2.1. Genetic Diversity and Differentiation
Sequence data from nine nuclear loci was obtained through direct sequencing and molecular cloning techniques. Overall aligned sequence length ranged from 454 to 1,326 bp, with an average size of 725 bp (6,525 bp total). Basic summary statistics are given in
Table 1. Average per-bp silent π and θ values, respectively, were 0.036 and 0.051 for
H. petiolaris, and 0.041 and 0.048 for
H. neglectus. The large majority of the genetic variation is distributed within species with very little divergence between species, as indicated by the average net divergence across loci, which was 0.14% (range 0–0.46%). A summary of the four types of substitutions described in Wakeley and Hey [
16] are given in
Table 2. These substitutions are the number of sites polymorphic in each species, in both species, and the number of differences that are fixed between species. The degree of variation within each species is again showcased using these statistics, with an average of 45 and 23 polymorphisms unique to
H. petiolaris and
H. neglectus, respectively (based on pairwise sequence diversity levels within each species, the difference between species in the number of unique polymorphisms reflects sample size differences rather than overall genetic diversity differences). Surprisingly, none of the 847 polymorphisms in the overall dataset is distributed as a fixed difference between
H. petiolaris and
H. neglectus.
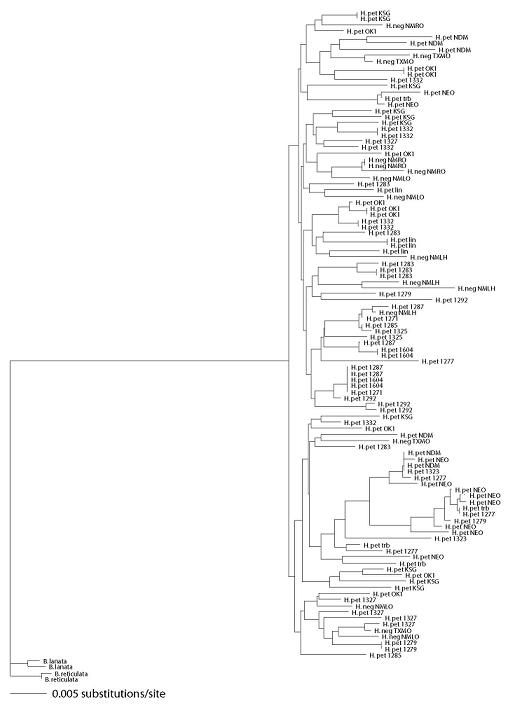
In AMOVAs, an average of 70.7% of variation was found within regions (P values for all nine loci <0.0001), compared to 25.9% variation found among regions (P values across loci range from <0.0001 to 0.0031) within species and 3.4% variation found between species (P values range across loci from 0.001 to 0.504; three of nine loci significant at the 0.05 level). In neighbor joining trees using Bahiopsis lanata and B. reticulata as outgroups, H. petiolaris and H. neglectus haplotypes had a polyphyletic relationship at all nine loci. Neighbor joining trees and 50% majority-rule bootstrap consensus neighbor joining trees for all nine loci are given in Supplementary File S1.
Results for Tajima’s D and Fu’s Fs tests are given in
Table 3. In
H. petiolaris, two loci were significantly negative using Tajima’s test, and all nine loci were significant using Fu’s test. The number of significant results for Tajima and Fu’s tests decreased to zero and eight, respectively, after Bonferroni correction. For
H. neglectus, no loci were significant for Tajima’s test, but seven loci were significant for Fu’s test (four after Bonferroni correction). Fs is considered more sensitive to population size change, with negative values indicating population growth, while D is considered more sensitive to selection [
17]. Given that, our results are most consistent with selective neutrality at all loci and population growth in both species. HKA tests were not significant across all loci, also indicating selective neutrality.
