Insight into a Novel p53 Single Point Mutation (G389E) by Molecular Dynamics Simulations

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
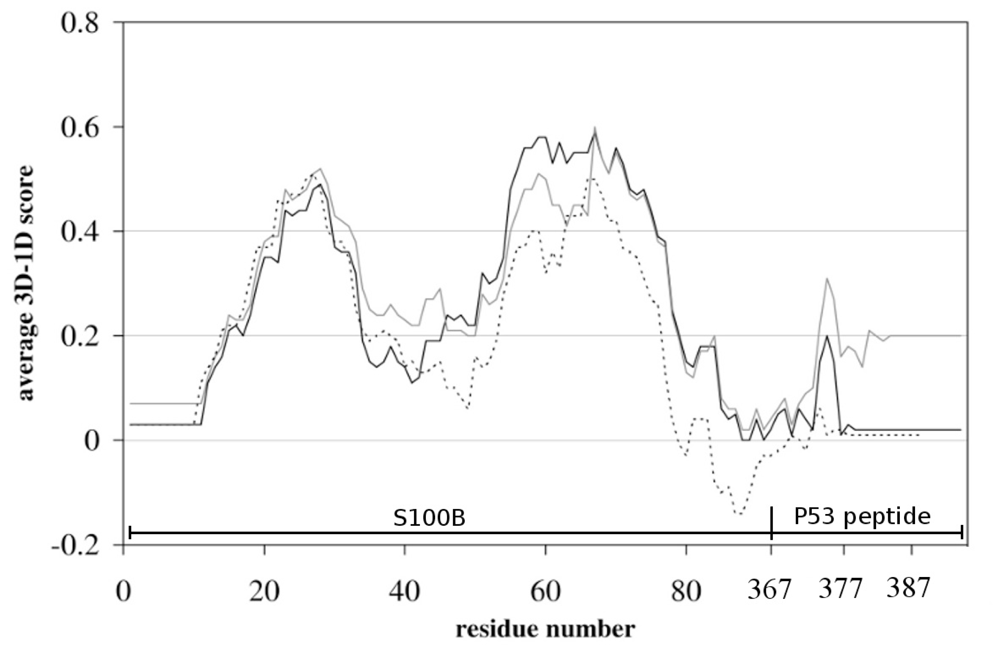
2.1. Molecular Modeling
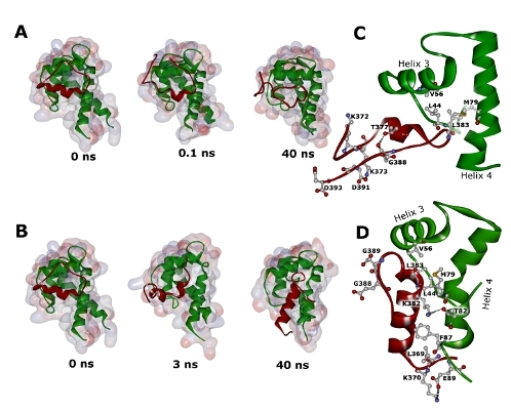
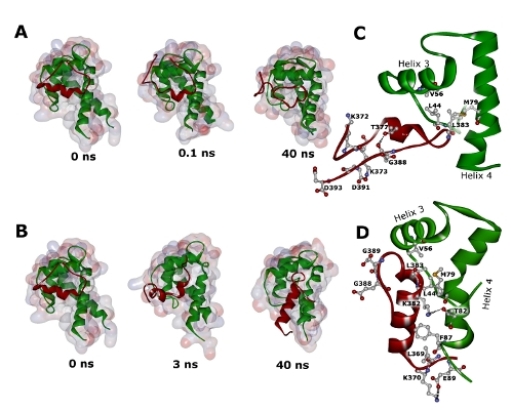
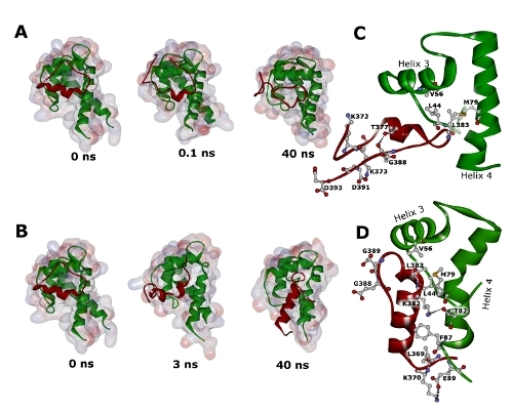
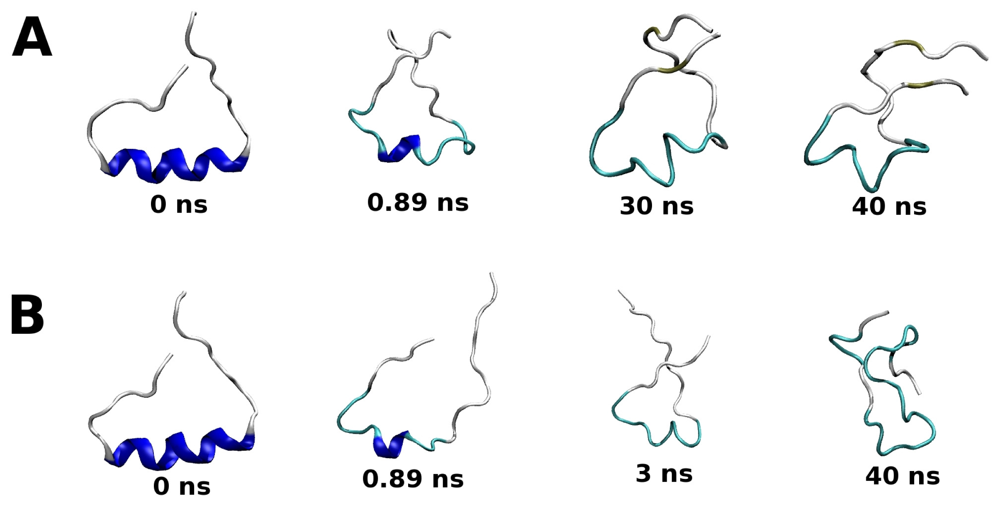
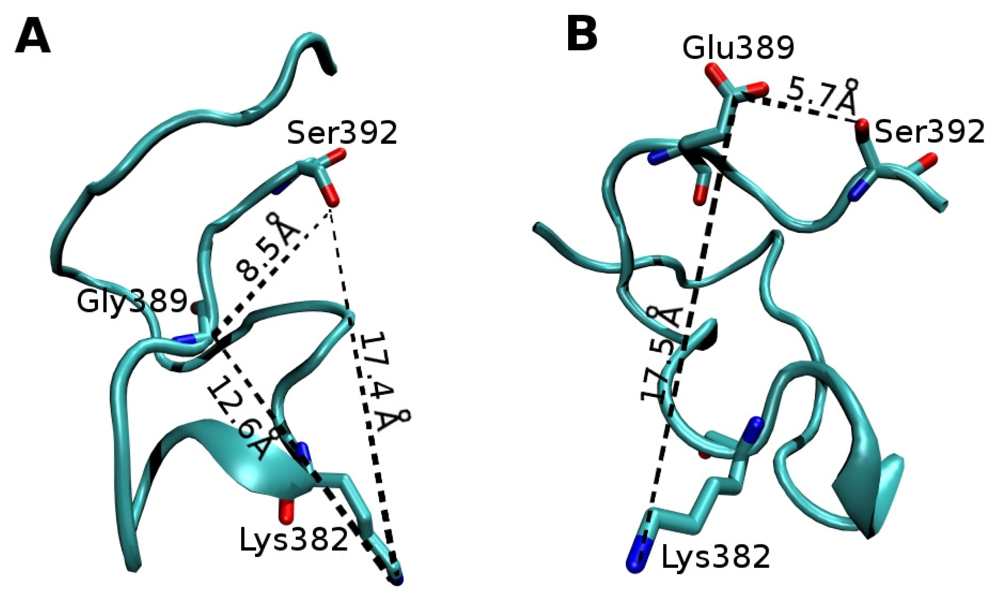
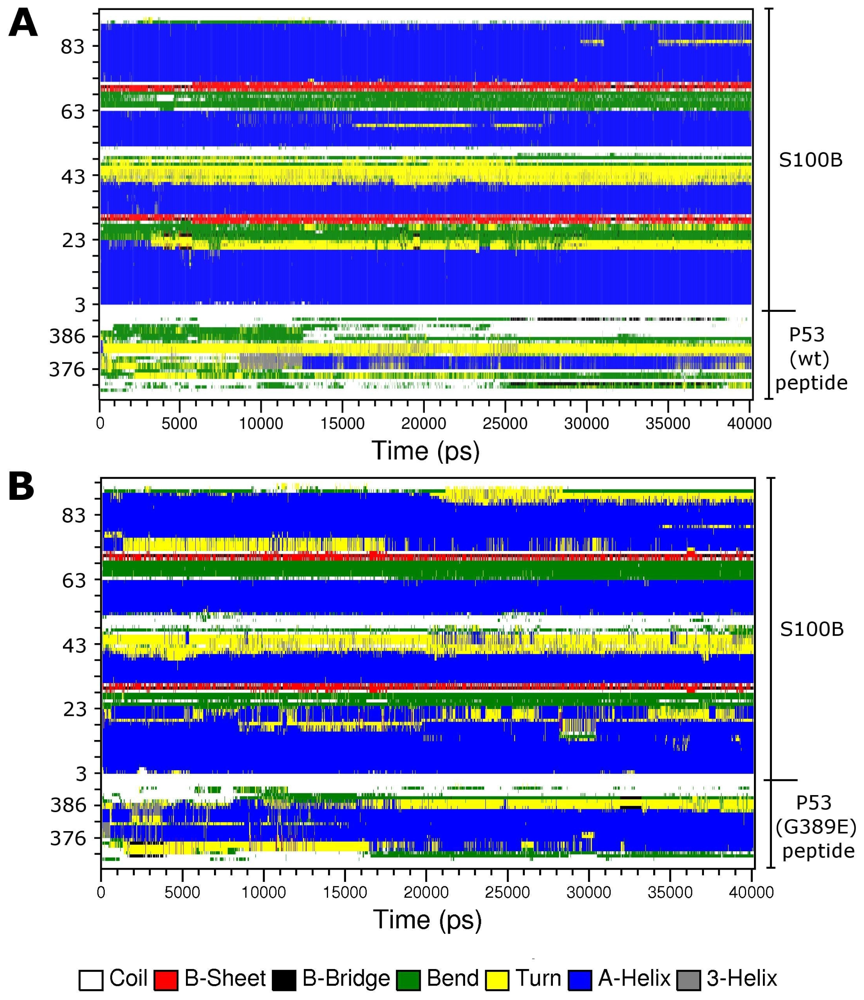
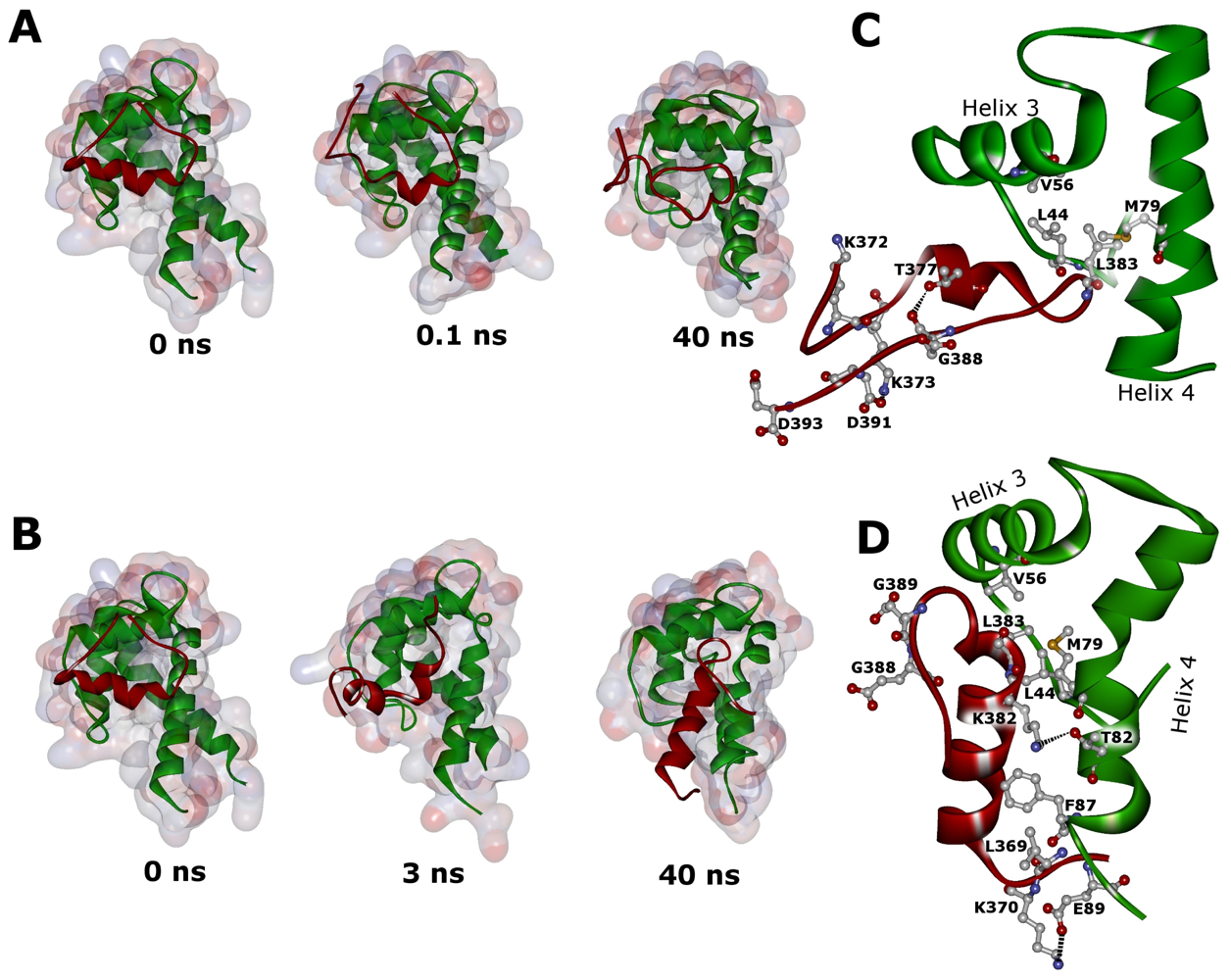
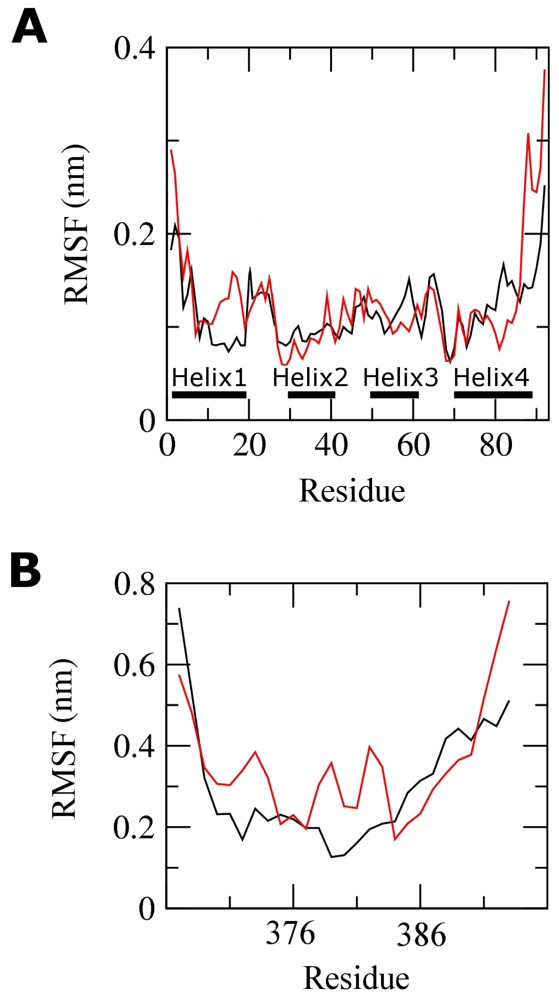
2.2. Molecular Dynamics Simulations of WT and G389E Unbound Peptides
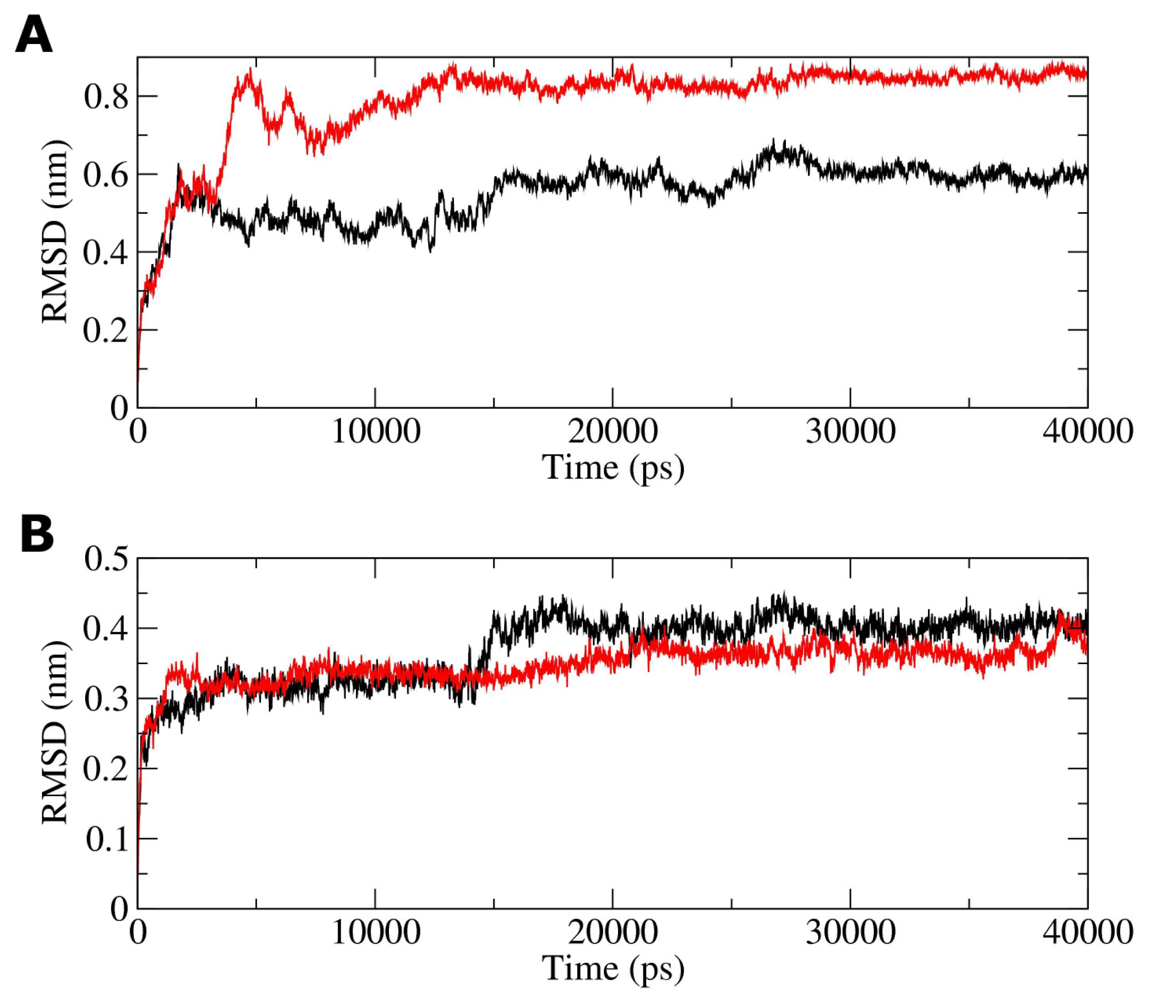
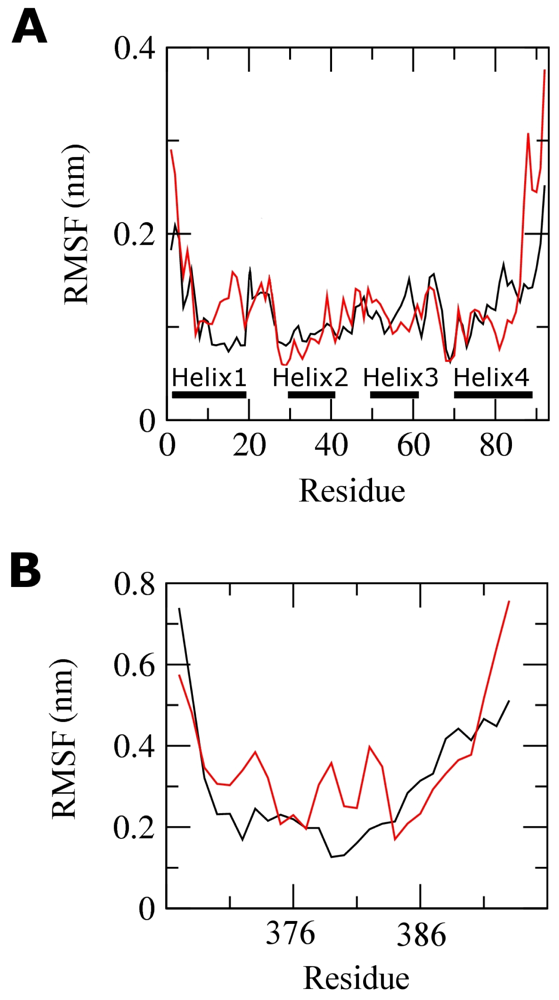
2.3. Molecular Dynamics Simulations of WT and G389E Peptides Bound to S100B
3. Experimental Section
3.1. Molecular Modeling
3.2. Molecular Dynamics Simulations
4. Conclusions
References
- Dyson, H; Wright, P. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol 2002, 12, 54–60. [Google Scholar]
