Folding, Stability and Shape of Proteins in Crowded Environments: Experimental and Computational Approaches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Crowded conditions in vitro and in silico and protein models
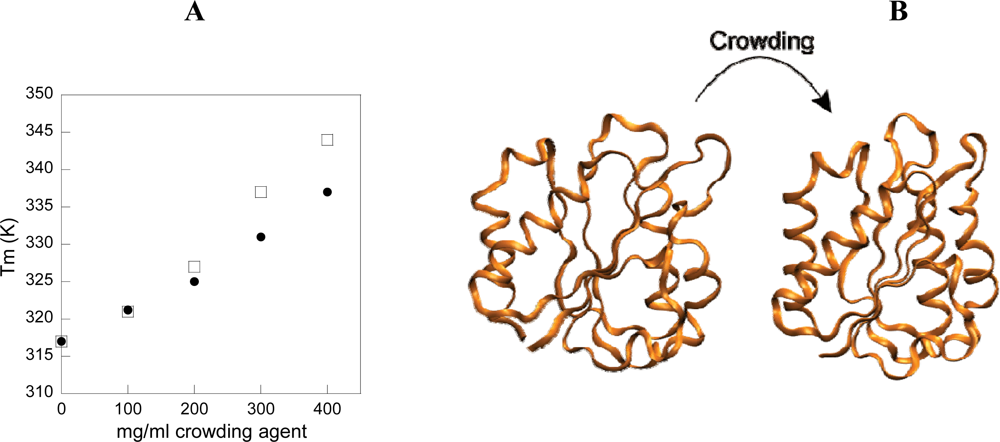
3. Effect of crowding on equilibrium properties of a spherical protein
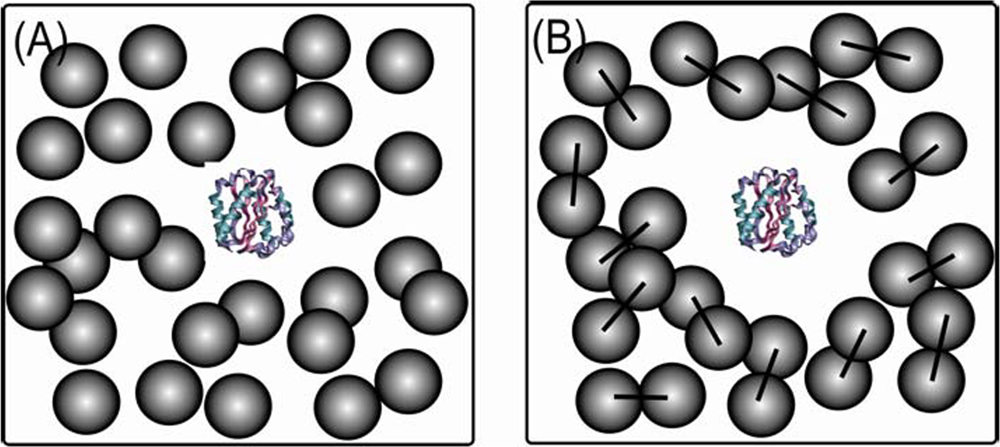
4. Shape of crowding agent influences folding routes
5. Dramatic effect of crowding on shape of a prolate protein
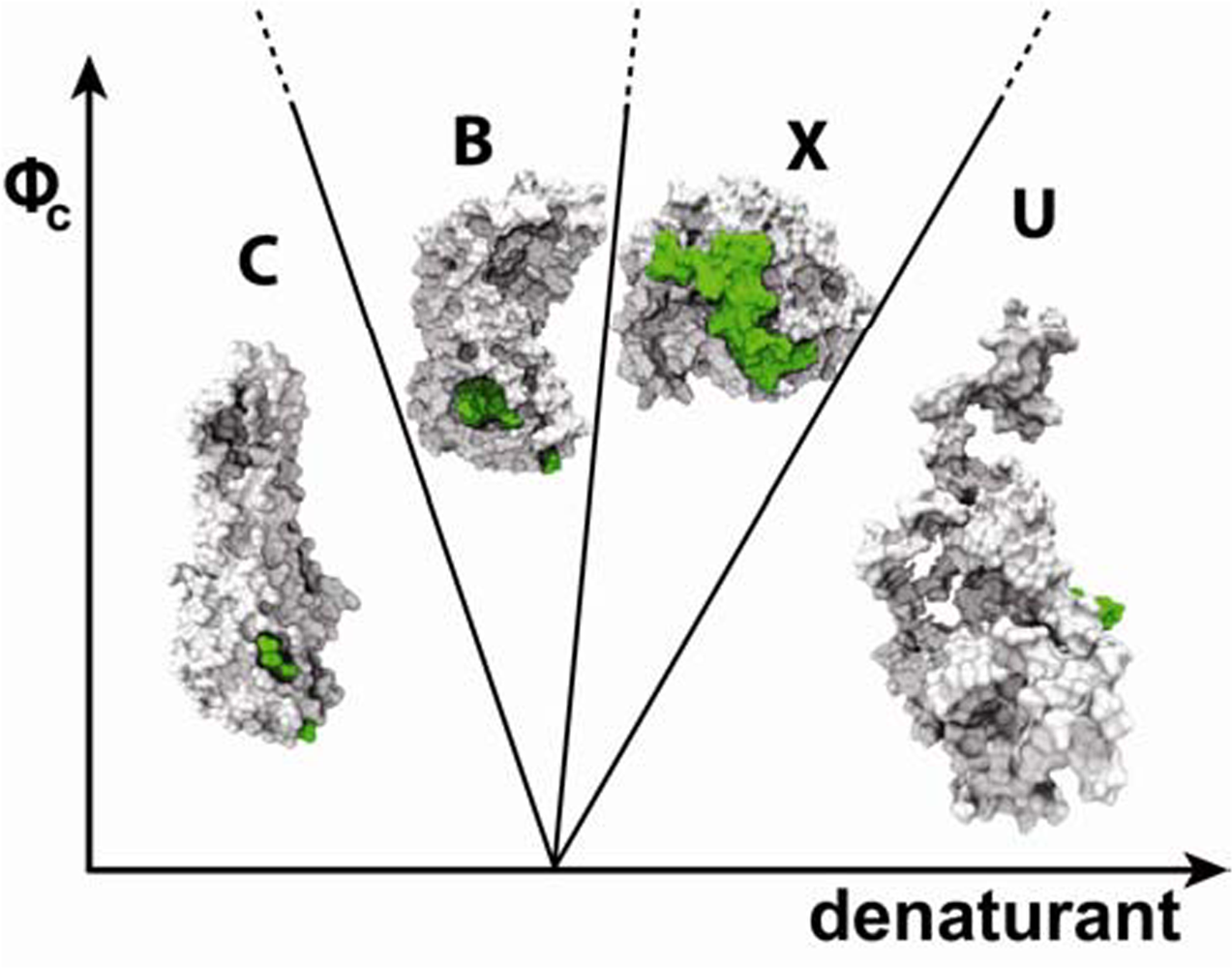
6. Role of shape in defining protein-folding speed
7. Conclusions
Appendix
a) Coarse-grained models
b) Energy functions for coarse-grained side chain interactions
c) Simulation techniques
Acknowledgments
References and Notes
- van den Berg, B; Ellis, RJ; Dobson, CM. Effects of macromolecular crowding on protein folding and aggregation. Embo J 1999, 18, 6927–33. [Google Scholar]
- Rivas, G; Ferrone, F; Herzfeld, J. Life in a crowded world. EMBO Rep 2004, 5, 23–27. [Google Scholar]
- Record, MT; Courtenay, ES; Cayley, S; Guttman, HJ. Biophysical compensation mechanism buffering E.coli protein-nucleic acid interactions against changing environment. Trends Biochem. Sci 1998, 23, 190–194. [Google Scholar]
- Ellis, RJ; Minton, AP. Join the crowd. Nature 2003, 425, 27–28. [Google Scholar]
- Minton, AP. Excluded volume as a determinant of macromolecular structure and reactivity. Biopolymers 1981, 20, 2093–2120. [Google Scholar]
- Zhou, H-X; Rivas, G; Minton, AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys 2008, 37, 375–397. [Google Scholar]
- Winzor, DJ; Wills, PR. Molecular crowding effects of linear polymers in protein solutions. Biophys. Chem 2006, 119, 186–195. [Google Scholar]
