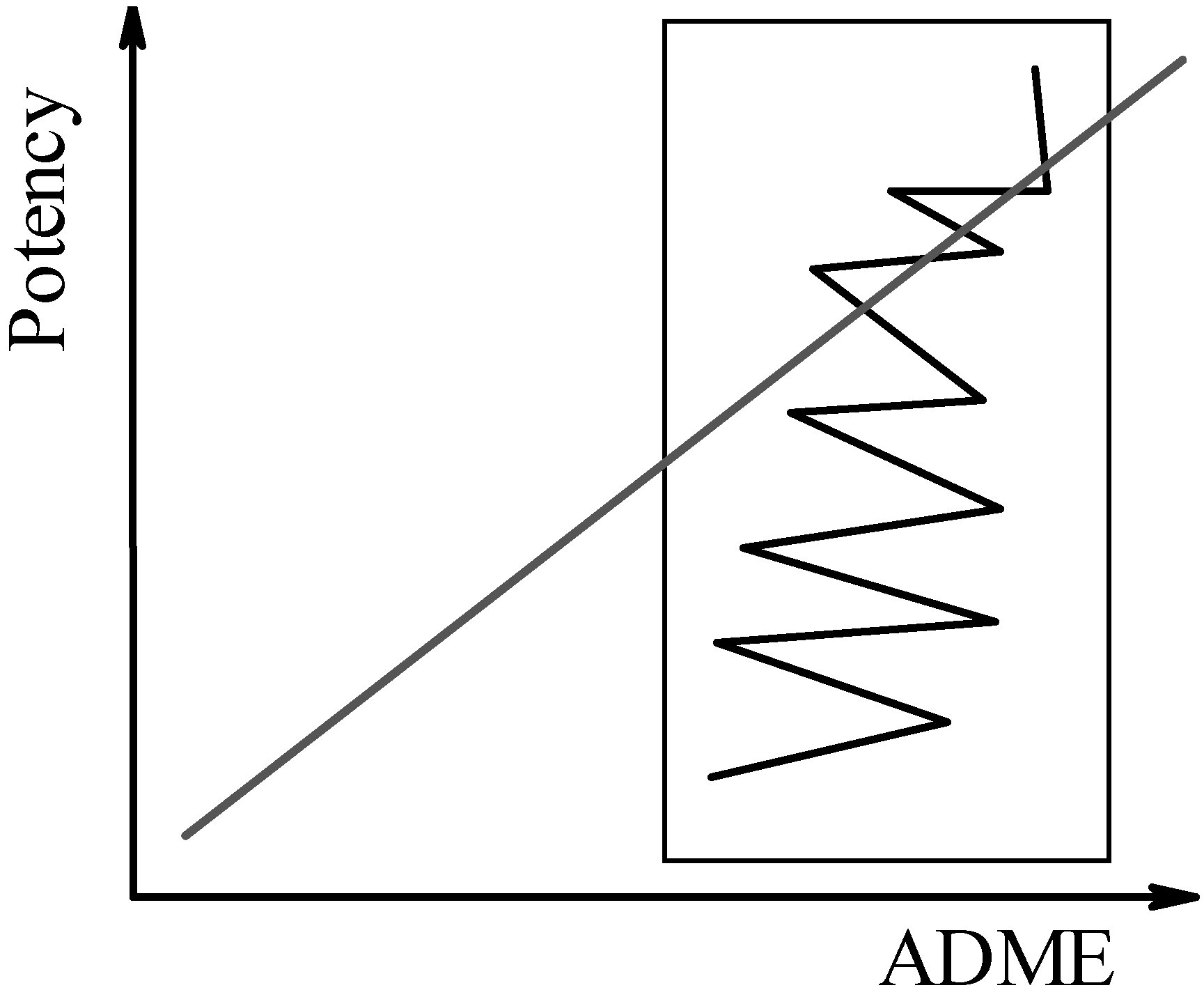
In the second scenario, ADME filters are enforced on the entire virtual screening library, prior to performing the docking step. This way, ADME properties are optimised at the onset, which is not unlike the current strategy being pursued in the pharmaceutical industry [10,21,22,23,35,36]. Various physico-chemical descriptors, e.g., PSA, the octanol/water partition coefficient estimated at pH 7.4 (logD74), solubility, or predicted pharmacokinetic properties, e.g., passive transcellular permeability in the intestine or in the brain, are used to filter out those compounds that do not meet the user-defined crieria. For orally available compounds, logD74 between –1 and 4, or PSA < 140 Å2 are generally acceptable whereas brain penetration is more restrictive (e.g., logD74 between +1 and 4, or PSA < 60 Å2). Docking and scoring would then be applied only on those compounds that meet these filtering criteria. In this scenario, one has to preserve the molecular determinants responsible for good ADME properties, while modifying the structure in order to achieve good affinity and selectivity. This may result in reduced ADME properties, but the process is less time-consuming, before optimal structures (Figure 2) are found.
