Screening of Peptide Ligands for Pyrroloquinoline Quinone Glucose Dehydrogenase Using Antagonistic Template-Based Biopanning

Abstract
:1. Introduction
2. Results and Discussion
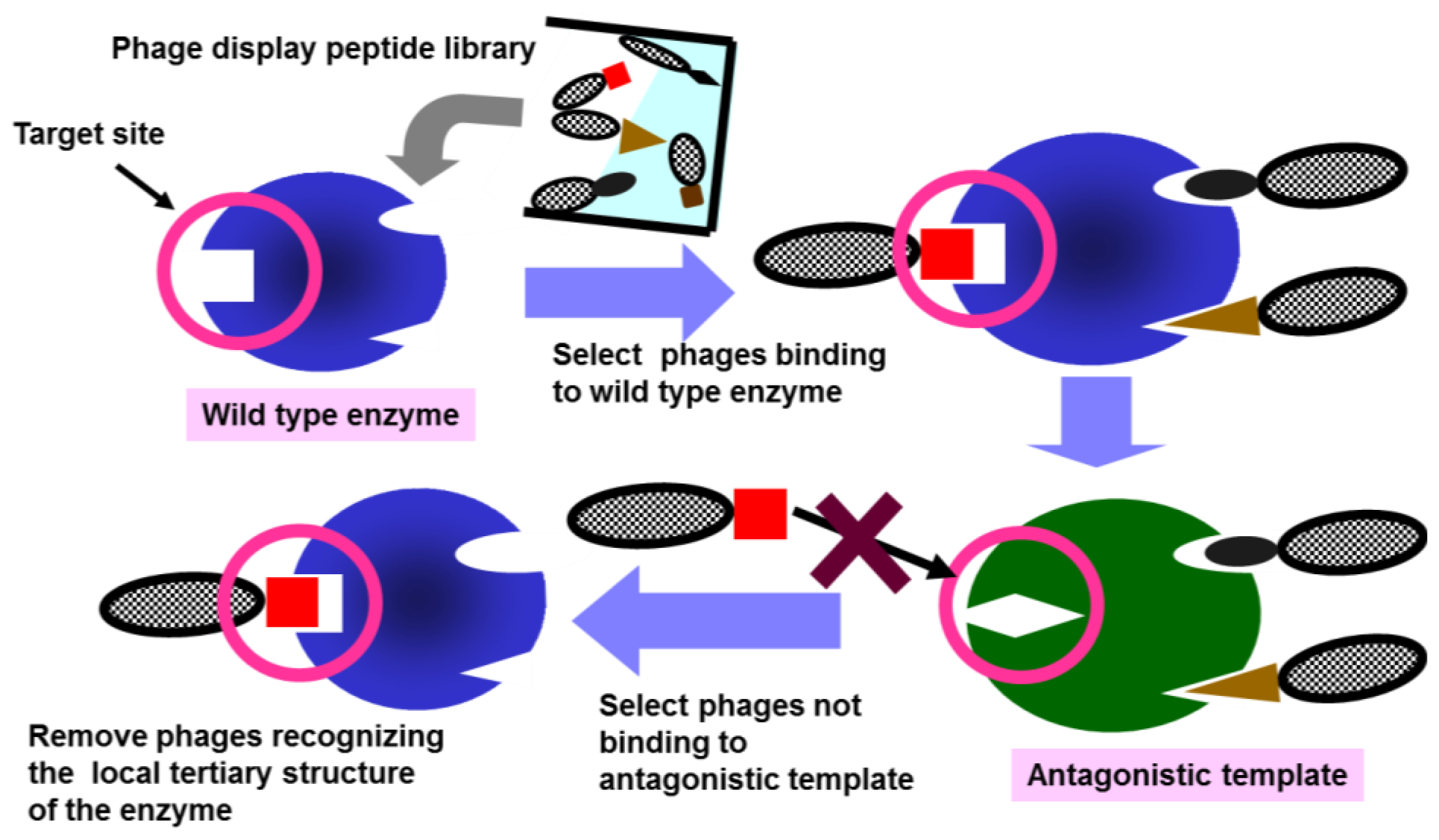
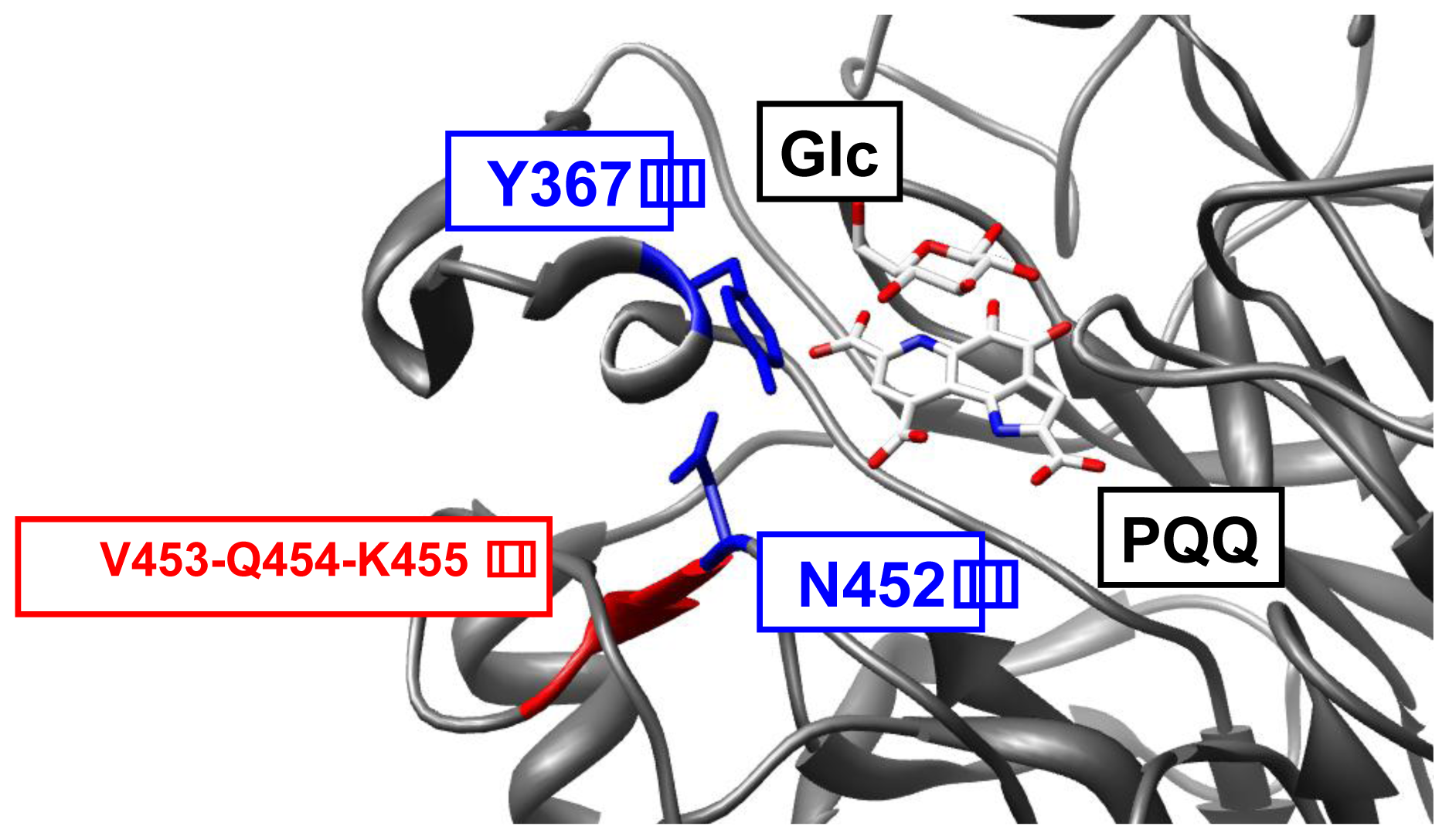
2.1. Construction of Antagonistic Templates
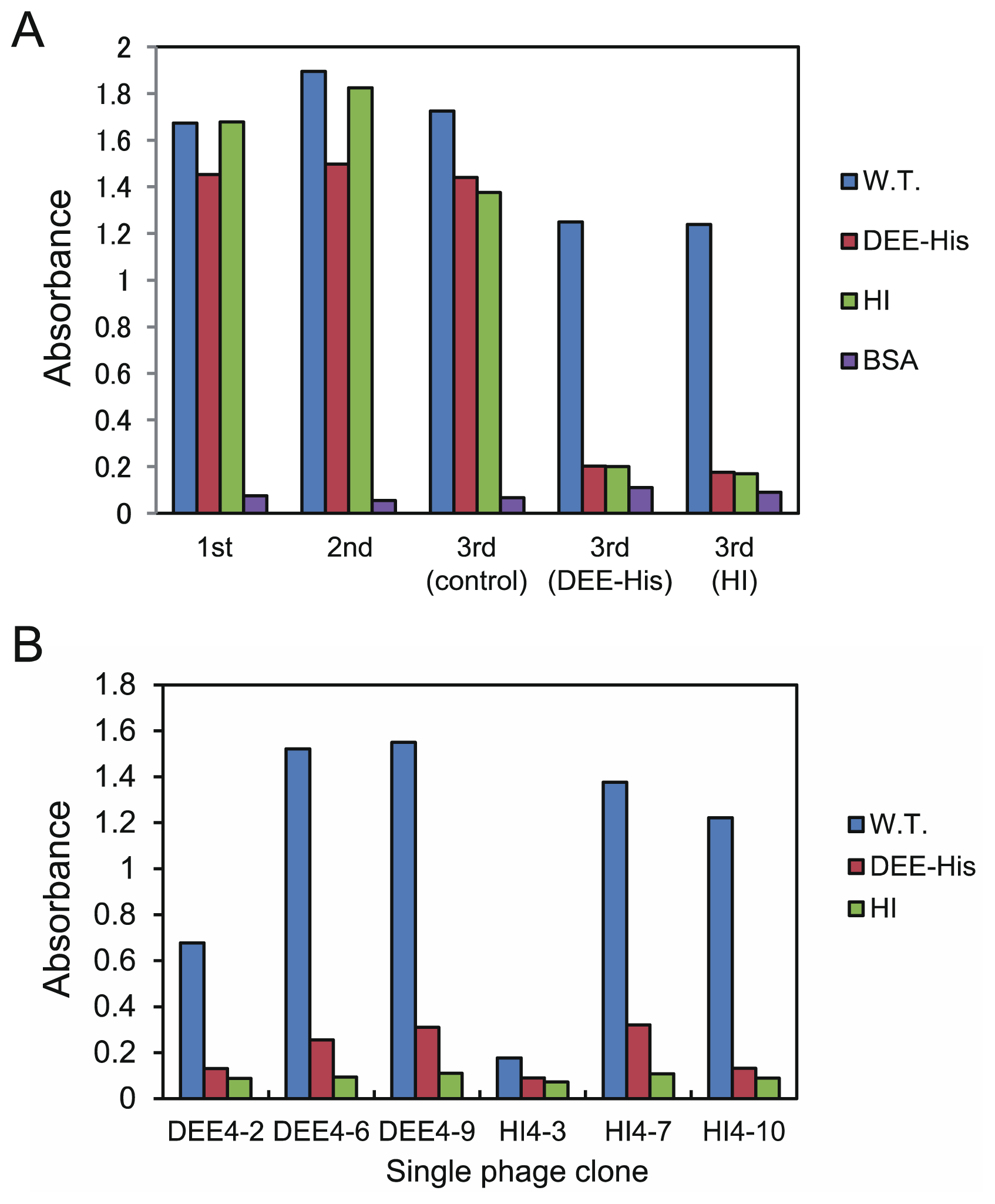
2.2. Antagonistic Template-Based Biopanning
2.3. Binding Assay for the Synthesized Peptides
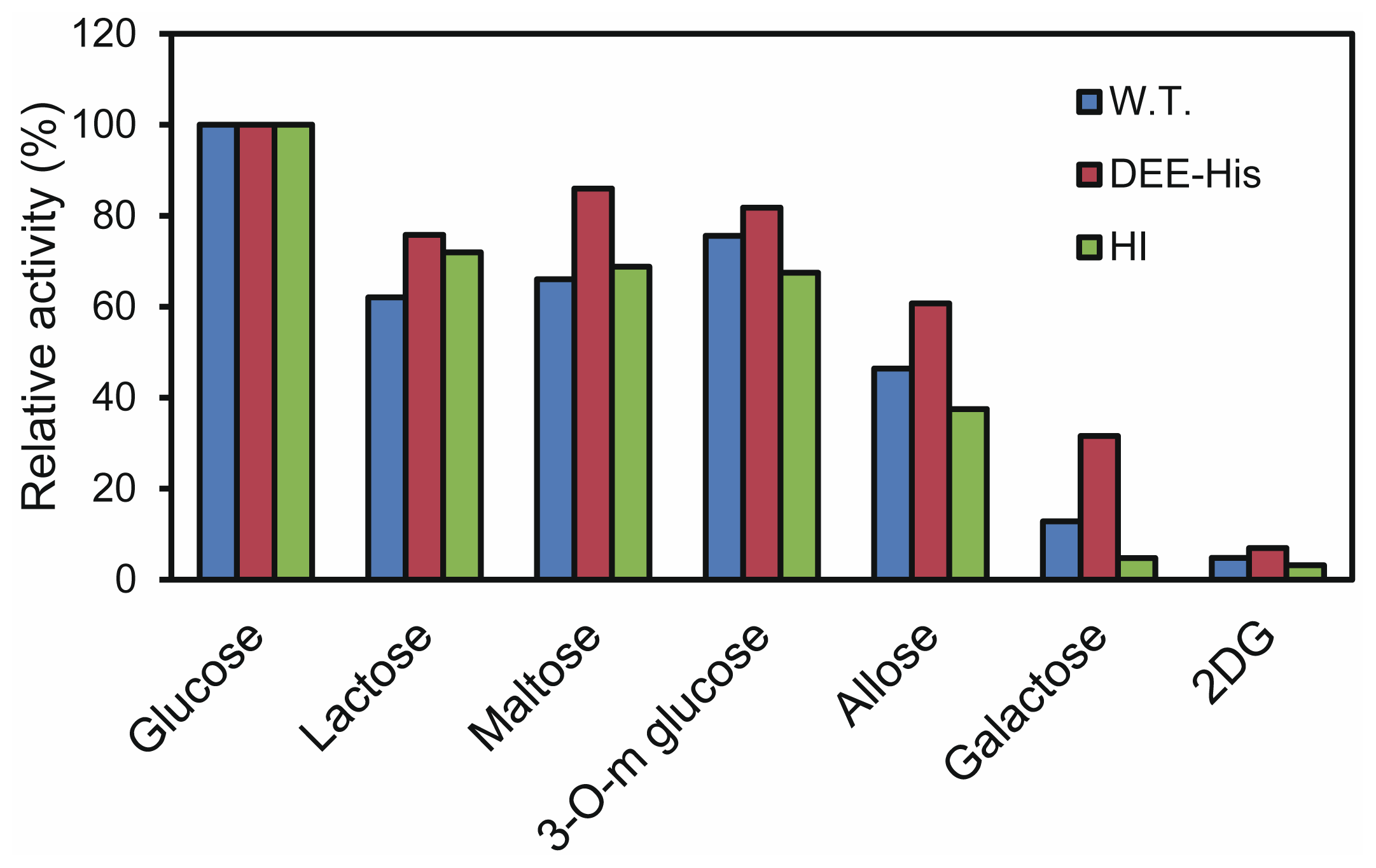
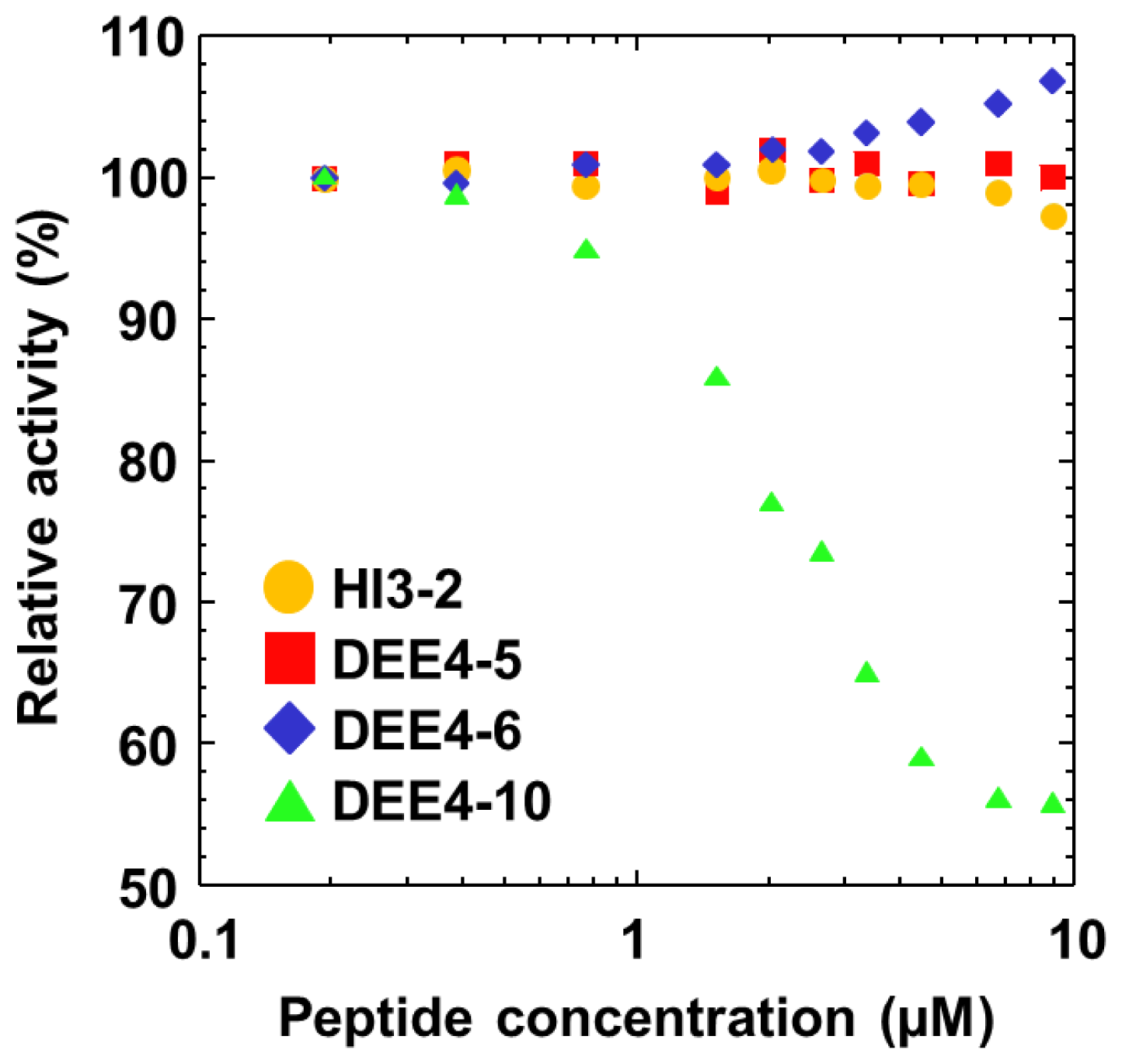
2.4. Inhibition Assay for the Synthesized Peptides
3. Experimental Section
3.1. Preparation of C-Terminal His-Tagged GDH-B (GB-His) and Antagonistic Templates
3.2. Screening Procedures
3.3. Phage ELISA
3.4. Assay for the Binding of the Synthesized Peptide to Wild-Type GDH-B and to the Antagonistic Templates
3.5. Inhibition Assay for the Synthesized Peptide and Wild-Type GDH-B
4. Conclusions
Supplementary Information
ijms-14-23244-s001.pdfConflicts of Interest
References
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making antibodies by phage display technology. Annu. Rev. Immunol 1994, 12, 433–455. [Google Scholar]
- Sidhu, S.S.; Lowman, H.B.; Cunningham, B.C.; Wells, J.A. Phage display for selection of novel binding peptides. Methods Enzymol 2000, 328, 333–363. [Google Scholar]
