Solid-Phase Synthesis of Methyl N-(pyrimidin-2-yl)glycinate

Chemistry Department, Moscow State University, Moscow 119899, Russia
*
Author to whom correspondence should be addressed.
Molecules 2003, 8(6), 467-471; https://doi.org/10.3390/80600467
Submission received: 13 May 2003
/
Revised: 17 June 2003
/
Accepted: 17 June 2003
/
Published: 30 June 2003
(This article belongs to the Special Issue Selected Papers from the Second Eurasian Meeting on Heterocyclic Chemistry: Heterocycles in Organic and Combinatorial Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A versatile method for the preparation of N-heteroaryl substituted aminoacid derivatives is suggested. The method avoids facile diketopiperazine formation.
Introduction
Aminoacid derivatives, in particular N-substituted α-aminoacids, are important synthetical precursors and building blocks in combinatorial chemistry. It is sometimes very difficult to obtain such N-substituted acids, especially for the cases when a heterocyclic substituent is attached to the amino group. Attempting to prepare ethyl N-(pyrimidin-2-yl)glycinate via reaction of 2-chloropyrimidine and ethyl glycinate we observed predominant formation of diketopiperazine. An alternative strategy – alkylation of the endocyclic nitrogen of 2-aminopyrimidine with chloroacetic acid (or its esters) followed by Dimroth rearrangement (which usually is a safe route to 2-alkylaminopyrimidines) – also failed due to intramolecular self-condensation between the 2-amino group and the acetic acid fragment. An alternative strategy is therefore required to prepare such a target.
Results and Discussion
Our aim was to synthesize N-(pyrimidin-2-yl)glycinate (I). This compound can be formally considered as a 2-alkylaminopyrimidine, and there are two main synthetic routes for this class of compounds (Scheme 1).
Scheme 1.

The first route (A) is the Dimroth rearrangement of 1-alkylpyrimidinium salts, which in turn can be easily prepared by alkylation of the endocyclic nitrogen atom of 2-aminopyrimidine (one would expect that for the target molecule I such an alkylating agent might correspond to an α-halogenated acetic acid derivative). The second strategy (B) is a nucleophilic substitution in a pyrimidine ring with a good leaving group in the alpha position by amines.
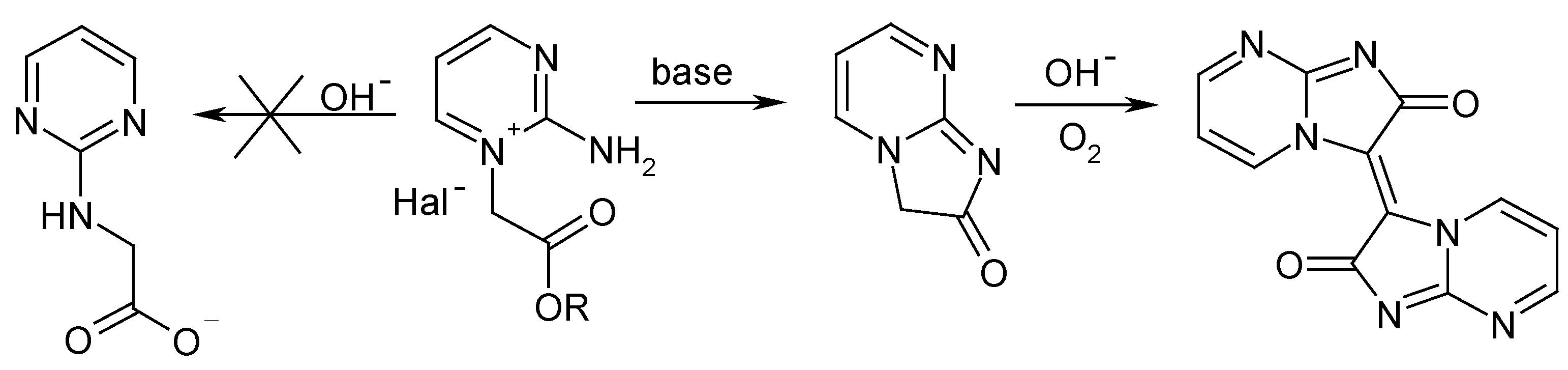
We have attempted to apply strategy A by alkylating 2-aminopyrimidine with ethyl bromoacetate and sodium chloroacetate. Both salts were obtained, and their structures were proven by NMR. However, our attempts to perform the Dimroth rearrangement gave undesirable compounds with deep coloration. It was assumed that the expected intramolecular self-condensation between the 2-amino group and the acetic acid fragment occurred, probably followed by further oxidative dimerization under basic conditions (Scheme 2):
Scheme 2.

