Density Functional Theory (DFT) Study of Triphenylamine-Based Dyes for Their Use as Sensitizers in Molecular Photovoltaics


 ,
, 
Abstract
:1. Introduction
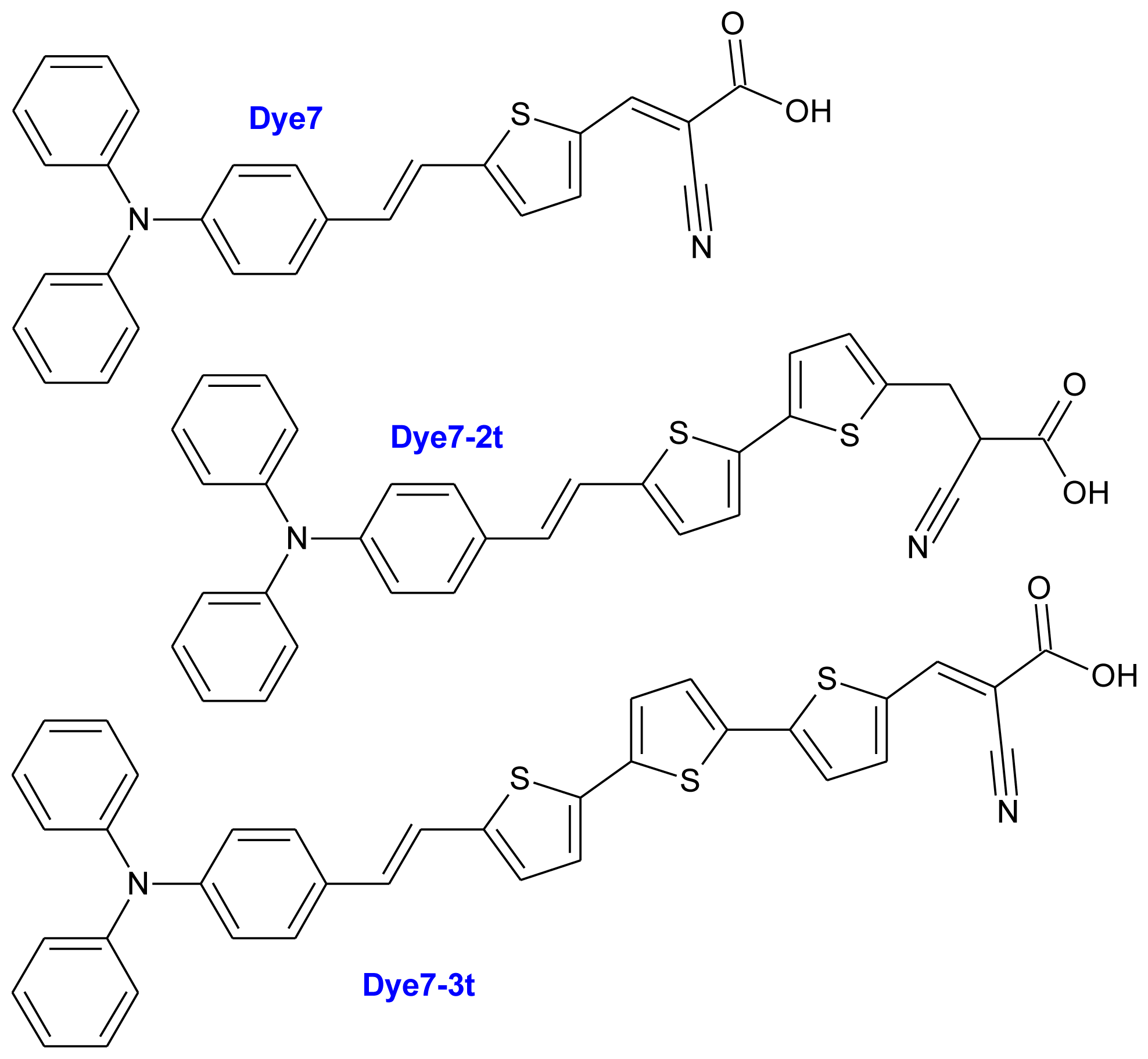
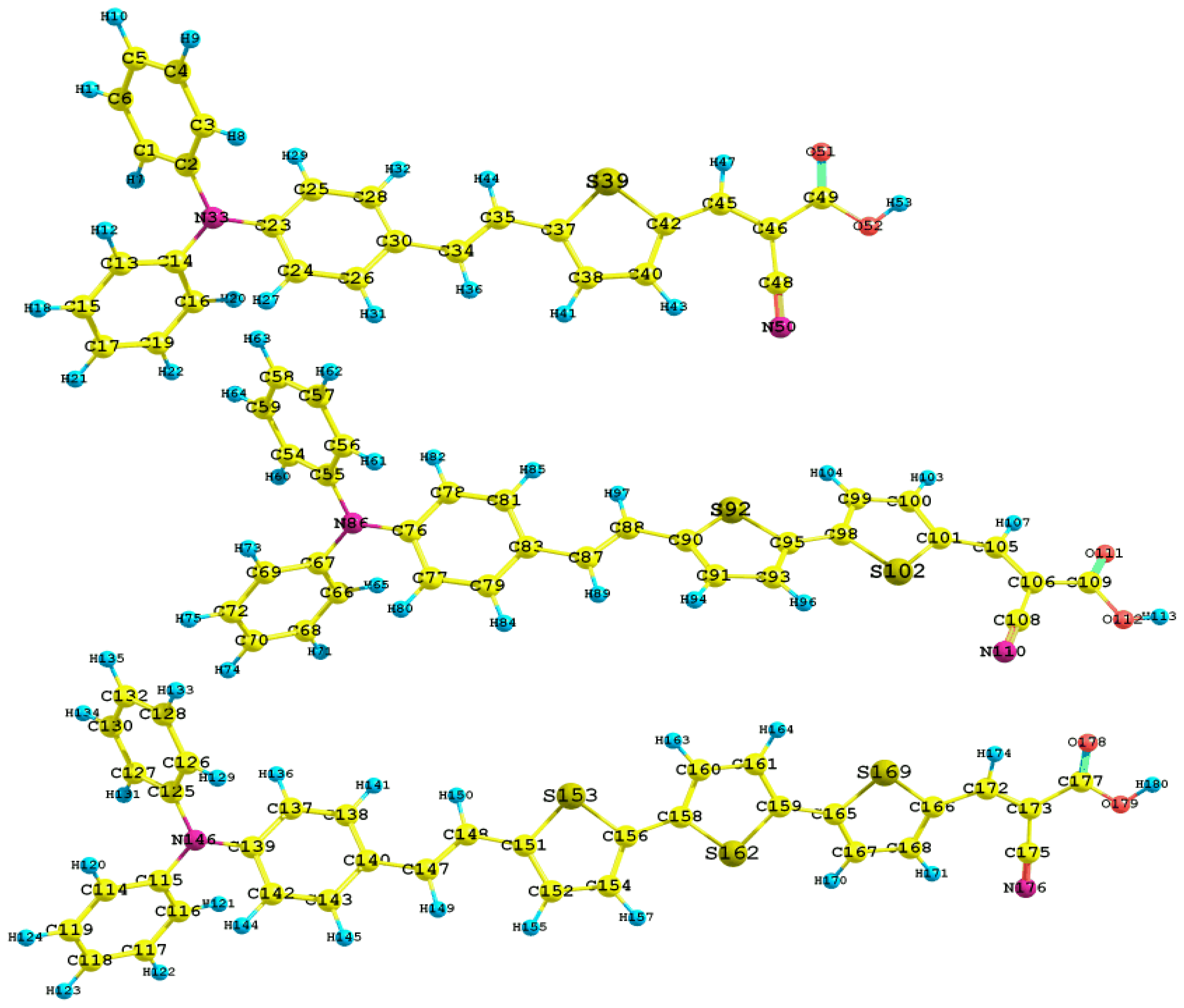
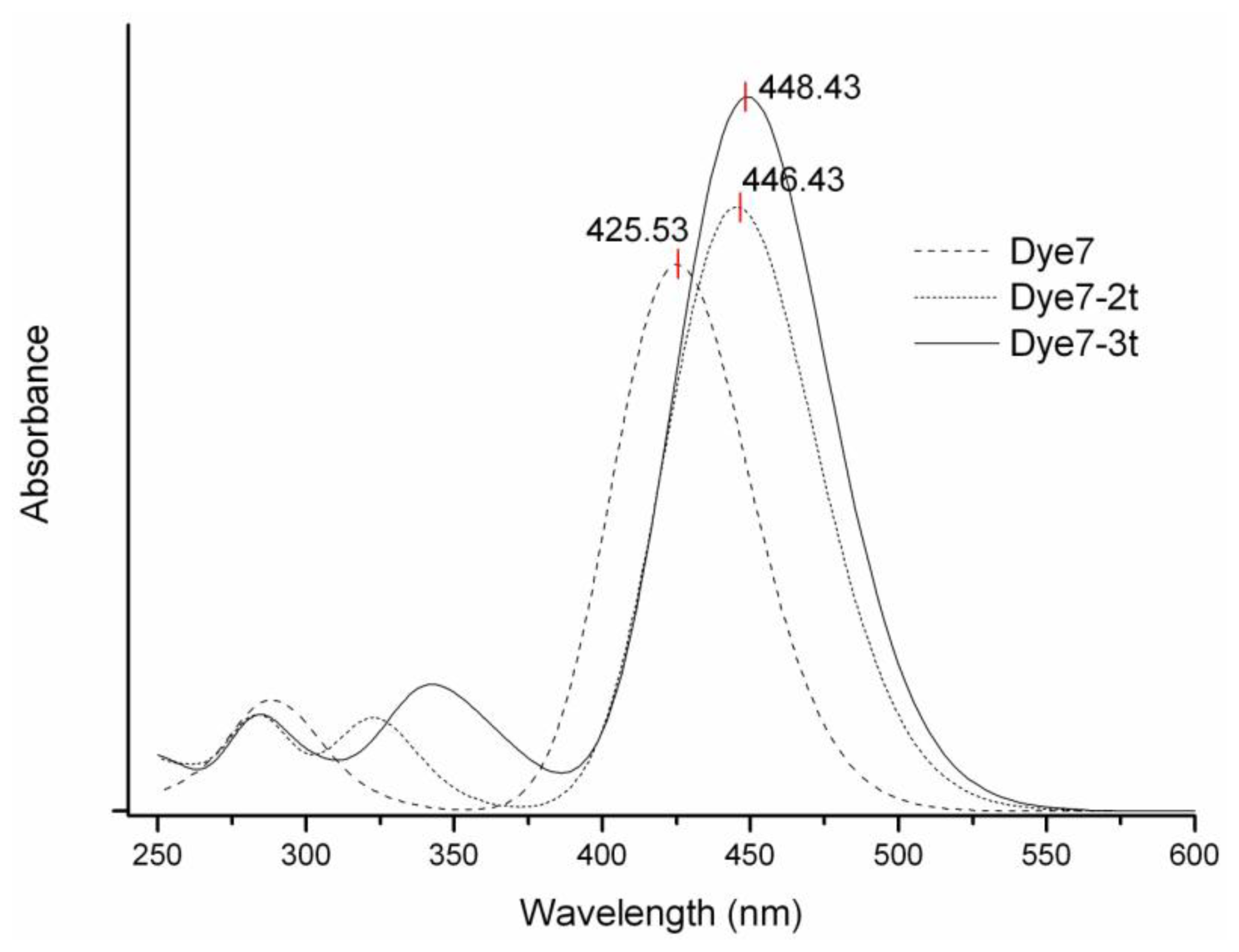
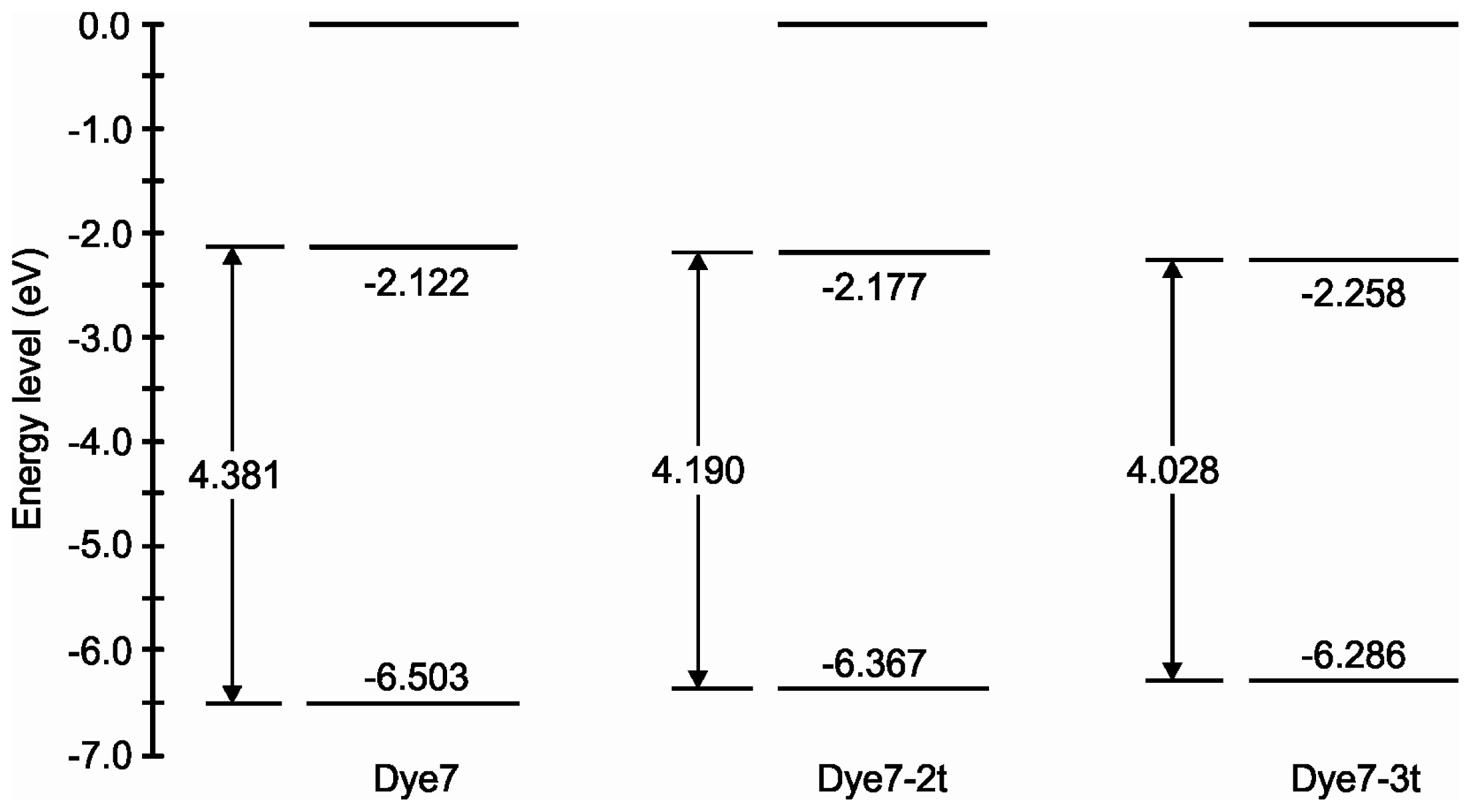
2. Results and Discussion
3. Experimental Section
4. Conclusions
Acknowledgments
References
- Nazeeruddin, M.K. Michael graetzel festschrift, a tribute for his 60th birthday. Coord. Chem. Rev 2004, 248, 1161–1164. [Google Scholar]
- Bisquert, J.; Cahen, D.; Hodes, G.; Rühle, S.; Zaban, A. Physical chemical principles of photovoltaic conversion with nanoparticulate, mesoporous dye-sensitized solar cells. J. Phys. Chem. B 2004, 108, 8106–8118. [Google Scholar]
- Saito, Y.; Fukuri, N.; Senadeera, R.; Kitamura, T.; Wada, Y.; Yanagida, S. Solid state dye sensitized solar cells using in situ polymerized PEDOTs as hole conductor. Electrochem. Commun 2004, 6, 71–74. [Google Scholar]
- Qiu, F.L.; Fisher, A.C.; Walker, A.B.; Peter, L.M. The distribution of photoinjected electrons a dye-sensitized nanocrystalline TiO2 solar cell modelled by a boundary element method. Electrochem. Commun 2003, 5, 711–716. [Google Scholar]
- Asbury, J.B.; Ellingson, R.J.; Ghosh, H.N.; Ferrere, S.; Nozik, A.J.; Lian, T. Femtosecond IR study of excited-state relaxation and electron-injection dynamics of Ru(dcbpy)2(NCS)2 in solution and on nanocrystalline TiO2 and Al2O3 thin films. J. Phys. Chem. B 1999, 103, 3110–3119. [Google Scholar]
- Werner, J.H. Second and Third Generation Photovoltaics-Dreams and Reality. Advances in Solid State Physics 2004, 44, 172–172. [Google Scholar]
- Xie, G.; Lin, J.; Wu, J.; Lan, Z.; Li, Q.; Xiao, Y.; Yue, G.; Yue, H.; Huang, M. Application of upconversion luminescence in dye-sensitized solar cells. Chin. Sci. Bull 2011, 56, 96–101. [Google Scholar]