Figure 2.
Second scenario: The optimisation of binding affinity (potency) is performed starting from an ADME-filtered region (box) of the medicinal chemistry space.
Figure 2.
Second scenario: The optimisation of binding affinity (potency) is performed starting from an ADME-filtered region (box) of the medicinal chemistry space.

This approach, based on the “computational alert” at Pfizer [10], is based on the expectation that, among the (virtual) molecules screened for good ADME properties, there will be compounds with high affinity. It should be pointed out that, even if successful in ruling out potential problems with passive permeability, this strategy is unlikely to address active mechanisms unless more advanced predictive models are developed. However, it will be less effective in the event that no virtual hits are found in the docking and scoring step, since compounds with potentially good affinity are likely to be discarded due to more modest performance in terms of predicted pharmacokinetic properties.
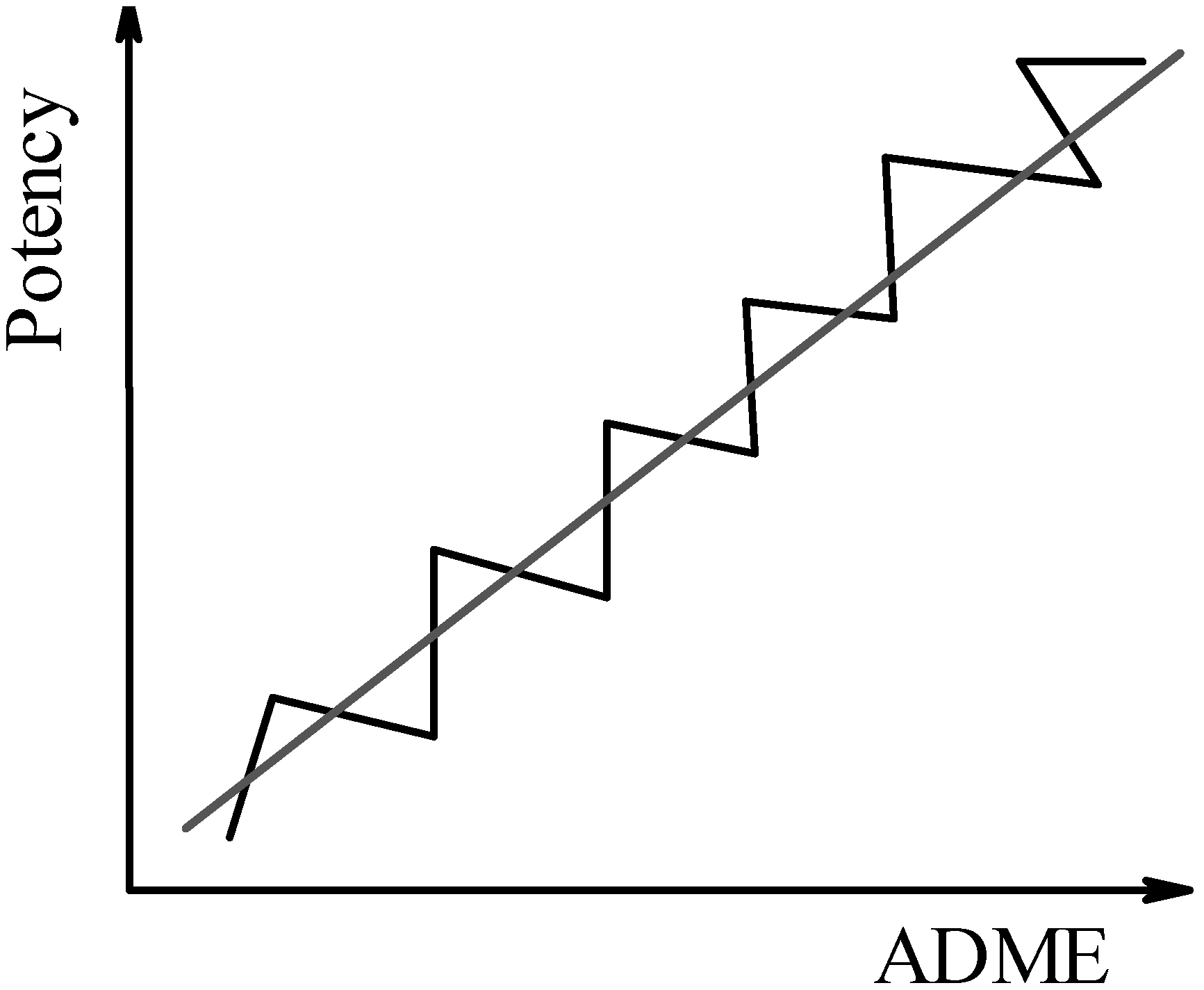
In the third scenario, one would simultaneously monitor changes that influence binding affinity and ADME properties, as previously suggested [29]. The real difficulty consists in addressing the appropriate molecular determinants that define the desired compound characteristics, in a consistent manner. For example, hydrophobicity and hydrogen bonds are known to contribute to both affinity and (passive) permeability - but the question remains, in what proportion are they contributing in each case? However, with an integrated software framework that can monitor ligand (or library) alterations in the “fitness landscape”, it is expected that progressive increments in affinity and pharmacokinetic properties are obtained (Figure 3).
The proof of concept of such an approach has already been addressed [29,37]. Briefly, the VolSurf program [38], extensively validated in modeling ADME properties [39,40,41], was successfully used to model the binding affinity of a ligand-diverse, protein-diverse data set [42], and the binding affinity of a ligand-diverse, single-target data set [43]. While all three scenarios rely on the quality and accuracy of the experimental data used to derive the predictive models (for both scoring and ADME), the third one in particular is expected to be less vulnerable to prediction errors (see Figure 4). If applied interactively, this integrated approach is likely to converge on interesting compounds more rapidly. This could be a less resource-demanding strategy.
Figure 3.
Third scenario: Binding affinity (potency) and ADME properties are optimised simultaneously.
Figure 3.
Third scenario: Binding affinity (potency) and ADME properties are optimised simultaneously.