- Hoess, R.H. Protein design and phage display. Chem. Rev 2001, 101, 3205–3218. [Google Scholar]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv 2010, 28, 849–858. [Google Scholar]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar]
- Rebar, E.J.; Pabo, C.O. Zinc finger phage: Affinity selection of fingers with new DNA-binding specificities. Science 1994, 263, 671–673. [Google Scholar]
- Nomura, Y.; Sharma, V.; Yamamura, A.; Yokobayashi, Y. Selection of silk-binding peptides by phage display. Biotechnol. Lett 2011, 33, 1069–1073. [Google Scholar]
- Whaley, S.R.; English, D.S.; Hu, E.L.; Barbara, P.F.; Belcher, A.M. Selection of peptides with semiconductor binding specificity for directed nanocrystal assembly. Nature 2000, 405, 665–668. [Google Scholar]
- Brown, S. Metal-recognition by repeating polypeptides. Nat. Biotechnol 1997, 15, 269–272. [Google Scholar]
- Koivunen, E.; Gay, D.A.; Ruoslahti, E. Selection of peptides binding to the alpha 5 beta 1 integrin from phage display library. J. Biol. Chem 1993, 268, 20205–20210. [Google Scholar]
- Nagai, Y.; Tucker, T.; Ren, H.; Kenan, D.J.; Henderson, B.S.; Keene, J.D.; Strittmatter, W.J.; Burke, J.R. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J. Biol. Chem 2000, 275, 10437–10442. [Google Scholar]