- Argazzi, R.; Larramona, G.; Contado, C.; Bignozzi, C.A. Preparation and photoelectrochemical characterization of a red sensitive osmium complex containing 4,4′,″-tricarboxy-2,2′:6′,2′-terpyridine and cyanide ligands. J. Photochem. Photobiol. A Chem 2004, 164, 15–21. [Google Scholar]
- Koyama, Y.; Miki, T.; Wang, X.-F.; Nagae, H. Dye-sensitized solar cells based on the principles and materials of photosynthesis: mechanisms of suppression and enhancement of photocurrent and conversion efficiency. Int. J. Mol. Sci 2009, 10, 4575–4622. [Google Scholar]
- Martens, T.; Munters, T.; Goris, L.; D’Haen, J.; Schouteden, K.; D’Olieslaeger, M.; Lutsen, L.; Vanderzande, D.; Geens, W.; Poortmans, J.; et al. Nanostructured organic pn junctions towards 3D photovoltaics. Appl. Phys. A Mater. Sci. Process 2004, 79, 27–30. [Google Scholar]
- Chen, J.-G.; Chen, C.-Y.; Wu, S.-J.; Li, J.-Y.; Wu, C.-G.; Ho, K.-C. On the photophysical and electrochemical studies of dye-sensitized solar cells with the new dye CYC-B1. Sol. Energy Mater. Solar Cells 2008, 92, 1723–1727. [Google Scholar]
- Heimer, T.A.; Heilweil, E.J.; Bignozzi, C.A.; Meyer, G.J. Electron injection, recombination, and halide oxidation dynamics at dye-sensitized metal oxide interfaces. J. Phys. Chem. A 2000, 104, 4256–4262. [Google Scholar]
- Figgemeier, E.; Hagfeldt, A. Are dye-sensitized nano-structured solar cells stable? An overview of device testing and component analyses. Int. J. Photoenergy 2004, 6, 127–140. [Google Scholar]
- Grätzel, M. Nanocrystalline Electronic Junctions. In Studies in Surface Science and Catalysis; Prashant, V.K., Dan, M., Eds.; Elsevier: Amsterdam, Netherlands, 1997; Volume 103, pp. 353–375. [Google Scholar]
- Grätzel, M. Dye-sensitized solar cells. J. Photochem. Photobiol. C Photochem. Rev 2003, 4, 145–153. [Google Scholar]
- Kamat, P.V.; Haria, M.; Hotchandani, S. C60 cluster as an electron shuttle in a ru(ii)-polypyridyl sensitizer-based photochemical solar cell. J. Phys. Chem. B 2004, 108, 5166–5170. [Google Scholar]
- O’Regan, B.; Gratzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar]