- Zimmerman, SB; Minton, AP. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct 1993, 22, 27–65. [Google Scholar]
- Minton, AP. Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: Macromolecular crowding and protein stability revisited. Biophys. J 2005, 88, 971–985. [Google Scholar]
- Zhou, HX. Loops, linkages, rings, catenanes, cages, and crowders: Entropy based strategies for stabilizing proteins. Acc. Chem. Res 2004, 37, 123–130. [Google Scholar]
- Hermans, J. Excluded-volume theory of polymer-protein interactions based on polymer chain statistics. J. Chem. Phys 1982, 77, 2193–2203. [Google Scholar]
- Eisenberg, H. Thermodynamics and the structure of biological macromolecules. Eur. J. Biochem 1990, 187, 7–22. [Google Scholar]
- Cheung, MS; Klimov, D; Thirumalai, D. Molecular crowding enhances native state stability and refolding rates. Proc. Natl. Acad. Sci. USA 2005, 102, 4753–4758. [Google Scholar]
- Laurent, TC; Ogston, AG. The interaction between polysaccharides and other macromolecules. 4. The osmotic pressure of mixtures of serum albumin and hyaluronic acid. Biochem. J 1963, 89, 249–253. [Google Scholar]
- Laurent, TC. Enzyme reactions in polymer media. Eur. J. Biochem 1971, 21, 498–506. [Google Scholar]
- van den Berg, B; Wain, R; Dobson, CM; Ellis, RJ. Macromolecular crowding perturbs protein refolding kinetics: Implications for folding inside the cell. Embo. J 2000, 19, 3870–3875. [Google Scholar]
- Uversky, VN; Bower, KS; Li, J; Fink, AL. Accelerated alpha-synuclein fibrillation in crowded milieu. FEBS Lett 2002, 515, 99–103. [Google Scholar]
- Ai, X; Zhou, Z; Bai, Y; Choy, W-Y. 15N NMR spin relaxation dispersion study of the molecular crowding effects on protein folding under native conditions. J. Am. Chem. Soc 2006, 128, 3916–3917. [Google Scholar]
