Apigenin Protects Endothelial Cells from Lipopolysaccharide (LPS)-Induced Inflammation by Decreasing Caspase-3 Activation and Modulating Mitochondrial Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
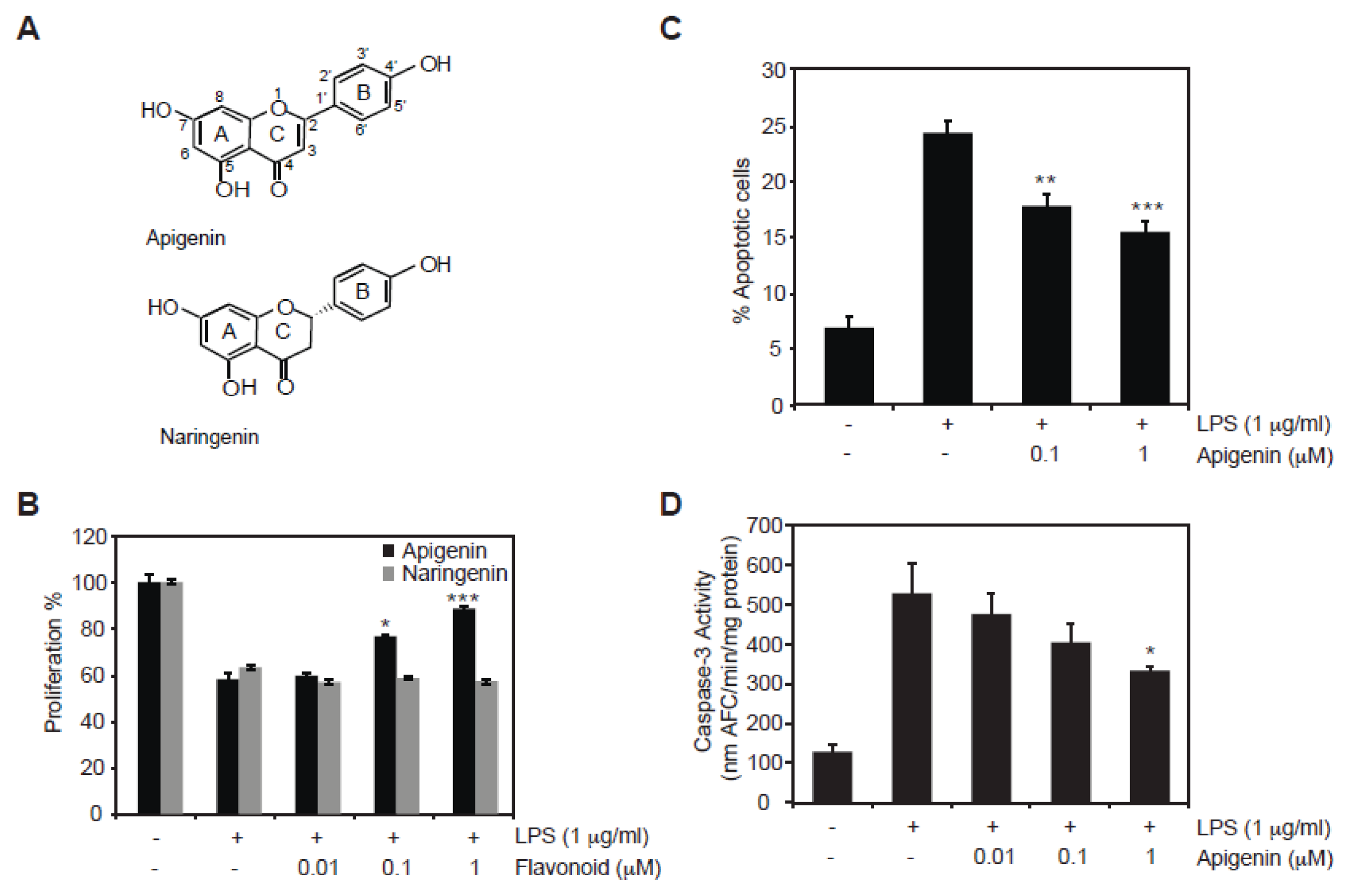