- Dawson, R; Müller, L; Dehner, A; Klein, C; Kessler, H; Buchner, J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol 2003, 332, 1131–1141. [Google Scholar]
- Vogelstein, B; Lane, D; Levine, A. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar]
- Brachmann, R; Yu, K; Eby, Y; Pavletich, N; Boeke, J. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J 1998, 17, 1847–1859. [Google Scholar]
- Kato, S; Han, S; Liu, W; Otsuka, K; Shibata, H; Kanamaru, R; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci USA 2003, 100, 8424–8429. [Google Scholar]
- Ahn, J; Prives, C. The C-terminus of p53: The more you learn the less you know. Nat. Struct. Biol 2001, 8, 730–732. [Google Scholar]
- Hupp, T; Lane, D. Allosteric activation of latent p53 tetramers. Curr. Biol 1994, 4, 865–875. [Google Scholar]
- Anderson, M; Woelker, B; Reed, M; Wang, P; Tegtmeyer, P. Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: Implications for regulation. Mol. Cell. Biol 1997, 17, 6255–6264. [Google Scholar]
- Hupp, T; Meek, D; Midgley, C; Lane, D. Regulation of the specific DNA binding function of p53. Cell 1992, 71, 875–886. [Google Scholar]
- Nichols, N; Matthews, K. Human p53 phosphorylation mimic, S392E, increases nonspecific DNA affinity and thermal stability. Biochemistry 2002, 41, 170–178. [Google Scholar]
- Sakaguchi, K; Sakamoto, H; Lewis, M; Anderson, C; Erickson, J; Appella, E; Xie, D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997, 36, 10117–10124. [Google Scholar]
- Kim, Y; Park, B; Kim, D; Kim, W; Kim, S; Oh, K; Lim, J; Kim, J; Park, C; Park, S. Modification of serine 392 is a critical event in the regulation of p53 nuclear export and stability. FEBS Lett 2004, 572, 92–98. [Google Scholar]
- Soussi, T; Béroud, C. Significance of TP53 mutations in human cancer: A critical analysis of mutations at CpG dinucleotides. Hum. Mutat 2003, 21, 192–200. [Google Scholar]