- Goldman, E.R.; Pazirandeh, M.P.; Mauro, J.M.; King, K.D.; Frey, J.C.; Anderson, G.P. Phage-displayed peptides as biosensor reagents. J. Mol. Recognit 2000, 13, 382–387. [Google Scholar]
- Samoylov, A.M.; Samoylova, T.I.; Pathirana, S.T.; Globa, L.P.; Vodyanoy, V.J. Peptide biosensor for recognition of cross-species cell surface markers. J. Mol. Recognit 2002, 15, 197–203. [Google Scholar]
- Wu, P.; Leinonen, J.; Koivunen, E.; Lankinen, H.; Stenman, U.H. Identification of novel prostate-specific antigen-binding peptides modulating its enzyme activity. Eur. J. Biochem 2000, 267, 6212–6220. [Google Scholar]
- Chakravarty, S.; Mitra, N.; Queitsch, I.; Surolia, A.; Varadarajan, R.; Dubel, S. Protein stabilization through phage display. FEBS Lett 2000, 476, 296–300. [Google Scholar]
- Yoshida, H.; Yagi, Y.; Ikebukuro, K.; Sode, K. Improved substrate specificity of water-soluble pyrroloquinoline quinone glucose dehydrogenase by a peptide ligand. Biotechnol. Lett 2003, 25, 301–305. [Google Scholar]
- White, R.R.; Shan, S.; Rusconi, C.P.; Shetty, G.; Dewhirst, M.W.; Kontos, C.D.; Sullenger, B.A. Inhibition of rat corneal angiogenesis by a nuclease-resistant RNA aptamer specific for angiopoietin-2. Proc. Natl. Acad. Sci. USA 2003, 100, 5028–5033. [Google Scholar]