- Cao, J.; Sun, J.-Z.; Hong, J.; Yang, X.-G.; Chen, H.-Z.; Wang, M. Direct observation of microscopic photoinduced charge redistribution on TiO2 film sensitized by chloroaluminum phthalocyanine and perylenediimide. Appl. Phys. Lett 2003, 83, 1896–1898. [Google Scholar]
- Zhang, C.R.; Liu, Z.J.; Chen, Y.H.; Chen, H.S.; Wu, Y.Z.; Yuan, L.H. DFT and TDDFT study on organic dye sensitizers D5, DST and DSS for solar cells. J. Mol. Struct. THEOCHEM 2009, 899, 86–93. [Google Scholar]
- Ham, H.W.; Kim, Y.S. Theoretical study of indoline dyes for dye-sensitized solar cells. Thin Solid Films 2010, 518, 6558–6563. [Google Scholar]
- Zhang, C.-R.; Liu, Z.-J.; Chen, Y.-H.; Chen, H.-S.; Wu, Y.-Z.; Feng, W.; Wang, D.-B. DFT and TD-DFT study on structure and properties of organic dye sensitizer TA-St-CA. Curr. Appl. Phys 2010, 10, 77–83. [Google Scholar]
- Ruiz-Anchondo, T.; Flores-Holguín, N.; Glossman-Mitnik, D. Natural carotenoids as nanomaterial precursors for molecular photovoltaics: A computational DFT study. Molecules 2010, 15, 4490–4510. [Google Scholar]
- De Angelis, F. Direct vs. indirect injection mechanisms in perylene dye-sensitized solar cells: A DFT/TDDFT investigation. Chem. Phys. Lett 2010, 493, 323–327. [Google Scholar]
- Zhang, F.; Luo, Y.-H.; Song, J.-S.; Guo, X.-Z.; Liu, W.-L.; Ma, C.-P.; Huang, Y.; Ge, M.-F.; Bo, Z.; Meng, Q.-B. Triphenylamine-based dyes for dye-sensitized solar cells. Dyes Pigment 2009, 81, 224–230. [Google Scholar]
- Casanova, D.; Rotzinger, F.P.; Grätzel, M. Computational study of promising organic dyes for high-performance sensitized solar cells. J. Chem. Theory Comput 2010, 6, 1219–1227. [Google Scholar]
- El-Shishtawy, R.M. Functional dyes, and some hi-tech applications. Int. J. Photoenergy 2009, 2009, 1–21. [Google Scholar]
- Sahu, D.; Padhy, H.; Patra, D.; Kekuda, D.; Chu, C.-W.; Chiang, I.H.; Lin, H.-C. Synthesis and application of H-Bonded cross-linking polymers containing a conjugated pyridyl H-Acceptor side-chain polymer and various carbazole-based H-Donor dyes bearing symmetrical cyanoacrylic acids for organic solar cells. Polymer 2010, 51, 6182–6192. [Google Scholar]