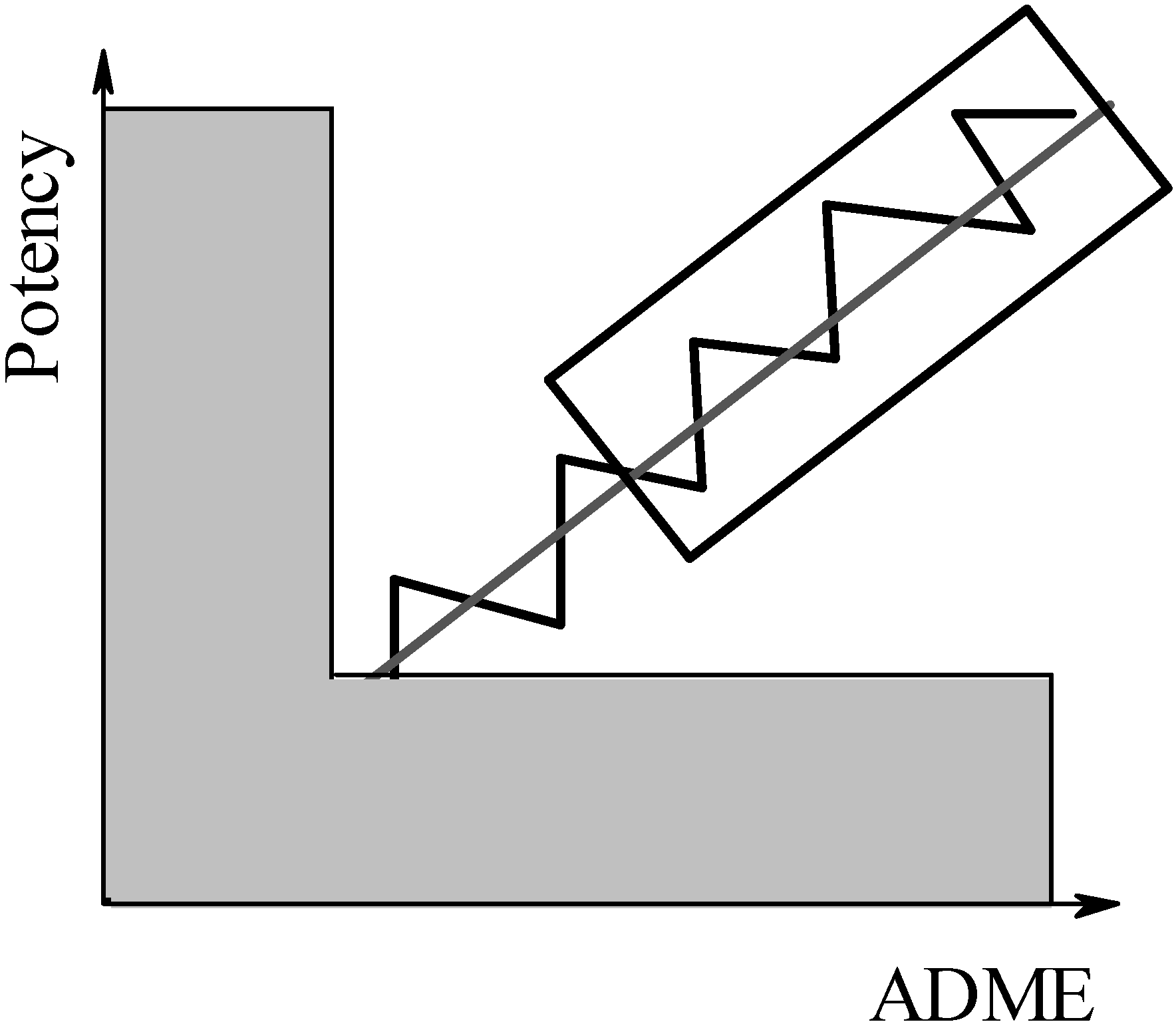
Figure 4.
Zooming in on compounds of interest: Molecules located in the shaded area are eliminated in the early stages of virtual screening, using various (pre)filtering schemes. Molecules located in the boxed area are then investigated with more accurate methods in a second step.
Figure 4.
Zooming in on compounds of interest: Molecules located in the shaded area are eliminated in the early stages of virtual screening, using various (pre)filtering schemes. Molecules located in the boxed area are then investigated with more accurate methods in a second step.