- Seiwert, S.D.; Stines Nahreini, T.; Aigner, S.; Ahn, N.G.; Uhlenbeck, O.C. RNA aptamers as pathway-specific MAP kinase inhibitors. Chem. Biol 2000, 7, 833–843. [Google Scholar]
- Andreola, M.L.; Pileur, F.; Calmels, C.; Ventura, M.; Tarrago-Litvak, L.; Toulme, J.J.; Litvak, S. DNA aptamers selected against the HIV-1 RNase H display in vitro antiviral activity. Biochemistry 2001, 40, 10087–10094. [Google Scholar]
- Yribarren, A.S.; Thomas, D.; Friboulet, A.; Avalle, B. Selection of peptides inhibiting a beta-lactamase-like activity. Eur. J. Biochem 2003, 270, 2789–2795. [Google Scholar]
- Stoop, A.A.; Craik, C.S. Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nat. Biotechnol 2003, 21, 1063–1068. [Google Scholar]
- Zhang, M.Y.; Shu, Y.; Phogat, S.; Xiao, X.; Cham, F.; Bouma, P.; Choudhary, A.; Feng, Y.R.; Sanz, I.; Rybak, S.; et al. Broadly cross-reactive HIV neutralizing human monoclonal antibody Fab selected by sequential antigen panning of a phage display library. J. Immunol. Methods 2003, 283, 17–25. [Google Scholar]