- Qin, H.; Wenger, S.; Xu, M.; Gao, F.; Jing, X.; Wang, P.; Zakeeruddin, S.M.; Grätzel, M. An organic sensitizer with a fused dithienothiophene unit for efficient and stable dye-sensitized solar cells. J. Am. Chem. Soc 2008, 130, 9202–9203. [Google Scholar]
- Yum, J.-H.; Hagberg, D.P.; Moon, S.-J.; Karlsson, K.M.; Marinado, T.; Sun, L.; Hagfeldt, A.; Nazeeruddin, M.K.; Grätzel, M. A light-resistant organic sensitizer for solar-cell applications. Angew. Chem 2009, 121, 1604–1608. [Google Scholar]
- Zhang, G.; Bala, H.; Cheng, Y.; Shi, D.; Lv, X.; Yu, Q.; Wang, P. High efficiency and stable dye-sensitized solar cells with an organic chromophore featuring a binary p-conjugated spacer. Chem. Commun 2009, 16, 2198–2200. [Google Scholar]
- Im, H.; Kim, S.; Park, C.; Jang, S.-H.; Kim, C.-J.; Kim, K.; Park, N.-G.; Kim, C. High performance organic photosensitizers for dye-sensitized solar cells. Chem. Commun 2010, 46, 1335–1337. [Google Scholar]
- Zeng, W.; Cao, Y.; Bai, Y.; Wang, Y.; Shi, Y.; Zhang, M.; Wang, F.; Pan, C.; Wang, P. Efficient dye-sensitized solar cells with an organic photosensitizer featuring orderly conjugated ethylenedioxythiophene and dithienosilole blocks. Chem. Mater 2010, 22, 1915–1925. [Google Scholar]
- Duan, T.; Fan, K.; Fu, Y.; Zhong, C.; Chen, X.; Peng, T.; Qin, J. Triphenylamine-based organic dyes containing a 1,2,3-triazole bridge for dye-sensitized solar cells via a “Click” reaction. Dyes Pigment 2012, 94, 28–33. [Google Scholar]
- Hara, K.; Sato, T.; Katoh, R.; Furube, A.; Yoshihara, T.; Murai, M.; Kurashige, M.; Ito, S.; Shinpo, A.; Suga, S.; et al. Novel conjugated organic dyes for efficient dye-sensitized solar cells. Adv. Funct. Mater 2005, 15, 246–252. [Google Scholar]