Conclusions
The strategy of screening a limited number of diverse marketed drugs, for which the ADME properties have already been well understood in humans, is already being implemented at different companies, e.g., Prestwick Chemical Inc. [44,45] and CEREP [46]. Both Lipinski [27] and Wermuth [44] suggest that screening compounds with well-understood ADME properties (RO5-compliant) is a winning strategy. This should be extended to virtual screening as well. The third scenario is likely to be more useful in the absence of (experimental) hits from already-existing drugs. We have previously argued [47] that preference should be given to low molecular weight, low hydrophobicity (CLOGP) molecules, when designing leadlike combinatorial libraries. Confirmed [48] from an independent dataset [49], our initial cut-off values (MW < 350 and CLOGP < 3.5) have been revised (MW < 450 and CLOGP < 4.5), since they appeared to be too restrictive [50]. Virtual screening strategies should follow similar guidelines, by observing the parsimony principle (less is better) with regards to molecular complexity: If one can choose between two classes of compounds with similar properties, one should choose the simpler ones for further lead optimisation [51].
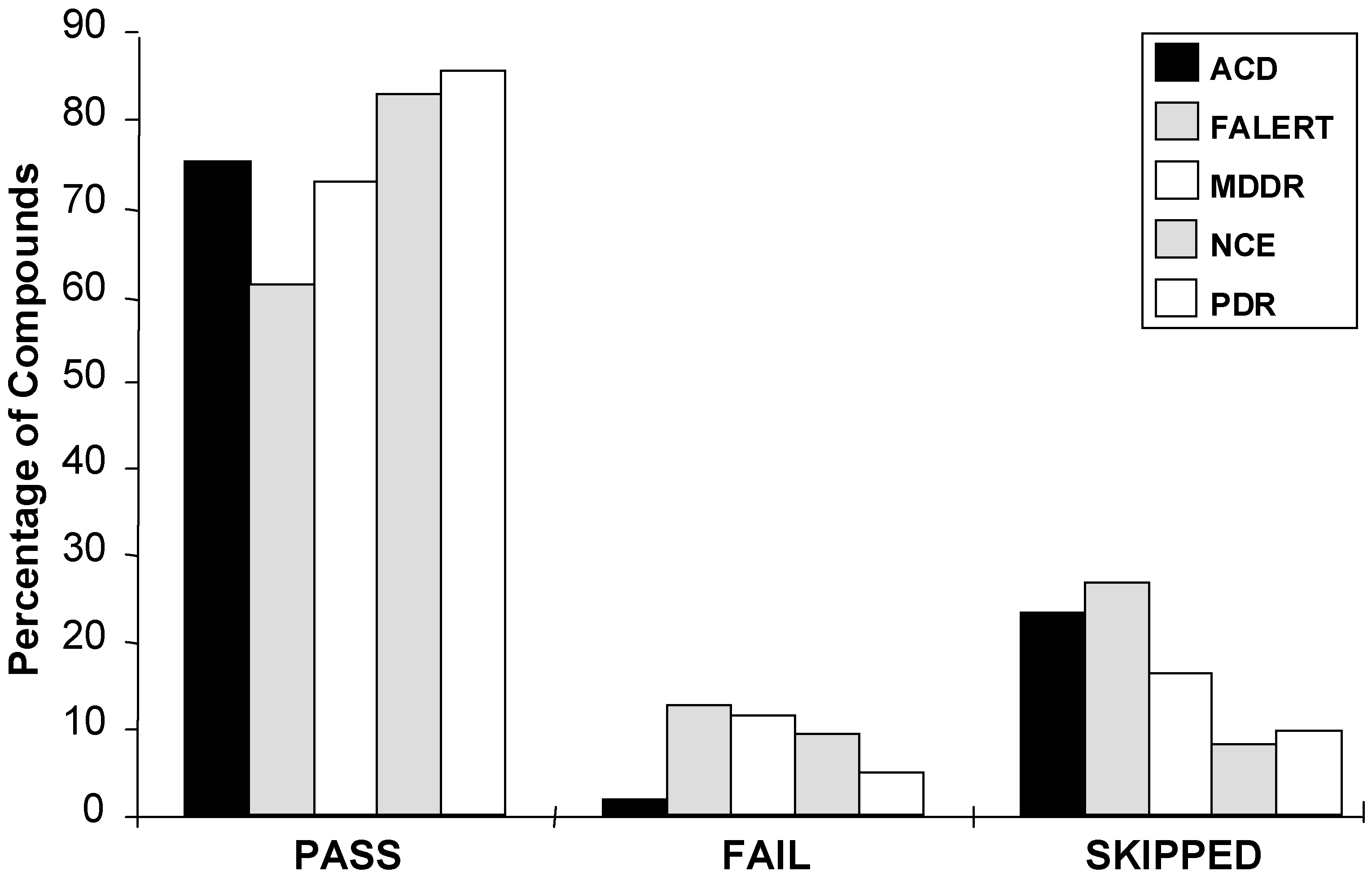
Figure 5.