- Meiring, M.S.; Litthauer, D.; Harsfalvi, J.; van Wyk, V.; Badenhorst, P.N.; Kotze, H.F. In vitro effect of a thrombin inhibition peptide selected by phage display technology. Thromb. Res 2002, 107, 365–371. [Google Scholar]
- Niu, W.; Jiang, N.; Hu, Y. Detection of proteins based on amino acid sequences by multiple aptamers against tripeptides. Anal. Biochem 2007, 362, 126–135. [Google Scholar]
- Yagi, Y.; Terada, K.; Noma, T.; Ikebukuro, K.; Sode, K. In silico panning for a non-competitive peptide inhibitor. BMC Bioinforma 2007, 8, 11. [Google Scholar]
- Abe, K.; Kobayashi, N.; Sode, K.; Ikebukuro, K. Peptide ligand screening of alpha-synuclein aggregation modulators by in silico panning. BMC Bioinforma 2007, 8, 451. [Google Scholar]
- Oubrie, A.; Rozeboom, H.J.; Dijkstra, B.W. Active-site structure of the soluble quinoprotein glucose dehydrogenase complexed with methylhydrazine: A covalent cofactor-inhibitor complex. Proc. Natl. Acad. Sci. USA 1999, 96, 11787–11791. [Google Scholar]
- Oubrie, A.; Rozeboom, H.J.; Kalk, K.H.; Duine, J.A.; Dijkstra, B.W. The 1.7 Å crystal structure of the apo form of the soluble quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus reveals a novel internal conserved sequence repeat. J. Mol. Biol 1999, 289, 319–333. [Google Scholar]