- Patel, CN; Noble, SM; Weatherly, GT; Tripathy, A; Winzor, DJ; Pielak, GJ. Effects of molecular crowing by saccharides on alpha-chymotrypsin dimerization. Protein Sci 2002, 11, 997–1003. [Google Scholar]
- Davis-Searles, PR; Saunders, AJ; Erie, DA; Winzor, DJ; Pielak, GJ. Interpreting the effect of small uncharged solutes on protein-folding equilibria. Annu. rev. Biophys. Biomol. Struct 2001, 30, 271–306. [Google Scholar]
- Schnell, S; Turner, TE. Reaction kinetics in intracellular environments with macromolecular crowding: Simulations and rate laws. Prog. Biophys. Mol. Biol 2004, 85, 235–260. [Google Scholar]
- Sasahara, K; McPhie, P; Minton, AP. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol 2003, 326, 1227–1237. [Google Scholar]
- Qu, Y; Bolen, D. Efficacy of macromolecular crowding in forcing proteins to fold. Biophys Chem 2002, 101–102, 155–165. [Google Scholar]
- Dedmon, MM; Patel, CN; Young, GB; Pielak, GJ. FlgM gains structure in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 12681–12684. [Google Scholar]
- Edmond, E; Ogston, AG. An approach to the study of phase separation in ternary aqueous systems. Biochem. J 1968, 109, 569–576. [Google Scholar]
- Shearwin, KE; Winzor, DJ. Thermodynamic nonideality in macromolecular solutions. Eur. J. Biochem 1990, 190, 523–529. [Google Scholar]
- Jones, K; Guidry, J; Wittung-Stafshede, P. The largest protein observed to fold by two-state kinetic mechanism does not obey contact-order correlation. J. Am. Chem. Soc 2003, 125, 9096–9097. [Google Scholar]
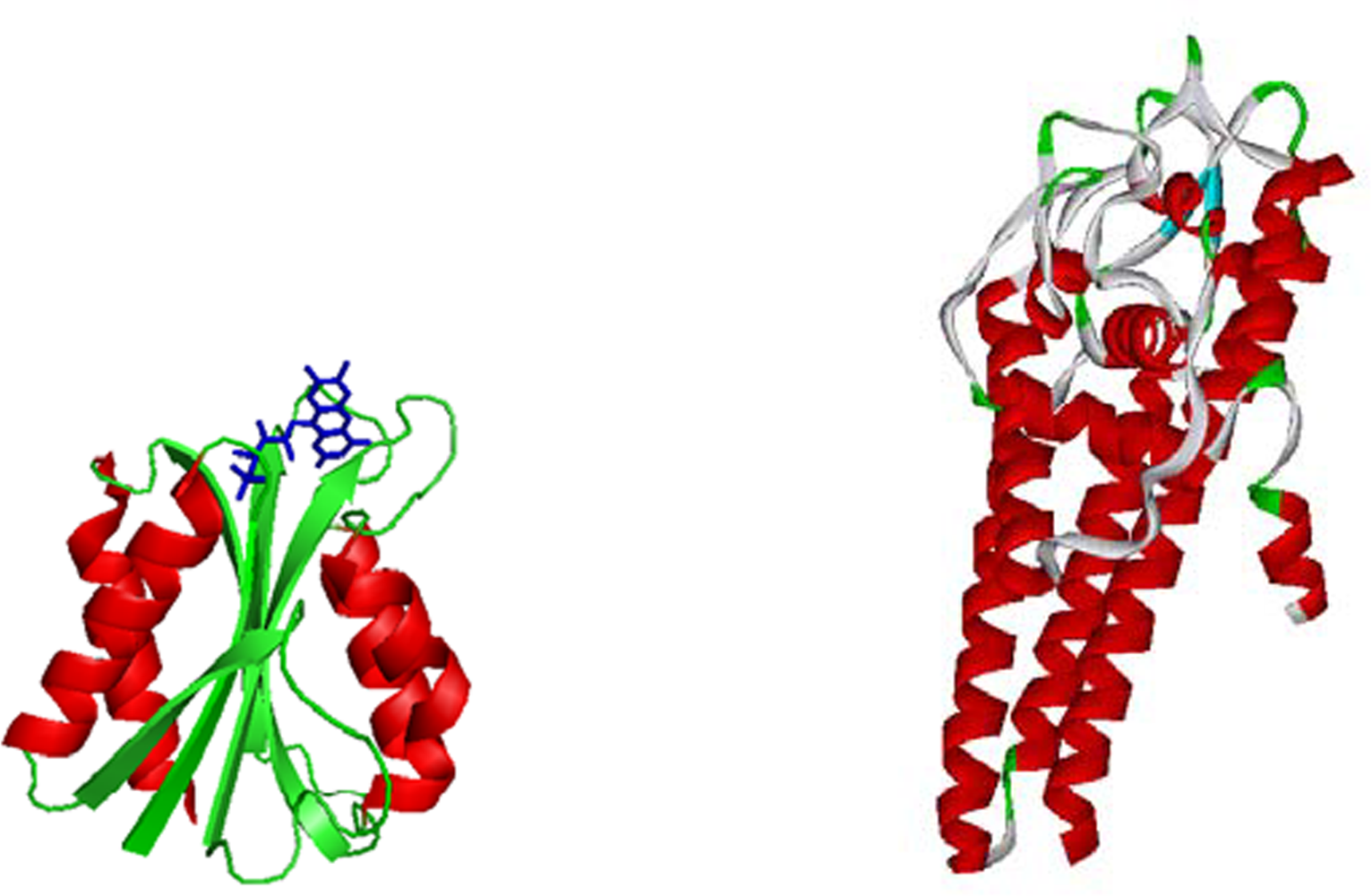