- Hara, K.; Wang, Z.-S.; Sato, T.; Furube, A.; Katoh, R.; Sugihara, H.; Dan-oh, Y.; Kasada, C.; Shinpo, A.; Suga, S. Oligothiophene-containing coumarin dyes for efficient dye-sensitized solar cells. J. Phys. Chem. B 2005, 109, 15476–15482. [Google Scholar]
- Hagberg, D.P.; Marinado, T.; Karlsson, K.M.; Nonomura, K.; Qin, P.; Boschloo, G.; Brinck, T.; Hagfeldt, A.; Sun, L. Tuning the HOMO and LUMO energy levels of organic chromophores for dye sensitized solar cells. J. Org. Chem 2007, 72, 9550–9556. [Google Scholar]
- Justin Thomas, K.R.; Hsu, Y.-C.; Lin, J.T.; Lee, K.-M.; Ho, K.-C.; Lai, C.-H.; Cheng, Y.-M.; Chou, P.-T. 2,3-Disubstituted thiophene-based organic dyes for solar cells. Chem. Mater 2008, 20, 1830–1840. [Google Scholar]
- Li, Y.; Liu, S.; Zhao, X.; Chen, M.; Ma, F. Intramolecular charge transfer in the dye molecules containing bis-dimethylfluoreneaniline. J. Mol. Struct. THEOCHEM 2008, 867, 10–16. [Google Scholar]
- Balanay, M.P.; Kim, S.M.; Lee, M.J.; Lee, S.H.; Kim, D.H. Conformational analysis and electronic properties of 2-cyano-3-(thiophen-2-yl)acrylic acid in sensitizers for dye-sensitized solar cells: A theoretical study. Bull. Korean Chem. Soc 2009, 30, 2077–2082. [Google Scholar]
- Sánchez-Bojorge, N.-A.; Flores-Holguín, N.; Glossman-Mitnik, D.; Rodríguez-Valdez, L.M. Computational note on the chemical reactivity of pyrrole derivatives. J. Mol. Struct. THEOCHEM 2009, 912, 119–120. [Google Scholar]
- Gundlach, L.; Ernstorfer, R.; Willig, F. Ultrafast interfacial electron transfer from the excited state of anchored molecules into a semiconductor. Prog. Surf. Sci 2007, 82, 355–377. [Google Scholar]
- Nagy, Á. Density functional. Theory and application to atoms and molecules. Phys. Rep. 1998, 298, 1–79. [Google Scholar]
- Gaussian 09W Program, version 7.0; Gaussian, Inc: Wallingford, CT, USA, 2004.
- Green, M.A. Solar Cells: Operating Principles, Technology and Systems Applications; Prentice-Hall, Inc: Englewood Cliffs, NJ, USA, 1982. [Google Scholar]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods; Gaussian, Inc: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Flores-Holguín, N.; Rodríguez-Valdez, L.M.; Glossman-Mitnik, D. CHIH-DFT computational molecular characterization of phenanthro [9,10-c]-1,2,5-thiadiazole 1,1-dioxide. J. Mol. Struct. THEOCHEM 2008, 862, 60–65. [Google Scholar]