- Oubrie, A.; Rozeboom, H.J.; Kalk, K.H.; Olsthoorn, A.J.; Duine, J.A.; Dijkstra, B.W. Structure and mechanism of soluble quinoprotein glucose dehydrogenase. EMBO J 1999, 18, 5187–5194. [Google Scholar]
- Murzin, A.G. Structural principles for the propeller assembly of beta-sheets: The preference for seven-fold symmetry. Proteins 1992, 14, 191–201. [Google Scholar]
- Chen, C.K.; Chan, N.L.; Wang, A.H. The many blades of the beta-propeller proteins: Conserved but versatile. Trends Biochem. Sci 2011, 36, 553–561. [Google Scholar]
- Tachino, A.; Igarashi, S.; Sode, K. W-motif exchange between beta-propeller proteins. Protein J 2007, 26, 153–158. [Google Scholar]
- Igarashi, S.; Okuda, J.; Ikebukuro, K.; Sode, K. Molecular engineering of PQQGDH and its applications. Arch. Biochem. Biophys 2004, 428, 52–63. [Google Scholar]
- Hamamatsu, N.; Suzumura, A.; Nomiya, Y.; Sato, M.; Aita, T.; Nakajima, M.; Husimi, Y.; Shibanaka, Y. Modified substrate specificity of pyrroloquinoline quinone glucose dehydrogenase by biased mutation assembling with optimized amino acid substitution. Appl. Microbiol. Biotechnol 2006, 73, 607–617. [Google Scholar]
- Durand, F.; Limoges, B.; Mano, N.; Mavre, F.; Miranda-Castro, R.; Saveant, J.M. Effect of substrate inhibition and cooperativity on the electrochemical responses of glucose dehydrogenase. Kinetic characterization of wild and mutant types. J. Am. Chem. Soc 2011, 133, 12801–12809. [Google Scholar]