- Eicken, C; Sharma, V; Klabunde, T; Lawrenz, MB; Hardham, JM; Norris, SJ; Sacchettini, JC. Crystal structure of Lyme Disease Variable Surface Antigen VlsE of Borrelia burgdorferi. J. Biol. Chem 2002, 277, 21691–21696. [Google Scholar]
- Steensma, E; van Mierlo, CPM. Structural characterisation of apoflavodoxin shows that the location of the stable nucleus differs among proteins with a flavodoxin-like topology. J. Mol. Biol 1998, 282, 653–666. [Google Scholar]
- Muralidhara, BK; Chen, M; Ma, J; Wittung-Stafshede, P. Effect of inorganic phosphate on FMN binding and loop flexibility in Desulfovibrio desulfuricans apo-flavodoxin. J. Mol. Biol 2005, 349, 87–97. [Google Scholar]
- Muralidhara, BK; Wittung-Stafshede, P. Thermal unfolding of apo and holo Desulfovibrio desulfuricans Flavodoxin: Cofactor stablizes folded and intermediate states. Biochemistry 2004, 43, 12855–12864. [Google Scholar]
- Onuchic, JN; Luthey-Schulten, Z; Wolynes, PG. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem 1997, 48, 545–600. [Google Scholar]
- Bryngelson, JD; Wolynes, PG. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar]
- Onuchic, J; Wolynes, PG. Theory of protein folding. Curr. Opin. Struct. Biol 2004, 14, 70–75. [Google Scholar]
- Cheung, MS; Chavez, LL; Onuchic, JN. The Energy landscape for protein folding and possible connections to function. Polymer 2004, 45, 547–555. [Google Scholar]
- Stagg, L; Zhang, S-Q; Cheung, MS; Wittung-Stafshede, P. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc. Natl. Acad. Sci. USA 2007, 104, 18976–18981. [Google Scholar]
- Samiotakis, A; Homouz, D; Cheung, MS. SCAAL: A robust, accurate, and high-efficient all-atomistic protein reconstruction method from low-resolution protein models. submitted, 2008.