Comparative distribution of RO5 tests across different chemical databases: ACD, the Available Chemicals Directory, FALERT, Current Patents Fast-Alert, MDDR, MACCS Drug Data Report, NCE, New Chemical Entities, and PDR, the Physician’s Desk Reference (see [52] for details on these databases). The “skipped” column indicates molecules for which CLOGP could not be estimated.
Figure 5.
Comparative distribution of RO5 tests across different chemical databases: ACD, the Available Chemicals Directory, FALERT, Current Patents Fast-Alert, MDDR, MACCS Drug Data Report, NCE, New Chemical Entities, and PDR, the Physician’s Desk Reference (see [52] for details on these databases). The “skipped” column indicates molecules for which CLOGP could not be estimated.

Care should be exercised in applying the RO5 for virtual screening: RO5 criteria were developed by studying marketed drugs [10], not HTS leads. These criteria should not to be interpreted as a natural law, as they are based on a negative, not positive, selection: They point out where success is more probable, but do not warrant it. While RO5 criteria identify the boundary for the 90% percentile of drug-property distribution with respect to size (MW), hydrophobicity (CLOGP) and hydrogen-bond capacity, they do not imply drug marketability [52]: More than 140,000 compounds in the Available Chemicals Directory are inside that space (see Fig. 5). One needs to tighten these criteria when seeking lead structures [50]. In other words, the rule of five was designed to assist decision-makers in drug discovery: "Out of these 2, or 5000 (virtual) hits, which ones should I study further?" - and, based on the above, the answer is: "The ones that are RO5-compliant".
References and Notes
- Horrobin, D. F. Innovation in the pharmaceutical industry. J. Royal Soc. Med. 2000, 93, 341–345. [Google Scholar]
- Some typical issues: How long does it take to purchase those [unique] reagents? Are they still available? At what price/purity?
- Terrett, N. K.; Gardner, M.; Gordon, D. W.; Kobylecki, R. J.; Steele, J. Drug discovery by combinatorial chemistry - the development of a novel method for the rapid synthesis of single compounds. Chem. Eur. J. 1997, 3, 1917–1920. [Google Scholar]
- Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [Google Scholar]
- Franzen, R. The Suzuki, the Heck, and the Stille reaction; three versatile methods for the introduction of new C-C bonds on solid support. Can. J. Chem. 2000, 78, 957–962. [Google Scholar]
- Rishton, G. M. Reactive compounds and in vitro false positives in HTS. Drug Discovery Today 1997, 2, 382–384. [Google Scholar]
- Weber, L.; Wallbaum, S.; Broger, C.; Gubernator, K. Optimization of the biological activity of combinatorial compound libraries by a genetic algorithm. Angew. Chem., Int. Ed. 1995, 34, 2280–2282. [Google Scholar]
- Linusson, A.; Gottfries, J.; Lindgren, F.; Wold, S. Statistical molecular design of building blocks for combinatorial chemistry. J. Med. Chem. 2000, 43, 1320–1328. [Google Scholar]
- Anonymous. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. 2000. Available from http://www.fda.gov/cder/OPS/BCS_guidance.htm.
- Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Drews, J. Drug discovery: a historical perspective. Science 2000, 287, 1960–1964. [Google Scholar]
- Walters, W. P.; Stahl, M. T.; Murcko, M. A. Virtual screening - an overview. Drug Discovery Today 1998, 3, 160–178. [Google Scholar]
- Fox, S.; Farr-Jones, S.; Yund, M. A. High throughput screening for drug discovery: continually transitioning into new technology. J. Biomol. Screening 1999, 4, 183–186. [Google Scholar]
- Young, S.; Li, J. Virtual screening of focused combinatorial libraries. Innovations Pharm. Technol. 2000, 0(5), 24-26, 28. [Google Scholar]
- For example: I need to compute a molecule faster than chemists can make it (or faster than the HTS lab can screen it)
- Charifson, P. S.; Corkery, J. J.; Murcko, M. A.; Walters, W. P. Consensus scoring: A method for improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar]
- Stahl, M.; Bohm, H. J. Development of filter functions for protein-ligand docking. J. Mol. Graphics Modell. 1998, 16, 121–132. [Google Scholar]
- The QSAR paradigm for structure-permeability expresses the passive permeability as a function of hydrophobicity, molecular size, and hydrogen-bond capacity
- Van de Waterbeemd, H.; Camenisch, G.; Folkers, G.; Raevsky, O. A. Estimation of Caco-2 cell permeability using calculated molecular descriptors. Quant. Struct.-Act. Relat. 1996, 15, 480–490. [Google Scholar]
- Leo, A. Estimating LogPoct from structures. Chem. Rev. 1993, 5, 1281–1306, CLOGP is available from Daylight Chemical Information Systems, http://www.daylight.com. [Google Scholar]
- Clark, D. E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J. Pharm. Sci. 1999, 88, 807–814. [Google Scholar]
- Clark, D. E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J. Pharm. Sci. 1999, 88, 815–821. [Google Scholar]
- Kelder, J.; Grootenhuis, P. D. J.; Bayada, D. M.; Delbressine, L. P. C.; Ploemen, J. P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar]
- Olsson, T.; Oprea, T. I. Cheminformatics: a tool for decision-makers in drug discovery. Curr. Opin. Drug Discovery Dev. 2001, 4, 308–313. [Google Scholar]
- Oprea, T. I. Rapid estimation of hydrophobicity for virtual combinatorial library analysis. SAR QSAR Environ. Res. 2001, 12, 129–141. [Google Scholar]
- Rishton, G. Personal communication, 2001.
- Lipinski, C. A. Druglike properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar]
- Oprea, T.I.; Marshall, G. R. Receptor-based prediction of affinity. Perspectives in Drug Discovery and Design 1998, 9–11, 35–61. [Google Scholar]
- Oprea, T.I.; Zamora, I.; Svensson, P. Qvo vadis, scoring functions? Toward an integrated pharmacokinetic and binding Affinity Prediction Framework. In Combinatorial Library Design and Evaluation for Drug Design; Ghose, A. K., Viswanadhan, V. N., Eds.; Marcel Dekker Inc.: New York, 2001; pp. 233–266. [Google Scholar]
- Drews, J. Innovation deficit revisited: Reflections on the productivity of pharmaceutical R&D. Drug Discov. Today 1998, 3, 491–494. [Google Scholar]
- Davis, A. M.; Teague, S. J. Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew. Chem. Int. Ed. 1999, 38, 736–749. [Google Scholar]
- Palm, K.; Luthman, K.; Ungell, A.L.; Strandlund, G.; Artursson, P. Correlation of drug absorption with molecular surface properties. J. Pharm. Sci. 1996, 85, 32–39. [Google Scholar]
- Oprea, T. I.; Gottfries, J. Toward a minimalist model of oral drug absorbtion. J. Mol. Graphics Modell. 1999, 17, 261–274. [Google Scholar]
- Zamora, I.; Oprea, T. I.; Ungell, A. L. Prediction of Oral Drug Permeability. In Rational Approaches to Drug Design; Höltje, H. D., Sippl, W., Eds.; Prous Science Press: Barcelona, 2001; pp. 271–280. [Google Scholar]
- Darvas, F.; Dorman, G. Early integration of ADME/Tox parameters into the design process of combinatorial libraries. Chim. Oggi 1999, 17, 10–13. [Google Scholar]
- Pickett, S. D.; McLay, I.M.; Clark, D. E. Enhancing the hit-to-lead properties of lead optimization libraries. J. Chem. Inf. Comput. Sci. 2000, 40, 263–272. [Google Scholar]
- Zamora, I.; Oprea, T. I.; Cruciani, G.; Pastor, M.; Ungell, A. L. Surface descriptors for protein-ligand affinity prediction. J. Med. Chem. submitted.