- Igarashi, S.; Ohtera, T.; Yoshida, H.; Witarto, A.B.; Sode, K. Construction and characterization of mutant water-soluble PQQ glucose dehydrogenases with altered K(m) values—Site-directed mutagenesis studies on the putative active site. Biochem. Biophys. Res. Commun 1999, 264, 820–824. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [Google Scholar]
- Tokuriki, N.; Tawfik, D.S. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 2009, 459, 668–673. [Google Scholar]
- Cleton-Jansen, A.M.; Goosen, N.; Fayet, O.; van de Putte, P. Cloning, mapping, and sequencing of the gene encoding Escherichia coli quinoprotein glucose dehydrogenase. J. Bacteriol 1990, 172, 6308–6315. [Google Scholar]
- Sode, K.; Witarto, A.B.; Watanabe, K.; Noda, K.; Ito, S.; Tsugawa, W. Over expression of PQQ glucose dehydrogenase in Escherichia coli under holo enzyme forming condition. Biotechnol. Lett 1994, 16, 1265–1268. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Km (mM) | Vmax (U/mg protein) |
|---|---|---|
| GDH-B | 25 | 5200 |
| GB-His * | 24 | 3950 |
| HI | 18 | 170 |
| DEE-His | 91 | 28 |
| Name | Sequence | Kd (μM) | ||