- Homouz, D; Perham, M; Samiotakis, A; Cheung, MS; Wittung-Stafshede, P. Crowded, cell-like environment induces shape changes in aspherical protein. Proc. Natl. Acad. Sci. USA 2008, 105, 11754–11759. [Google Scholar]
- Minton, AP. Effect of a concentrated “inert” macromolecular cosolute on the stability of a globular protein with respect to denaturation by heat and by chaotropes: A statistical-thermodynamic model. Biophys. J 2000, 78, 101–109. [Google Scholar]
- Perham, M; Stagg, L; Wittung-Stafshede, P. Macromolecular crowding increases structural content of folded proteins. FEBS Lett 2007, 581, 5065–5069. [Google Scholar]
- Homouz, D; Stagg, L; Wittung-Stafshede, P; Cheung, MS. Macromolecular crowding modulates folding mechanism of α/β protein apoflavodoxin. Biophys. J 2009, 96, 671–680. [Google Scholar]
- Cheung, MS; Finke, JM; Callahan, B; Onuchic, JN. Exploring the interplay of topology and secondary structural formation in the protein folding problem. J. Phys. Chem. B 2003, 107, 11193–11200. [Google Scholar]
- Go, N; Abe, H. Randomness of the process of protein folding. Int. J. Pept. Protein Res 1983, 22, 622–632. [Google Scholar]
- Bueno, M; Ayuso-Tejedor, S; Sancho, J. Do proteins with similar folds have similar transition state structures? A diffuse transition state of the 169 residue apoflavodoxin. J. Mol. Biol 2006, 359, 813–824. [Google Scholar]
- Shea, JE; Onuchic, JN; Brooks, CL, 3rd. Exploring the origins of topological frustration: Design of a minimally frustrated model of fragment B of protein A. Proc. Natl. Acad. Sci. USA 1999, 96, 12512–12517. [Google Scholar]
- Clementi, C; Nymeyer, H; Onuchic, JN. Topological and energetic factors: What determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol 2000, 298, 937–953. [Google Scholar]
- Bowles, RK; Speedy, RJ. Cavities in the hard-sphere crystal and fluid. Mol. Phys 1994, 83, 113–125. [Google Scholar]
- Asakura, S; Oosawa, F. Interaction between particles suspended in solutions of macromolecules. J. Polym. Sci 1958, 33, 183–192. [Google Scholar]
- Plotkin, SS; Onuchic, JN. Investigation of routes and funnels in protein folding by free energy functional methods. Proc. Natl. Acad. Sci. USA 2000, 97, 6509–6514. [Google Scholar]
- Chavez, LL; Gosavi, S; Jennings, PA; Onuchic, JN. Multiple routes lead to the native state in the energy landscape of the beta-trefoil family. Proc. Natl. Acad. Sci. USA 2006, 103, 10254–10258. [Google Scholar]
- Liang, FT; Steere, AC; Marques, AR; Johnson, BJB; Miller, JN; Phillips, MT. Sensitive and specific serodiagnosis of Lyme disease by enzyme-linken immunosorbent assay with a peptide based on an immunodominant conserved region of borrelia burgdorferi VlsE. J. Clin. Microbiol 1999, 37, 3990–3996. [Google Scholar]
- Manning, MC; Illangasekare, M; Woody, RW. Cirular dichroism studies of distorted alpha-helices, twisted beta-sheets, and beta turns. Biophys. Chem 1988, 31, 77–86. [Google Scholar]