- De Angelis, F.; Fantacci, S.; Sgamellotti, A. An integrated computational tool for the study of the optical properties of nanoscale devices: Application to solar cells and molecular wires. Theor. Chem. Acc. Theory Comput. Model. (Theor. Chimi. Acta) 2007, 117, 1093–1104. [Google Scholar]
- Weng, Y.-X.; Wang, Y.-Q.; Asbury, J.B.; Ghosh, H.N.; Lian, T. Back electron transfer from TiO2 nanoparticles to FeIII(CN)63−: Origin of non-single-exponential and particle size independent dynamics. J. Phys. Chem. B 1999, 104, 93–104. [Google Scholar]
- Sharma, S.K.; Inamdar, A.I.; Im, H.; Kim, B.G.; Patil, P.S. Morphology dependent dye-sensitized solar cell properties of nanocrystalline zinc oxide thin films. Solid State Ion 2011, 509, 2127–2131. [Google Scholar]
- Lewards, E.G. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Norwell, MA, USA, 2003. [Google Scholar]
- Young, D. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models; John Wiley & Sons: Chichester, UK, 2002. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput 2006, 2, 364–382. [Google Scholar]
- Burke, K.; Werschnik, J.; Gross, E.K.U. Time-dependent density functional theory: Past, present, and future. J. Chem. Phys 2005, 123, 062206:1–062206:9. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys 1998, 109, 8218–8224. [Google Scholar]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett 1996, 256, 454–464. [Google Scholar]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys 1998, 108, 4439–4449. [Google Scholar]
- Gorelsky, S.I. SWizard Program. Available online: http://www.sg-chem.net/ accessed on 4 February 2012.
- Stewart, J.J.P. MOPAC2009. Stewart Computational Chemistry. Available online: http://OpenMOPAC.net accessed on 4 February 2012.