|---|---|---|---|---|
| GDH-B | DEE-His | HI | ||
| DEE4–5 | LGDSSNSQVSLN | 13 | N.B. | N.B. |
| DEE4–6 | SDLSPIQSLSAI | 2.5 | N.B. | 230 |
| DEE4–10 | NSTHHHHFATIW | 0.7 | N.B. | N.B. |
| HI3-2 | ELITNSETTQWF | 2.5 | N.B. | 88 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abe, K.; Yoshida, W.; Terada, K.; Yagi-Ishii, Y.; Ferri, S.; Ikebukuro, K.; Sode, K. Screening of Peptide Ligands for Pyrroloquinoline Quinone Glucose Dehydrogenase Using Antagonistic Template-Based Biopanning. Int. J. Mol. Sci. 2013, 14, 23244-23256. https://doi.org/10.3390/ijms141223244
Abe K, Yoshida W, Terada K, Yagi-Ishii Y, Ferri S, Ikebukuro K, Sode K. Screening of Peptide Ligands for Pyrroloquinoline Quinone Glucose Dehydrogenase Using Antagonistic Template-Based Biopanning. International Journal of Molecular Sciences. 2013; 14(12):23244-23256. https://doi.org/10.3390/ijms141223244
Chicago/Turabian StyleAbe, Koichi, Wataru Yoshida, Kotaro Terada, Yukiko Yagi-Ishii, Stefano Ferri, Kazunori Ikebukuro, and Koji Sode. 2013. "Screening of Peptide Ligands for Pyrroloquinoline Quinone Glucose Dehydrogenase Using Antagonistic Template-Based Biopanning" International Journal of Molecular Sciences 14, no. 12: 23244-23256. https://doi.org/10.3390/ijms141223244