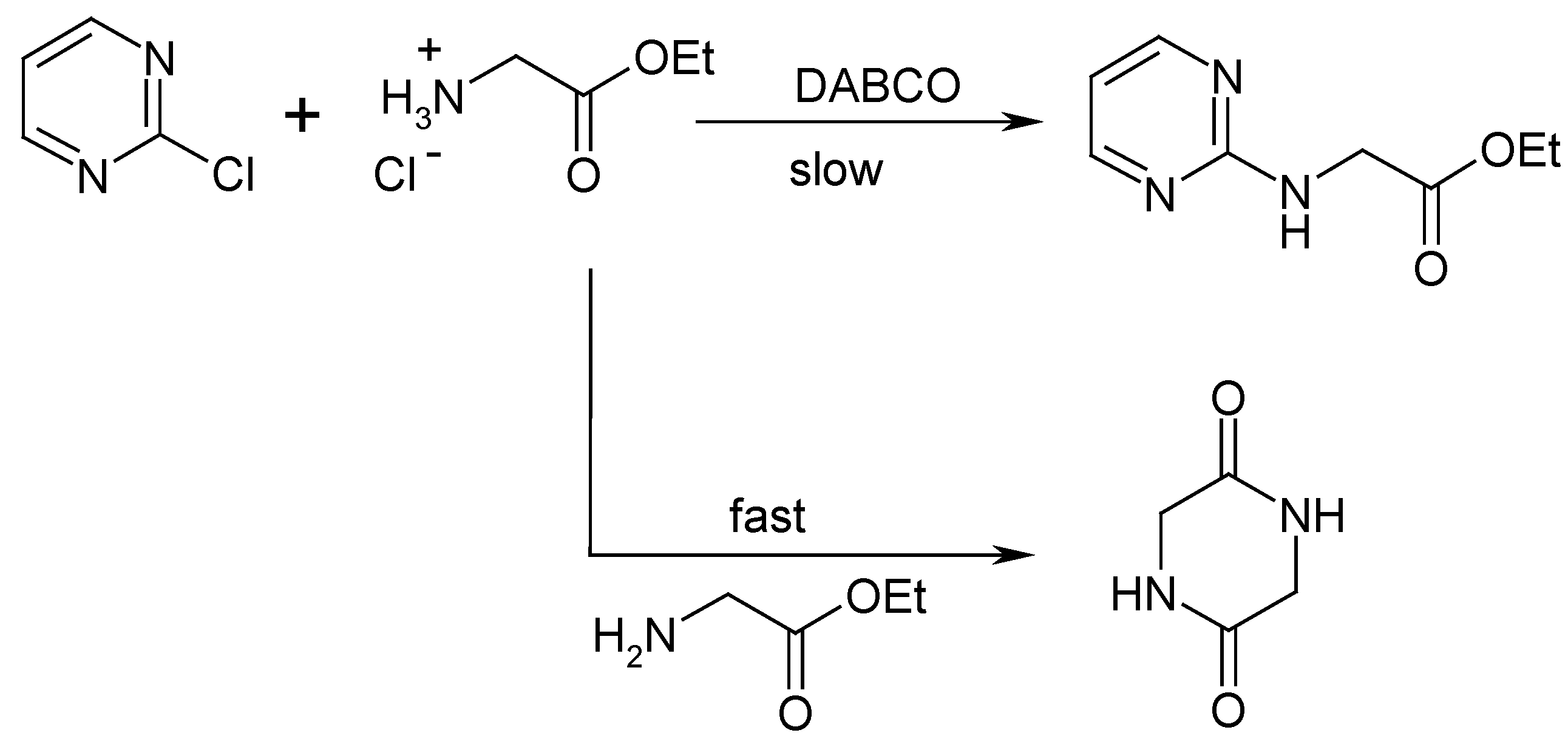
The second route to prepare the target compound I is the nucleophilic substitution in the pyrimidine ring bearing a good leaving group in the alpha position by amines. Such nucleophilic substitutions in pyrimidine rings are well-studied reactions [1]. We tried to apply this reaction to 2-chloropyrimidine using ethyl glycinate chlorohydrate in the presence of strong base (Scheme 3). Unfortunately, the expected substitution proceeds quite slowly, in contrast to the quick self-reaction of ethyl glycinate, and consequently we observed predominant formation of diketopiperazine.