- Stewart, J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model 2007, 13, 1173–1213. [Google Scholar]
- Gorelsky, S.I. AOMix Program. Available online: http://www.sg-chem.net accessed on 4 February 2012.
- Gorelsky, S.I.; Lever, A.B.P. Electronic structure and spectra of ruthenium diimine complexes by density functional theory and INDO/S. Comparison of the two methods. J. Organomet. Chem 2001, 635, 187–196. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye7 | Value | Dye7-2t | Value | Dye7-3t | Value |
|---|---|---|---|---|---|
| C2-N33 | 1.418 | C55-N86 | 1.416 | C125-N146 | 1.417 |
| C14-N33 | 1.418 | C67-N86 | 1.417 | C115-N146 | 1.416 |
| C23-N33 | 1.404 | C76-N86 | 1.407 | C139-N149 | 1.408 |
| C2-C3 | 1.398 | C55-C56 | 1.399 | C125-C126 | 1.399 |
| C15-C17 | 1.394 | C70-C72 | 1.394 | C118-C119 | 1.394 |
| C30-C34 | 1.460 | C83-C87 | 1.463 | C140-C147 | 1.463 |
| C35-C37 | 1.452 | C88-C90 | 1.454 | C148-C151 | 1.454 |
| C42-C45 | 1.430 | C101-C105 | 1.427 | C166-C172 | 1.431 |
| C2-N33-C23 | 120.3 | C55-N86-C76 | 120.4 | C125-N146-C139 | 120.2 |
| C14-N33-C23 | 120.6 | C67-N86-C76 | 120.1 | C115-N146-C139 | 120.3 |
| C30-C34-C35 | 126.6 | C83-C87-C88 | 126.6 | C140-C147-C148 | 126.3 |
| C42-C45-C46 | 129.2 | C101-C105-C106 | 130.2 | C166-C172-C173 | 129.0 |
| C2-N33-C23-C25 | 35.16 | C55-N86-C76-C78 | 37.71 | C125-N146-C139-C137 | 37.52 |
| C23-N33-C14-C16 | 44.61 | C66-C67-N86-C76 | 44.26 | C116-C115-N146-C139 | 42.86 |
| C45-C46-C49-O51 | 0.21 | C105-C106-C109-O111 | 0.22 | C172-C173-C177-O178 | 0.27 |
| Number | nm | eV | (f) | Assignment; H = HOMO, L = LUMO, L + 1 = LUMO + 1, etc. |
|---|---|---|---|---|
| 1 | 425.5 | 2.91 | 1.6808 | S H-0- > L + 0 (+73%) H-1- > L + 0 (8%) |
| 2 | 320.5 | 3.87 | 0.0361 | S H-1- > L + 0 (+55%) H-0- > L + 1 (19%) |
| 3 | 301.4 | 4.11 | 0.0797 | S H-0- > L + 2 (+42%) H-0- > L + 1 (+27%) H-1- > L + 0 (+10%) H-0- > L + 0 (+7%) |
| 4 | 294.4 | 4.21 | 0.1563 | S H-0- > L + 2 (+40%) H-0- > L + 1 (30%) H-0- > L + 0 (7%) H-1- > L + 0 (6%) |
| 5 | 281.2 | 4.41 | 0.2058 | S H-0- > L + 3 (+80%) H-1- > L + 3 (+9%) |
| 6 | 266.4 | 4.65 | 0.0517 | S H-6- > L + 0 (+44%) H-7- > L + 0 (14%) H-4- > L + 0 (+13%) H-1- > L + 1 (10%) H-0- > L + 4 (+6%) |
| 7 | 259.4 | 4.78 | 0.0365 | S H-0- > L + 5 (+40%) H-0- > L + 4 (15%) H-7- > L + 0 (11%) |
| Number | nm | eV | (f) | Assignment; H = HOMO, L = LUMO, L + 1 = LUMO + 1, etc. |
|---|---|---|---|---|
| 1 | 446.4 | 2.78 | 1.8564 | S H-0- > L + 0 (+67%) H-1- > L + 0 (22%) H-0- > L + 1 (+7%) |
| 2 | 322.8 | 3.84 | 0.2720 | S H-0- > L + 1 (+41%) H-0- > L + 0 (25%) H-1- > L + 0 (19%) H-1- > L + 1 (6%) |
| 3 | 299.8 | 4.14 | 0.0368 | S H-0- > L + 2 (+65%) H-0- > L + 3 (14%) H-1- > L + 2 (+8%) |
| 4 | 286.8 | 4.32 | 0.0514 | S H-1- > L + 1 (+36%) H-2- > L + 0 (22%) H-8- > L + 0 (14%) H-0- > L + 3 (11%) H-5- > L + 0 (+6%) |
| 5 | 283.2 | 4.38 | 0.2221 | S H-0- > L + 4 (+77%) H-1- > L + 4 (+17%) |
| 6 | 258.6 | 4.79 | 0.0496 | S H-0- > L + 5 (+28%) H-8- > L + 0 (+12%) H-5- > L + 0 (+11%) H-0- > L + 7 (7%) |
| 7 | 248.9 | 4.98 | 0.0668 | S H-0- > L + 6 (+42%) H-0- > L + 3 (12%) H-1- > L + 6 (+10%) |