- Concolino, P; Satta, M; Santonocito, C; Carrozza, C; Rocchetti, S; Ameglio, F; Giardina, E; Zuppi, C; Capoluongo, E. Linkage between I172N mutation, a marker of 21-hydroxylase deficiency, and a single nucleotide polymorphism in Int6 of CYP21B gene: A genetic study of Sardinian family. Clin. Chim Acta 2006, 364, 298–302. [Google Scholar]
- Pan, Y; Ma, B; Venkataraghavan, R; Levine, A; Nussinov, R. In the quest for stable rescuing mutants of p53: Computational mutagenesis of flexible loop L1. Biochemistry 2005, 44, 1423–1432. [Google Scholar]
- Karplus, M. Molecular dynamics of biological macromolecules: A brief history and perspective. Biopolymers 2003, 68, 350–358. [Google Scholar]
- Rustandi, R; Baldisseri, D; Weber, D. Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat. Struct. Biol 2000, 7, 570–574. [Google Scholar]
- Lin, J; Yang, Q; Yan, Z; Markowitz, J; Wilder, P; Carrier, F; Weber, D. Inhibiting S100B restores p53 levels in primary malignant melanoma cancer cells. J. Biol. Chem 2004, 279, 34071–34077. [Google Scholar]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008, 9, 40–47. [Google Scholar]