Scheme 3.

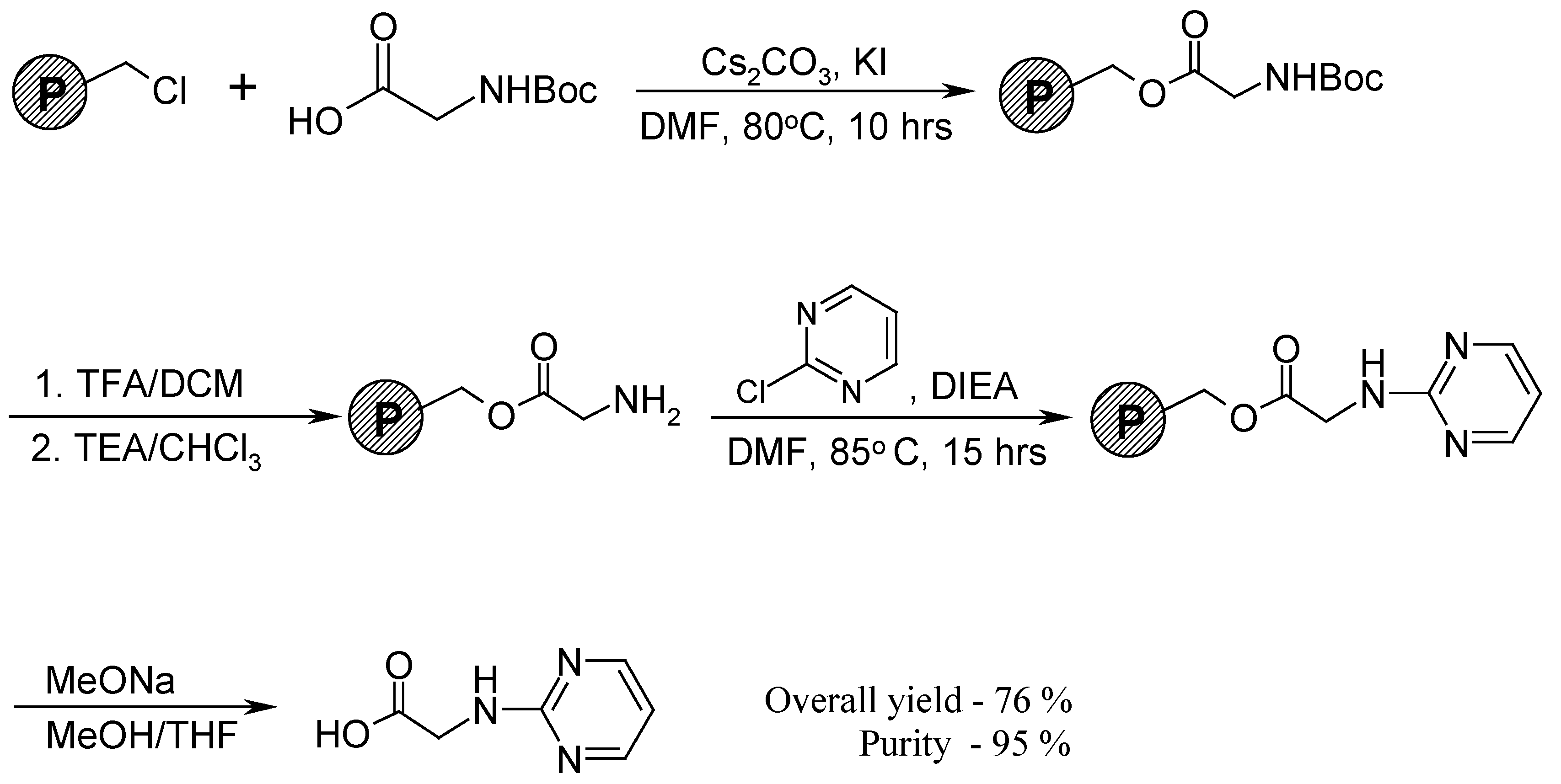
Attempting to resolve the problem, we have applied a solid-phase synthetic approach (Scheme 4). At first Boc-protected glycine (in the form of its cesium salt) was attached onto Merrifield resin under standard conditions [2] in order to prevent possible self-condensation. Solid phase supported glycine was deprotected and successfully involved in the reaction with 2-chloropyrimidine. The degree of conversion was monitored by the ninhydrin test. Finally, the product was cleaved from the Merrifield resin in the presence of sodium methoxide in the methanol/tetrahydrofuran solution [3].