| 8 | 239.1 | 5.19 | 0.1066 | S H-1- > L + 3(+15%) H-2- > L + 1(+11%) H-0- > L + 8(8%) H-0- > L + 7(+8%) H-9- > L + 0(8%) H-0- > L + 9(+8%) |
| 9 | 231.1 | 5.36 | 0.0553 | S H-4- > L + 0(+32%) H-4- > L + 1(+15%) H-0- > L + 7(+10%) H-7- > L + 0(10%) H-7- > L + 1(6%) |
| Number | nm | eV | (f) | Assignment; H = HOMO, L = LUMO, L + 1 = LUMO + 1, etc. |
|---|---|---|---|---|
| 1 | 448.4 | 2.76 | 2.1948 | S H-0- > L + 0 (+47%) H-1- > L + 0 (26%) H-0- > L + 1 (+12%) |
| 2 | 364.3 | 3.40 | 0.1536 | S H-0- > L + 1 (+49%) H-1- > L + 0 (+19%) H-0- > L + 2 (6%) H-2- > L + 0 (+6%) |
| 3 | 339.3 | 3.65 | 0.3332 | S H-0- > L + 0 (+47%) H-1- > L + 0 (+21%) H-0- > L + 1 (12%) H-2- > L + 0 (+9%) H-1- > L + 1 (+5%) |
| 4 | 309.6 | 4.00 | 0.0758 | S H-1- > L + 1 (+39%) H-0- > L + 2 (19%) H-2- > L + 0 (12%) |
| 5 | 300.2 | 4.13 | 0.0462 | S H-0- > L + 3 (+70%) H-1- > L + 3 (+13%) H-1- > L + 1 (+12%) |
| 6 | 284.2 | 4.36 | 0.2165 | S H-0- > L + 4 (+69%) H-1- > L + 4 (+18%) |
| 7 | 278.2 | 4.46 | 0.0348 | S H-0- > L + 2 (+29%) H-1- > L + 2 (27%) H-1- > L + 1 (+15%) H-0- > L + 1 (+6%) |
| 8 | 255.8 | 4.85 | 0.0597 | S H-10- > L + 0 (+32%) H-9- > L + 0 (13%) H-6- > L + 0 (8%) |
| 9 | 243.6 | 5.09 | 0.0752 | S H-0- > L + 10 (13%) H-2- > L + 1 (+12%) H-11- > L + 0 (+11%) H-0- > L + 5 (+10%) H-5- > L + 0 (+9%) H-6- > L + 0 (+6%) |
| 10 | 234.0 | 5.30 | 0.0496 | S H-2- > L + 2 (+13%) H-1- > L + 5 (+10%) H-0- > L + 11 (10%) H-9- > L + 0 (+6%) |
| Molecule | μ (Debye) | α (Bohr3) | pKa |
|---|---|---|---|
| Dye7 | 7.15 | 479.04 | −0.17 |
| Dye7-2t | 6.42 | 578.60 | −0.24 |
| Dye-3t | 7.13 | 684.24 | −0.39 |
| Molecule | Site for Electrophilic Attack | Site for Nucleophilic Attack |
|---|---|---|
| Dye7 | N33 | C45 |
| Dye7-2t | N86 | C105 |
| Dye7-3t | N146 | C172 |
| Molecule | Conceptual DFT | ||||
|---|---|---|---|---|---|
| I (eV) | A (eV) | χ (eV) | η (eV) | ω (eV) | |
| Dye7 | 6.844 | 1.672 | 4.258 | 2.586 | 3.506 |
| Dye7-2t | 6.693 | 1.748 | 4.220 | 2.473 | 3.602 |
| Dye7-3t | 6.625 | 1.826 | 4.226 | 2.399 | 3.721 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baldenebro-López, J.; Castorena-González, J.; Flores-Holguín, N.; Almaral-Sánchez, J.; Glossman-Mitnik, D. Density Functional Theory (DFT) Study of Triphenylamine-Based Dyes for Their Use as Sensitizers in Molecular Photovoltaics. Int. J. Mol. Sci. 2012, 13, 4418-4432. https://doi.org/10.3390/ijms13044418
Baldenebro-López J, Castorena-González J, Flores-Holguín N, Almaral-Sánchez J, Glossman-Mitnik D. Density Functional Theory (DFT) Study of Triphenylamine-Based Dyes for Their Use as Sensitizers in Molecular Photovoltaics. International Journal of Molecular Sciences. 2012; 13(4):4418-4432. https://doi.org/10.3390/ijms13044418
Chicago/Turabian StyleBaldenebro-López, Jesús, José Castorena-González, Norma Flores-Holguín, Jorge Almaral-Sánchez, and Daniel Glossman-Mitnik. 2012. "Density Functional Theory (DFT) Study of Triphenylamine-Based Dyes for Their Use as Sensitizers in Molecular Photovoltaics" International Journal of Molecular Sciences 13, no. 4: 4418-4432. https://doi.org/10.3390/ijms13044418