- VolSurf is available from Molecular Discovery Ltd. http://www.moldiscovery.com.
- Cruciani, G.; Crivori, P.; Carrupt, P. A.; Testa, B. Molecular fields in quantitative structure-permeation relationships: The VolSurf approach. J. Mol. Struct. (Theochem) 2000, 503, 17–30. [Google Scholar]
- Guba, W.; Cruciani, G. Molecular field-derived descriptors for the multivariate modeling of pharmacokinetic data. In Molecular Modeling and Prediction of Bioactivity; Gundertofte, K., Jørgensen, F. S., Eds.; New York; Kluwer Academic/Plenum Publishers, 2000; pp. 89–94. [Google Scholar]
- Crivori, P.; Cruciani, G.; Carrupt, P.A.; Testa, B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [Google Scholar]
- Head, R. D.; Smythe, M. L.; Oprea, T. I.; Waller, C.L.; Greene, S. M.; Marshall, G. R. VALIDATE: A new method for the receptor-based prediction of binding affinities of novel ligands. J. Am. Chem. Soc. 1996, 118, 3959–3969. [Google Scholar]
- Pastor, M.; Cruciani, G.; Watson, K.A. A strategy for the incorporation of water molecules present in a ligand binding site into a three-dimensional quantitative structure-activity relationship analysis. J. Med. Chem. 1997, 40, 4089–4102. [Google Scholar]
- Wermuth, C.G.; Clarence-Smith, K. Drug-like leads: bigger is not always better. Pharmaceutical News 2000, 7, 53–55. [Google Scholar]
- See http://www.prestwickchemical.com for details
- See http://www.cerep.fr/Cerep/Utilisateur/index.asp for details
- Teague, S. J.; Davis, A. M.; Leeson, P. D.; Oprea, T. I. The design of leadlike combinatorial libraries. Angew. Chem., Int. Ed. 1999, 38, 3743–3748. [Google Scholar]
- Hann, M. M.; Leach, A. R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar]
- Sneader, W. Drug prototypes and their exploitation; Chichester; John Wiley and Sons Ltd., 1996. [Google Scholar]
- Oprea, T. I.; Davis, A. M.; Teague, S. J.; Leeson, P. D. Is there a difference between leads and drugs? A historical perspective. J. Chem. Inf. Comput. Sci. 2001, 41, 1308–1315. [Google Scholar]
- Virtual screening cannot substitute creative thinking, serendipity and good experimental research. One should integrate experimental results into the process as early as possible
- Oprea, T. I. Property distribution of drug-related chemical databases. J. Comput. Aided Mol. Design 2000, 14, 251–264. [Google Scholar]
- Sample availability: Not applicable
© 2002 by Tudor Ionel Oprea ([email protected]). Reproduction is permitted for non commercial purposes
Share and Cite
MDPI and ACS Style
Oprea, T.I. Virtual Screening in Lead Discovery: A Viewpoint. Molecules 2002, 7, 51-62. https://doi.org/10.3390/70100051
AMA Style
Oprea TI. Virtual Screening in Lead Discovery: A Viewpoint. Molecules. 2002; 7(1):51-62. https://doi.org/10.3390/70100051
Chicago/Turabian StyleOprea, Tudor Ionel. 2002. "Virtual Screening in Lead Discovery: A Viewpoint" Molecules 7, no. 1: 51-62. https://doi.org/10.3390/70100051