Scheme 4.

Conclusions
We have found a solid-phase synthetic strategy to be advantageous for synthesis of the target compound I, compared to usual liquid phase processes.
Experimental
General
1H-NMR spectra were obtained using a Bruker AC 400 NMR spectrometer and were recorded at 360 MHz. Merrifield resin (2% crosslinked with divinylbenzene) was obtained from Reanal. All reagents were obtained from ACROS.
Synthetic procedure
Merrifield resin (1 g, 1.0 mmol) was swelled in DMF solution (6 mL) containing of Boc-protected glycine (0.525 g, 3 mmol), cesium carbonate (0.49 g, 1.5 mmol) and potassium iodide (0.17 g, 1 mmol). The mixture was constantly stirred and heated at 80oC for about 10 hours. The resin was washed successively with DMF, MeOH, DCM and dried in vacuo. Immobilized glycine was deprotected by suspending the resin (1.15 g) in a 1:1 mixture of TFA and DCM at room temperature for 0.5 hours [4]. The resin was washed sequentially with 1:1 DCM-DMF, DCM, MeOH, 1:10 TEA-CHCl3, and MeOH. In the next step dried resin (1.06 g) was heated in DMF solution containing 2-chloropyrimidine (1.137 g, 10 mmol) and DIEA (0.55 mL, 4 mmol) at 90oC for at least 15 hours. The resin turned to brown color and gave a negative ninhydrin test. After washing by DMF, DCM and drying we obtained 1.13 g of dark colored resin. Then this amount of resin was refluxed for 6 hours in a 4:1 mixture of THF-MeOH (5 mL) in the presence of sodium methoxide (0.5 mL of 0.2 M MeONa solution in MeOH). The resin was filtered and washed with THF (15 mL) and MeOH (10 mL, twice). After evaporation of solvents a solid residue was obtained (0.126 g) and analyzed without further purification. The overall yield was 76% and the purity of the product was evaluated as 95%. The structure was proven by its 1H-NMR spectra (CDCl3, 360 MHz), δ: 8.21-8.25 (2CH, d, J=7.6 Hz, 2H); 6.49-6.62 (CH, t, J=7.5 Hz, 1H); 5.45-5.60 (NH, bs, 1H); 4.13-4.16 (CH2CO2, d, J=5.2 Hz, 2H); 3.70 (CH3, s, 3H).
Acknowledgments
This work was supported by INTAS (Grant No. INTAS 00-0711).
References
- Illuminati, G. Nucleophilic heteroaromatic substitution. Adv. Heterocycl. Chem. 1964, Vol. 3, 275–283. [Google Scholar]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]
- Kobayashi, S.; Akiyama, R.; Kitagawa, H. Polymer-Supported α-Imino Acetates. Versatile Reagents for the Synthesis of alpha-Amino Acid Libraries. J. Comb. Chem. 2000, 2, 438–440. [Google Scholar]
- Mukaiyama, T. Oxidation-Reduction Condensation a New Method for Peptide Synthesis. Synth. Comm. 1972, 2, 243–265. [Google Scholar]
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Ermolat'ev, D.S.; Babaev, E.V. Solid-Phase Synthesis of Methyl N-(pyrimidin-2-yl)glycinate. Molecules 2003, 8, 467-471. https://doi.org/10.3390/80600467
AMA Style
Ermolat'ev DS, Babaev EV. Solid-Phase Synthesis of Methyl N-(pyrimidin-2-yl)glycinate. Molecules. 2003; 8(6):467-471. https://doi.org/10.3390/80600467
Chicago/Turabian StyleErmolat'ev, Denis S., and Eugene V. Babaev. 2003. "Solid-Phase Synthesis of Methyl N-(pyrimidin-2-yl)glycinate" Molecules 8, no. 6: 467-471. https://doi.org/10.3390/80600467