- Philipp, MT; Bowers, LC; Fawcett, PT; Jacobs, MB; Liang, FT; Marques, AR; Mitchell, PD; Purcell, JE; Ratterree, MS; Straubinger, RK. Antibody response to IR6, a conserved immunodominant region of the VlsE lipoprotein, wanes rapidly after antibiotic treatment of Borrelia burgdorderi infection in experimental animals and in humans. J. Infect. Dis 2001, 184, 870–878. [Google Scholar]
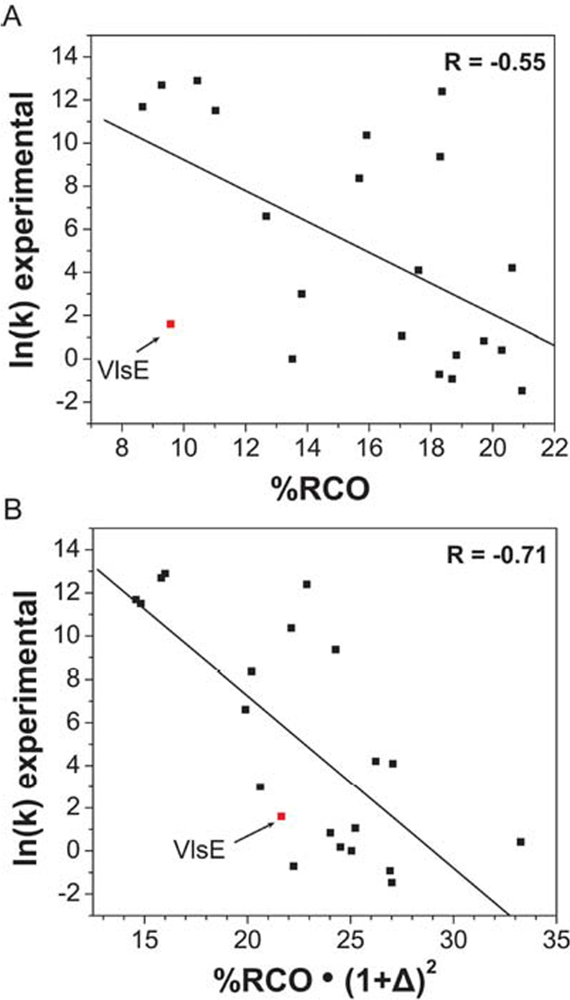
- Plaxco, KW; Simons, KT; Baker, D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol 1998, 277, 985–994. [Google Scholar]
- Ouyang, Z; Liang, J. Predicting protein folding rates from geometric contact and amino acid sequences. Protein Sci 2008, 17, 1256–1263. [Google Scholar]
- Du, F; Zhou, Z; Mo, ZY; Shi, JZ; Chen, J; Liang, Y. Mixed macromolecular crowding accelerates the refolding of rabbit muscle creatine kinase: Implications for protein folding in physiological environment. J. Mol. Biol 2006, 364, 469–482. [Google Scholar]
- Zhou, H-X. Effect of mixed macromolecular crowding agents on protein folding. Proteins 2008, 72, 1109–1113. [Google Scholar]
- Klimov, DK; Thirumalai, D. Native topology determines force-induced unfolding pathways in globular proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 7254–7259. [Google Scholar]
- Betancourt, MR; Thirumalai, D. Pair potentials for protein folding: Choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci 1999, 8, 361–369. [Google Scholar]
- Case, D; Pearlman, DA; Caldwell, JW; Cheatham, TE; Ross, WS; Simmerling, CL; Darden, TA; Merz, KM; Stanton, RV; Cheng, AL; et al. AMBER6, UCSF, 1999.
- Sugita, Y; Okamoto, Y. Replica-exchange molecular dynamics methods for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- Kumar, S; Bouzida, D; Swendsen, RH; Kollman, PA; Rosenberg, JM. The weighted histogram analysis method for free-energy calculations on biomolecules I. The method. J. Comput. Chem 1992, 13, 1011–1021. [Google Scholar]
- Chodera, JD; Swope, WC; Pitera, JW; Seok, C; Dill, KA. Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. J. Chem. Theory Comput 2007, 3, 26–41. [Google Scholar]




© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Samiotakis, A.; Wittung-Stafshede, P.; Cheung, M.S. Folding, Stability and Shape of Proteins in Crowded Environments: Experimental and Computational Approaches. Int. J. Mol. Sci. 2009, 10, 572-588. https://doi.org/10.3390/ijms10020572
Samiotakis A, Wittung-Stafshede P, Cheung MS. Folding, Stability and Shape of Proteins in Crowded Environments: Experimental and Computational Approaches. International Journal of Molecular Sciences. 2009; 10(2):572-588. https://doi.org/10.3390/ijms10020572
Chicago/Turabian StyleSamiotakis, Antonios, Pernilla Wittung-Stafshede, and Margaret S. Cheung. 2009. "Folding, Stability and Shape of Proteins in Crowded Environments: Experimental and Computational Approaches" International Journal of Molecular Sciences 10, no. 2: 572-588. https://doi.org/10.3390/ijms10020572