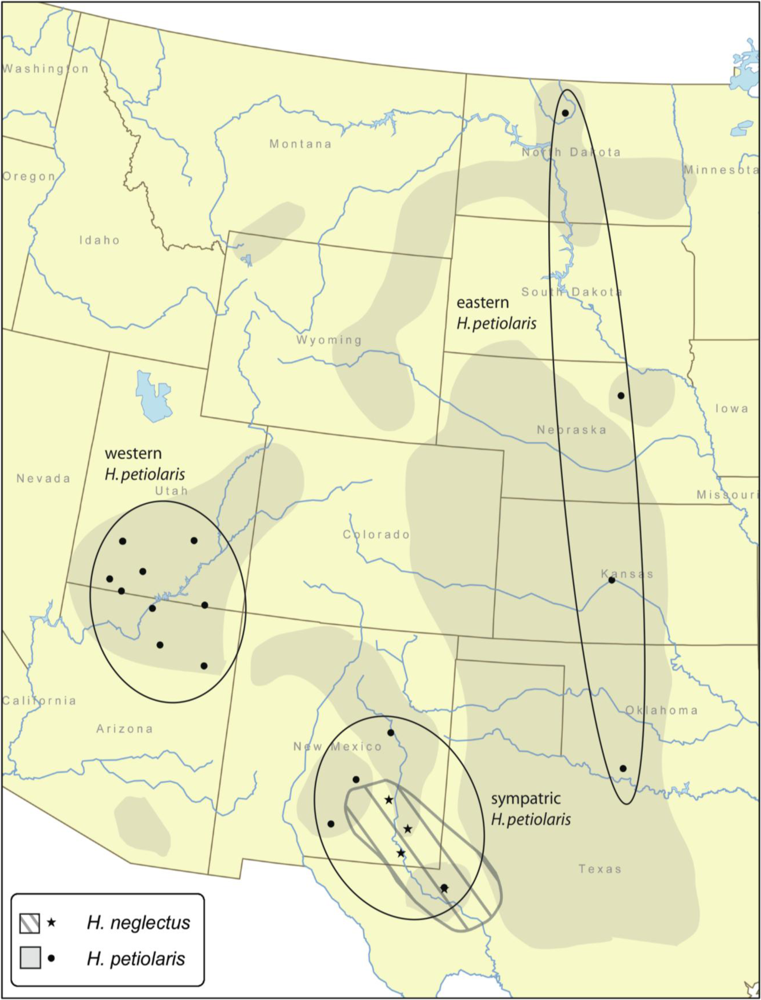
Mantel tests were performed on H. petiolaris populations and on all populations of both species together. Correlations between geographic and genetic distance averaged across loci are very low and non-significant both for H. petiolaris individually and for both species together (0.004 and 0.028, respectively). No individual locus shows a significant positive correlation for H. petiolaris individually, and a single locus, JLS2899, is significant for both species together.
2.2. Effective Population Sizes and Gene Flow Rates
Estimates of effective population sizes and long-term gene flow rates made using MIGRATE are given in
Tables 4 and
5, respectively. Modal population size estimates for the three
H. petiolaris regions range from 240,000 to 640,000. In contrast, the modal population size estimate for
H. neglectus is roughly 2.3 million; the low end of the 95% confidence interval is close to one million. This estimate is somewhat smaller than a previous estimate of the
H. petiolaris species-wide effective size [
18], but it is still far larger than would be expected for an isolated species with the geographic range and census size of
H. neglectus.
Likewise, gene flow estimates are not consistent with increased isolation between H. neglectus and H. petiolaris relative to isolation among regions within H. petiolaris. Modal gene flow estimates are Nem = 0.01 (the smallest value possible based on bin size), although confidence intervals are quite wide, with a lower bound of zero in all cases and upper bounds ranging from 3.8 to 5.5. Posterior distributions of gene flow estimates between H. neglectus and various H. petiolaris groups are broadly overlapping with posteriors for gene flow within H. petiolaris. When the six gene flow estimates involving H. neglectus are compared to the six estimates within H. petiolaris, the former have slightly higher average confidence interval upper bounds (4.57 vs. 4.29), median values (1.55 vs. 1.49) and mean values (0.96 vs. 0.78). In addition, although the confidence intervals are broadly overlapping, confidence interval upper bounds, median values, and mean values are slightly higher for interspecific comparisons involving the sympatric H. petiolaris region vs. interspecific comparisons involving the allopatric (eastern and western) H. petiolaris regions. This pattern is expected if the two species are exchanging genes in sympatry.
2.3. Species Status of H. neglectus
Defining distinct species has long been a goal of evolutionary biologists as well as natural historians. Although a modern consensus definition of a species is still a topic of debate [
19,
20], operational methods of identifying species have historically utilized morphological or ecological traits in addition to crossing data. Investigators have incorporated cytological and molecular data as it has become available, and the value of examining multiple sources of data is generally recognized [
1,
21,
22]. On occasion, the addition of molecular data has led to substantial reevaluation of species boundaries and relationships [e.g.,
23,
24].
Population genetic theory predicts that species with restricted ranges (and presumably concomitant small population sizes) should have less genetic variation than their widespread counterparts. This is expected mainly due to the increased importance of genetic drift in small populations [
25]. When comparing the narrowly distributed sunflower species
H. neglectus with its widespread sister species
H. petiolaris, we do not observe this pattern at any of the nine loci examined here. Because of the vast differences in species’ ranges and census population sizes, we expected far more genetic variation in
H. petiolaris. However, levels of genetic variation in
H. neglectus were comparable to (and in some cases
higher than) those in
H. petiolaris. In addition we found very little genetic divergence between the two species, as well as comparable levels of interspecific and intraspecific gene flow.
Three possible scenarios may explain the surprisingly high levels of genetic variation in H. neglectus. If H. neglectus is the product of a recent split from the more widespread H. petiolaris and H. neglectus populations are currently still undergoing lineage sorting, polyphyletic relationships among species may be expected. However given the restricted range of H. neglectus it is unlikely that the species would be able to sustain such high levels of variation that are observed for a significant length of time. Similarly, if H. neglectus was much more widespread than it is at present, high levels of variation may still be found in the much smaller species range. However, there is no evidence that this is the case. The values of Fu’s Fs and Tajima’s D presented here are more consistent with recent H. neglectus population growth rather than decline, making this scenario unlikely as well.
