Genome-Wide Identification and in Silico Analysis of Poplar Peptide Deformylases

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification and Characterization of PDF Genes in Populus
2.2. Revision of Poplar PDF Gene-encoding Proteins
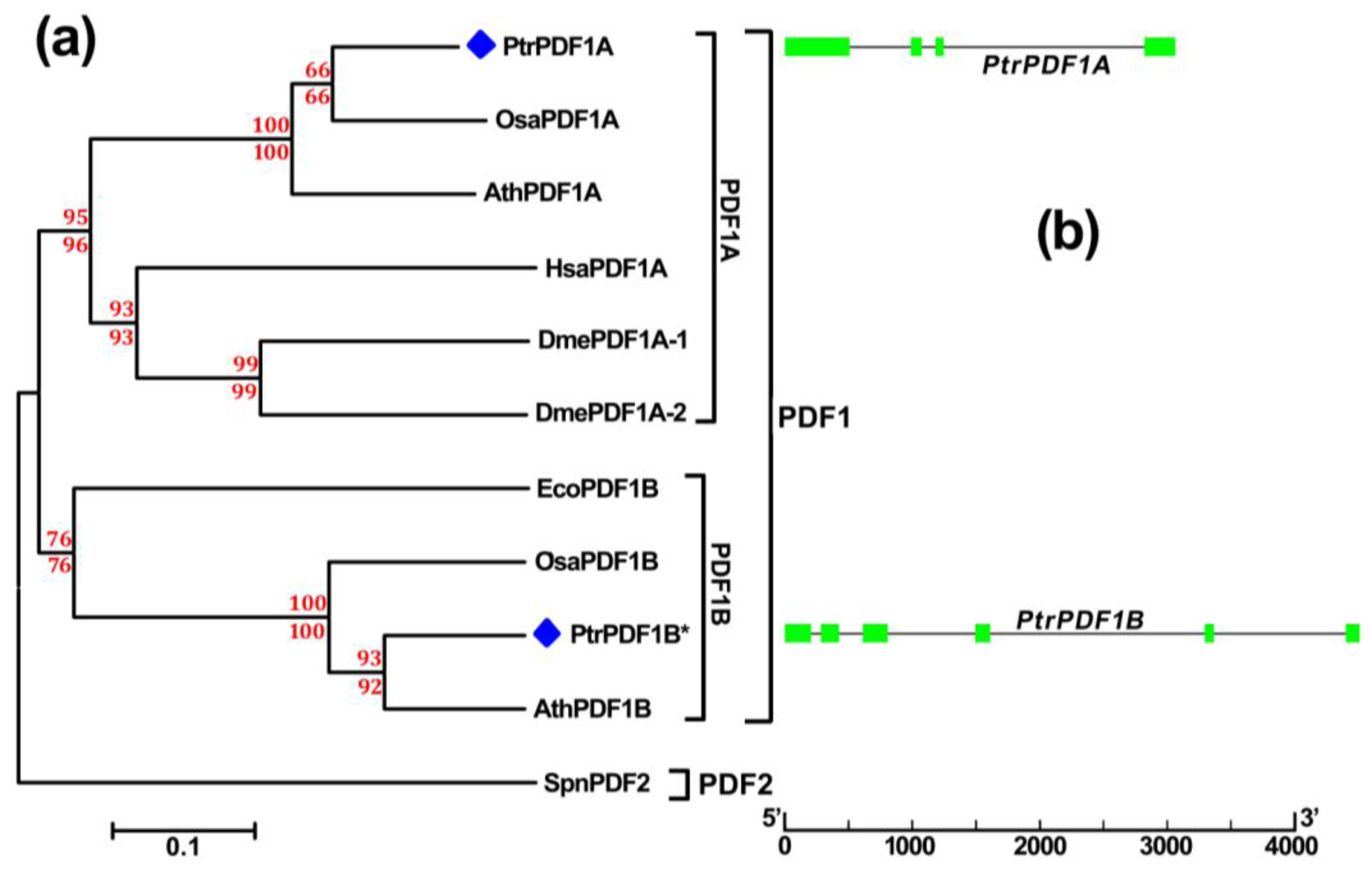
2.3. Divergence in Poplar PDF1s
2.4. Chromosome Location and Duplication of PDF1 Genes in Populus
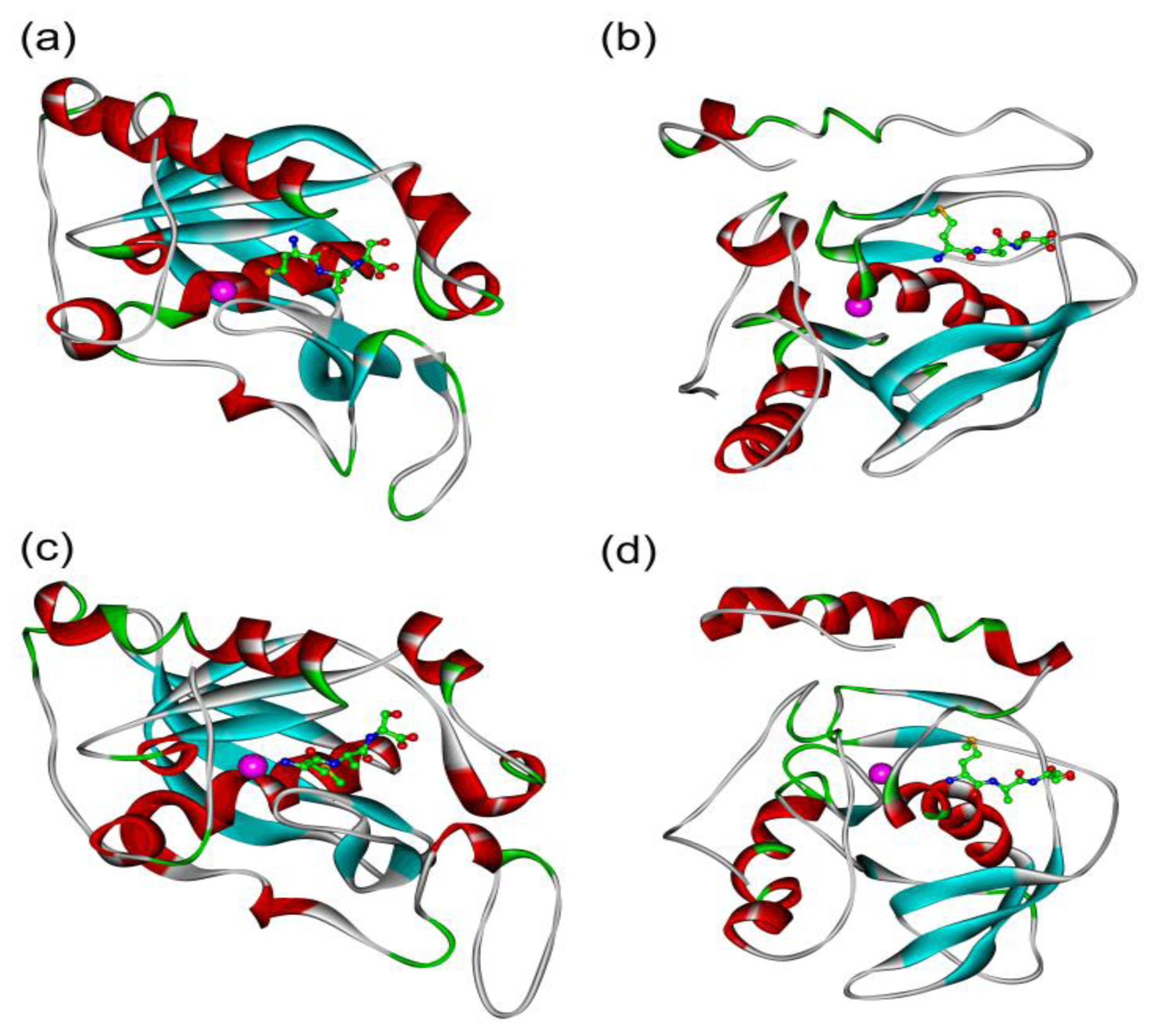
2.5. In Silico Simulation on the Poplar PDFs Reveal Analogous Activities with Their Individual Counterparts in Arabidopsis
3. Experimental Section
3.1. Identification of PDF Genes across Poplar Genome
3.2. Revision of Poplar PDF Proteins
3.3. Phylogenetic Analysis and Gene Structural Display
3.4. Chromosomal Location and in Silico Simulation
4. Conclusions
Acknowledgments
References
- Giglione, C.; Serero, A.; Pierre, M.; Boisson, B.; Meinnel, T. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. EMBO J 2000, 19, 5916–5929. [Google Scholar]
- Giglione, C.; Boularot, A.; Meinnel, T. Protein N-terminal methionine excision. Cell Mol. Life Sci 2004, 61, 1455–1474. [Google Scholar]
- Polevoda, B.; Sherman, F. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol 2003, 325, 595–622. [Google Scholar]
- Martinez, A.; Traverso, J.A.; Valot, B.; Ferro, M.; Espagne, C.; Ephritikhine, G.; Zivy, M.; Giglione, C.; Meinnel, T. Extent of N-terminal modifications in cytosolic proteins from eukaryotes. Proteomics 2008, 8, 2809–2831. [Google Scholar]