- Baudier, J; Delphin, C; Grunwald, D; Khochbin, S; Lawrence, J. Characterization of the tumor suppressor protein p53 as a protein kinase C substrate and a S100b-binding protein. Proc. Natl. Acad. Sci USA 1992, 89, 11627–11631. [Google Scholar]
- Wilder, P; Rustandi, R; Drohat, A; Weber, D. S100B(betabeta) inhibits the protein kinase C-dependent phosphorylation of a peptide derived from p53 in a Ca2+-dependent manner. Protein Sci 1998, 7, 794–798. [Google Scholar]
- Sali, A; Blundell, T. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234, 779–815. [Google Scholar]
- Lüthy, R; Bowie, J; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar]
- Laskowski, R; Rullmannn, J; MacArthur, M; Kaptein, R; Thornton, J. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol NMR 1996, 8, 477–486. [Google Scholar]
- Wilder, P; Lin, J; Bair, C; Charpentier, T; Yang, D; Liriano, M; Varney, K; Lee, A; Oppenheim, A; Adhya, S; Carrier, F; Weber, D. Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim. Biophys Acta 2006, 1763, 1284–1297. [Google Scholar]
- Whitlow, J; Varughese, J; Zhou, Z; Bartolotti, L; Li, Y. Computational screening and design of S100B ligand to block S100B-p53 interaction. J. Mol. Graph. Model 2009, 27, 969–977. [Google Scholar]
- Dzubiella, J. Salt-specific stability and denaturation of a short salt-bridge-forming alpha-helix. J. Am. Chem. Soc 2008, 130, 14000–14007. [Google Scholar]
- Luthy, R; Bowie, JU; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar]
- van Der Spoel, D; Lindahl, E; Hess, B; Groenhof, G; Mark, A; Berendsen, H. GROMACS: Fast, flexible, and free. J. Comput.Chem 2005, 26, 1701–1718. [Google Scholar]
- Darden, T; York, D; Pedersn, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Hess, B; Bekker, H; Berendsen, H; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Kabsch, W; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]







© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pirolli, D.; Carelli Alinovi, C.; Capoluongo, E.; Satta, M.A.; Concolino, P.; Giardina, B.; De Rosa, M.C. Insight into a Novel p53 Single Point Mutation (G389E) by Molecular Dynamics Simulations. Int. J. Mol. Sci. 2011, 12, 128-140. https://doi.org/10.3390/ijms12010128
Pirolli D, Carelli Alinovi C, Capoluongo E, Satta MA, Concolino P, Giardina B, De Rosa MC. Insight into a Novel p53 Single Point Mutation (G389E) by Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2011; 12(1):128-140. https://doi.org/10.3390/ijms12010128
Chicago/Turabian StylePirolli, Davide, Cristiana Carelli Alinovi, Ettore Capoluongo, Maria Antonia Satta, Paola Concolino, Bruno Giardina, and Maria Cristina De Rosa. 2011. "Insight into a Novel p53 Single Point Mutation (G389E) by Molecular Dynamics Simulations" International Journal of Molecular Sciences 12, no. 1: 128-140. https://doi.org/10.3390/ijms12010128