A more plausible explanation for the observed pattern is high levels of ongoing gene flow between the two nominal species. In his initial description of
H. neglectus, Heiser [
13] reported artificial hybrids with varying degrees of fertility (measured by pollen staining viability of 9–80%). In addition, apparent hybrids are not uncommon in areas of range overlap [
9,
10]. A previous study examining pollen viability in hybrids between
H. annuus and
H. petiolaris show drastically lower levels of viability [average 4.8%–26]. Despite these low F1 pollen viability levels, recent studies have shown evidence of long-term, ongoing introgression between
H. annuus and
H. petiolaris in both directions [
18,
27]. Given the relatively higher F1 pollen viabilities between
H. neglectus and
H. petiolaris, pollen sterility is unlikely to prevent significant introgression in sympatry.
Our modal gene flow estimates from MIGRATE among all regions in both species are surprisingly low, although confidence intervals are very broad and encompass very high gene flow levels as well (
Table 5). These results are inconsistent with higher estimates of gene flow within
H. petiolaris (N
em ∼ 1) previously reported [
28–
30]. The Bayesian implementation of MIGRATE can have a downward bias in gene flow estimates for a wide range of θ values, including the range of values estimated here [
31]; this may explain the discrepancy we see between estimates made using the different methods. However, we see no obvious reason why intraspecific estimates would be more strongly biased than interspecific estimates, so we do not expect the relative levels of interspecific
vs. intraspecific gene flow to be affected. The Bayesian implementation of MIGRATE estimates effective sizes well [substantially better than the maximum likelihood implementation–31], suggesting that our estimate of
H. neglectus’ effective size is reliable; as mentioned above this estimate is not consistent with an isolated species with a very small geographic range and census size, except under very unrealistic demographic conditions.
Within
H. petiolaris there are two recognized subspecies, ssp.
petiolaris and ssp.
fallax [
13], distinguished by some minor phenotypic differences as well as at least one chromosomal rearrangement; in addition, there is some cytological variation within ssp.
petiolaris [
32]. Generally, ssp.
petiolaris is found throughout the Great Plains up to the Rocky Mountains, while ssp.
fallax is found in the southwestern United States [
9]; the range of
H. neglectus overlaps or nearly overlaps with each
H. petiolaris subspecies. Crosses between cytological races of
H. petiolaris show varying degrees of fertility loss relative to crosses within races [
32]; interestingly, hybrids between
H. neglectus and both subspecies of
H. petiolaris show pollen viabilities within the range of variation seen in crosses between the
H. petiolaris subspecies and cytological races [
9,
13,
32].
Based on the comparable levels of genetic variation in both species and similar inter- and intraspecific gene flow and effective population size estimates from MIGRATE, the validity of
H. neglectus’ distinctness as a species should be more closely examined. Both species examined show high levels of genetic variation, which is in accordance with previous studies of
Helianthus species [
18]. Such high levels of variation in an endemic species may not be surprising only under a restrictive set of demographic assumptions (see above). Evidence for severe population size decline or extraordinarily high levels of gene flow are not borne out in the results presented here. The lack of clear genetic divergence or geographic barriers and similar levels of estimated intra and interspecific gene flow between species suggests that the gene pool of both
H. neglectus and
H. petiolaris is most appropriately viewed as one large collection of alleles.
A number of studies [
27,
33,
34] have recently shown that the genomes of the more distantly related
H. petiolaris and
H. annuus are highly permeable to gene flow, with large portions showing very little genetic divergence in the roughly two million years since the species’ initial divergence. Nonetheless, these two species remain morphologically and ecologically divergent through the action of multiple strong reproductive barriers. It is possible that a similar phenomenon is occurring here, although it is unlikely for several reasons. While there is some degree of reproductive isolation between
H. neglectus and
H. petiolaris due to chromosomal differentiation and possibly some genic factors, isolating barriers between these two species are far lower than barriers between
H. petiolaris and
H. annuus; in fact they are comparable to barriers between different chromosome races within
H. petiolaris. Second, while rates of introgression between
H. petiolaris and
H. annuus appear to be exceptionally high for two distinct species, they are still considerably lower than levels of gene flow within either species. Third,
H. neglectus and
H. petiolaris are far more similar to each other morphologically, ecologically, and chromosomally than are
H. petiolaris and
H. annuus, which differ by a minimum of 11 large-scale rearrangements [
35]. And fourth, both
H. annuus and
H. petiolaris are very widespread and have species-wide population census sizes well into the millions of individuals, consistent with their high levels of genetic diversity and large effective population sizes. In contrast,
H. neglectus is limited to a very small range, and while its census size is not known precisely, it is probably under our estimated effective size of 2.3 million. While it is possible under certain conditions for effective size to be larger than census size [
36], those conditions are not realistic in this case; the huge amount of genetic variation in
H. neglectus is not consistent with any substantial degree of genetic isolation.



{kind=link}
{kind=link}
{kind=link}