- Ross, S.; Giglione, C.; Pierre, M.; Espagne, C.; Meinnel, T. Functional and developmental impact of cytosolic protein N-terminal methionine excision in arabidopsis. Plant Physiol 2005, 137, 623–637. [Google Scholar]
- Pesaresi, P.; Gardner, N.A.; Masiero, S.; Dietzmann, A.; Eichacker, L.; Wickner, R.; Salamini, F.; Leister, D. Cytoplasmic N-terminal protein acetylation is required for efficient photosynthesis in Arabidopsis. Plant Cell 2003, 15, 1817–1832. [Google Scholar]
- Giglione, C.; Vallon, O.; Meinnel, T. Control of protein life-span by N-terminal methionine excision. EMBO J 2003, 22, 13–23. [Google Scholar]
- Dirk, L.; Schmidt, J.; Cai, Y.; Barnes, J.; Hanger, K.; Nayak, N.; Williams, M.; Grossman, R.; Houtz, R.; Rodgers, D. Insights into the substrate specificity of plant peptide deformylase, an essential enzyme with potential for the development of novel biotechnology applications in agriculture. Biochem. J 2008, 413, 417–427. [Google Scholar]
- Fieulaine, S.; Juillan-Binard, C.; Serero, A.; Dardel, F.; Giglione, C.; Meinnel, T.; Ferrer, J. The crystal structure of mitochondrial (Type 1A) peptide deformylase provides clear guidelines for the design of inhibitors specific for the bacterial forms. J. Biol. Chem 2005, 280, 42315–42324. [Google Scholar]
- Moon, S.; Giglione, C.; Lee, D.; An, S.; Jeong, D.; Meinnel, T.; An, G. Rice peptide deformylase PDF1B is crucial for development of chloroplasts. Plant Cell Physiol 2008, 49, 1536–1546. [Google Scholar]
- Escobar-Alvarez, S.; Goldgur, Y.; Yang, G.; Ouerfelli, O.; Li, Y.; Scheinberg, D. Structure and activity of human mitochondrial peptide deformylase, a novel cancer target. J. Mol. Biol 2009, 387, 1211–1228. [Google Scholar]
- Dirk, L.; Williams, M.; Houtz, R. Eukaryotic peptide deformylases. Nuclear-encoded and chloroplast-targeted enzymes in Arabidopsis. Plant Physiol 2001, 127, 97–107. [Google Scholar]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S.; Griffiths-Jones, S.; Howe, K.; Marshall, M.; Sonnhammer, E. The Pfam protein families database. Nucleic Acids Res 2002, 30, 276–280. [Google Scholar]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K. The Pfam protein families database. Nucleic Acids Res 2010, 38, D211–D222. [Google Scholar]
- Tuskan, G.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 2006, 35, D61–D65. [Google Scholar]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 2005, 33, D501–504. [Google Scholar]
- Basic Local Alignment Search Tool. National Center for Biotechnology Information (NCBI). Available online: http://blast.ncbi.nlm.nih.gov/ accessed on 5 April 2012.
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol 1987, 4, 406–425. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 2011, 28, 2731–2739. [Google Scholar]
- Guo, A.Y.; Zhu, Q.H.; Chen, X.; Luo, J.C. GSDS: A gene structure display server (in Chinese). Yi Chuan 2007, 29, 1023–1026. [Google Scholar]
- Abdel-Haleem, H. The origins of genome architecture. J. Hered 2007, 98, 633–634. [Google Scholar]
- The Genome Portal of the Department of Energy Joint Genome Institute. Available online: www.jgi.doe.gov/poplar accessed on 10 April 2012.
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J. The Pfam protein families database. Nucleic Acids Res 2012, 40, D290–D301. [Google Scholar]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform 2009, 23, 205–211. [Google Scholar]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symposium Series, Washington, DC, USA; 1999; pp. 95–98. [Google Scholar]
- Map Viewer. National Center for Biotechnology Information (NCBI). Available online: http://www.ncbi.nlm.nih.gov/projects/mapview/ accessed on 10 April 2012.
- Accelrys, InisghtII 2005; Accelrys Inc: San Diego, CA, USA, 2005.
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc 2008, 4, 1–13. [Google Scholar]
- Head, J.D.; Zerner, M.C. A Broyden–Fletcher–Goldfarb–Shanno optimization procedure for molecular geometries. Chem. Phys. lett 1985, 122, 264–270. [Google Scholar]
- Yang, Z.W.; Yang, G.; Zu, Y.G.; Fu, Y.J.; Zhou, L.J. The conformational analysis and proton transfer of the neuraminidase inhibitors: A theoretical study. Phys. Chem. Chem. Phys 2009, 11, 10035–10041. [Google Scholar]
- Yang, Z.; Nie, Y.; Yang, G.; Zu, Y.; Fu, Y.; Zhou, L. Synergistic effects in the designs of neuraminidase ligands: Analysis from docking and molecular dynamics studies. J. Theor. Biol 2010, 267, 363–374. [Google Scholar]
- Accrlrys, Affinity User Guide; Accelrys Inc: San Diego, CA, USA, 2005.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JGI NO. | Novel simplified nomenclature | Refseq protein ID | Refseq RNA ID (CDS) | Chromosome Location |
|---|---|---|---|---|
| 640630 | PtrPDF1A | XP_002298107.1 | XM_002298071.1 | LG_I: 9208431–9211542 (+) |
| 173925 | PtrPDF1B | XP_002300047.1 | XM_002300011.1 | LG_I: 21839768–21844235 (−) |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, C.-C.; Liu, B.-G.; Yang, Z.-W.; Li, C.-M.; Wang, B.-C.; Yang, C.-P. Genome-Wide Identification and in Silico Analysis of Poplar Peptide Deformylases. Int. J. Mol. Sci. 2012, 13, 5112-5124. https://doi.org/10.3390/ijms13045112
Liu C-C, Liu B-G, Yang Z-W, Li C-M, Wang B-C, Yang C-P. Genome-Wide Identification and in Silico Analysis of Poplar Peptide Deformylases. International Journal of Molecular Sciences. 2012; 13(4):5112-5124. https://doi.org/10.3390/ijms13045112
Chicago/Turabian StyleLiu, Chang-Cai, Bao-Guang Liu, Zhi-Wei Yang, Chun-Ming Li, Bai-Chen Wang, and Chuan-Ping Yang. 2012. "Genome-Wide Identification and in Silico Analysis of Poplar Peptide Deformylases" International Journal of Molecular Sciences 13, no. 4: 5112-5124. https://doi.org/10.3390/ijms13045112