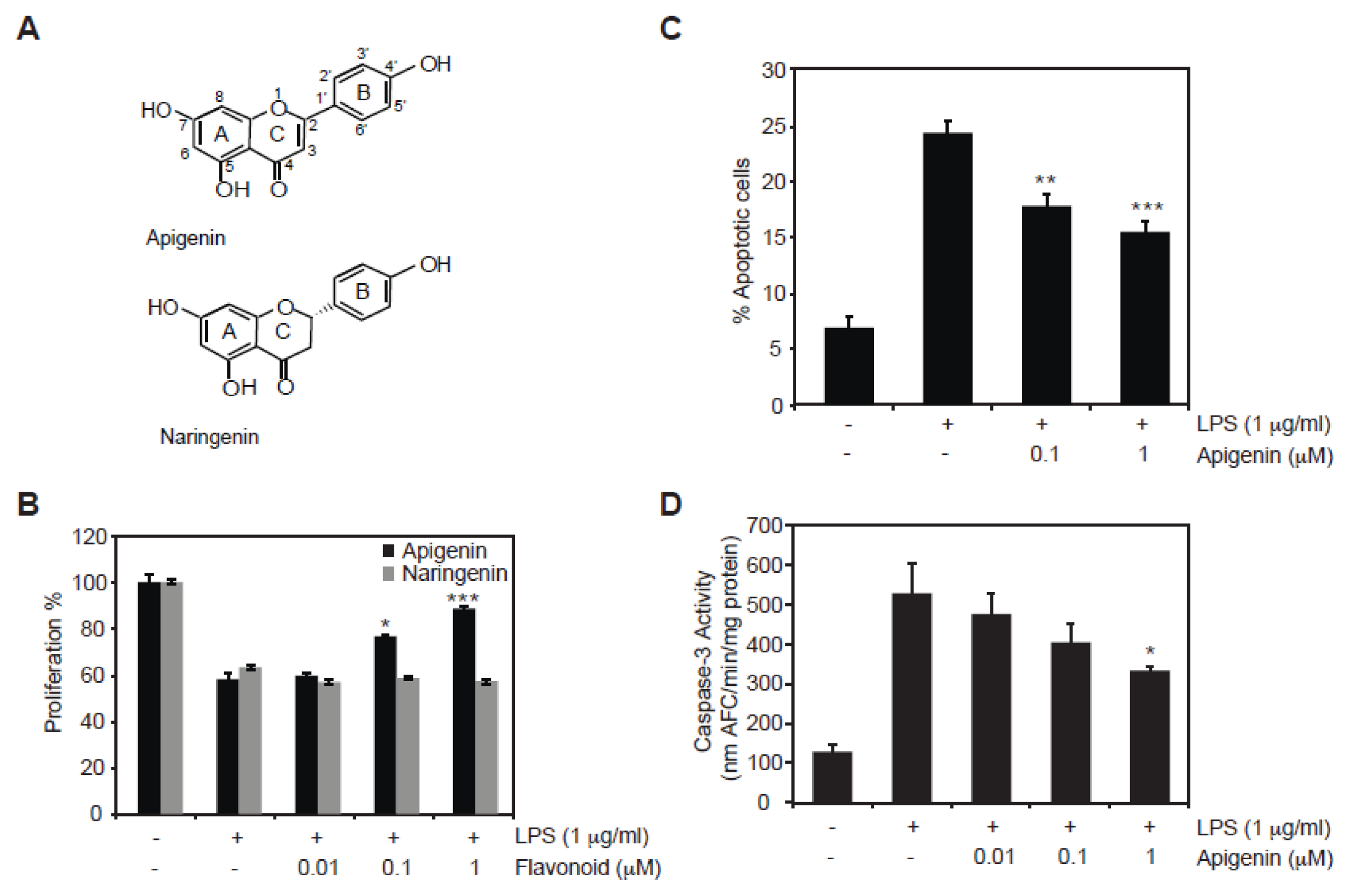
2.1. Protective Role of Apigenin against LPS-Induced Cell Death
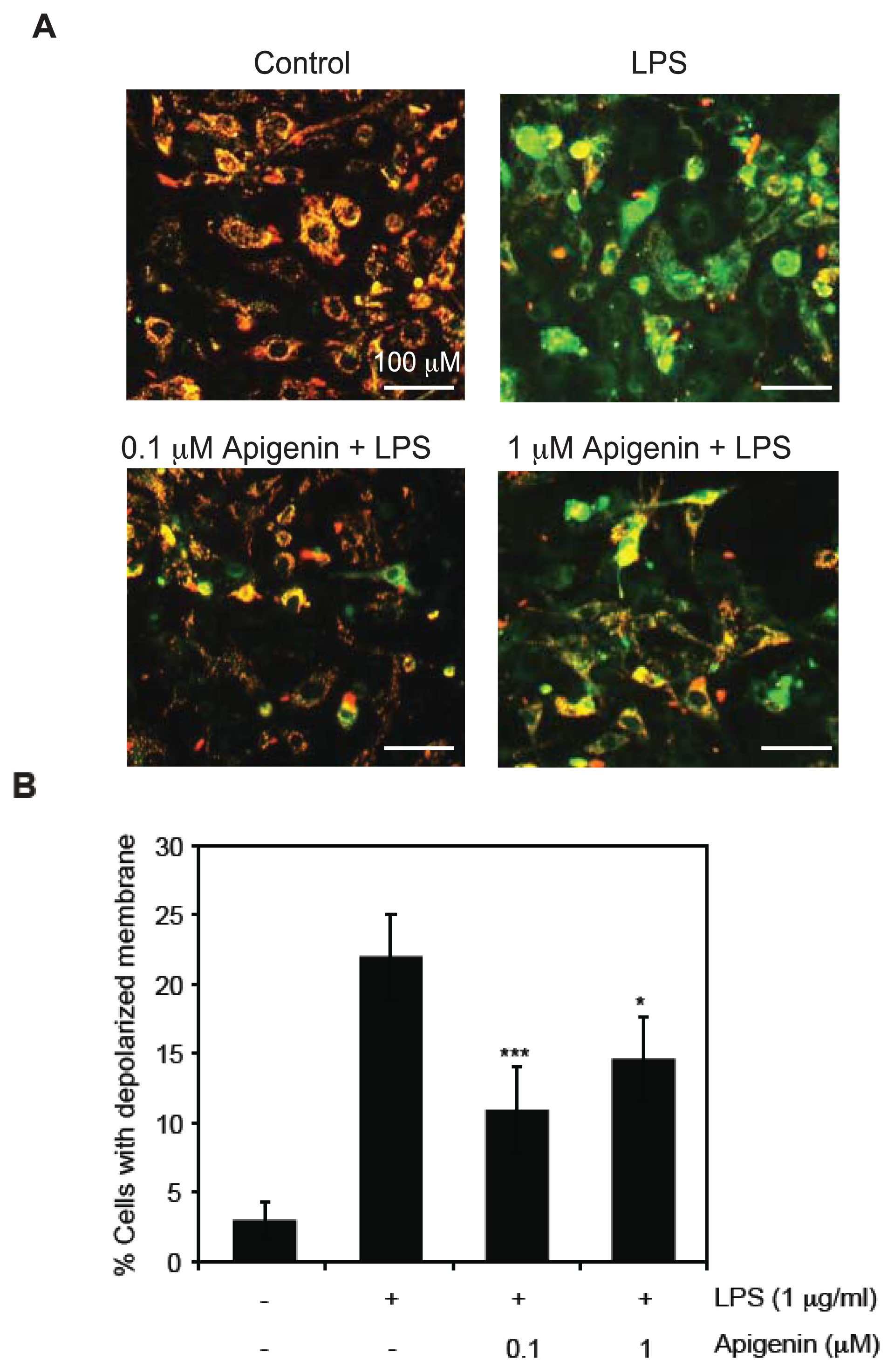
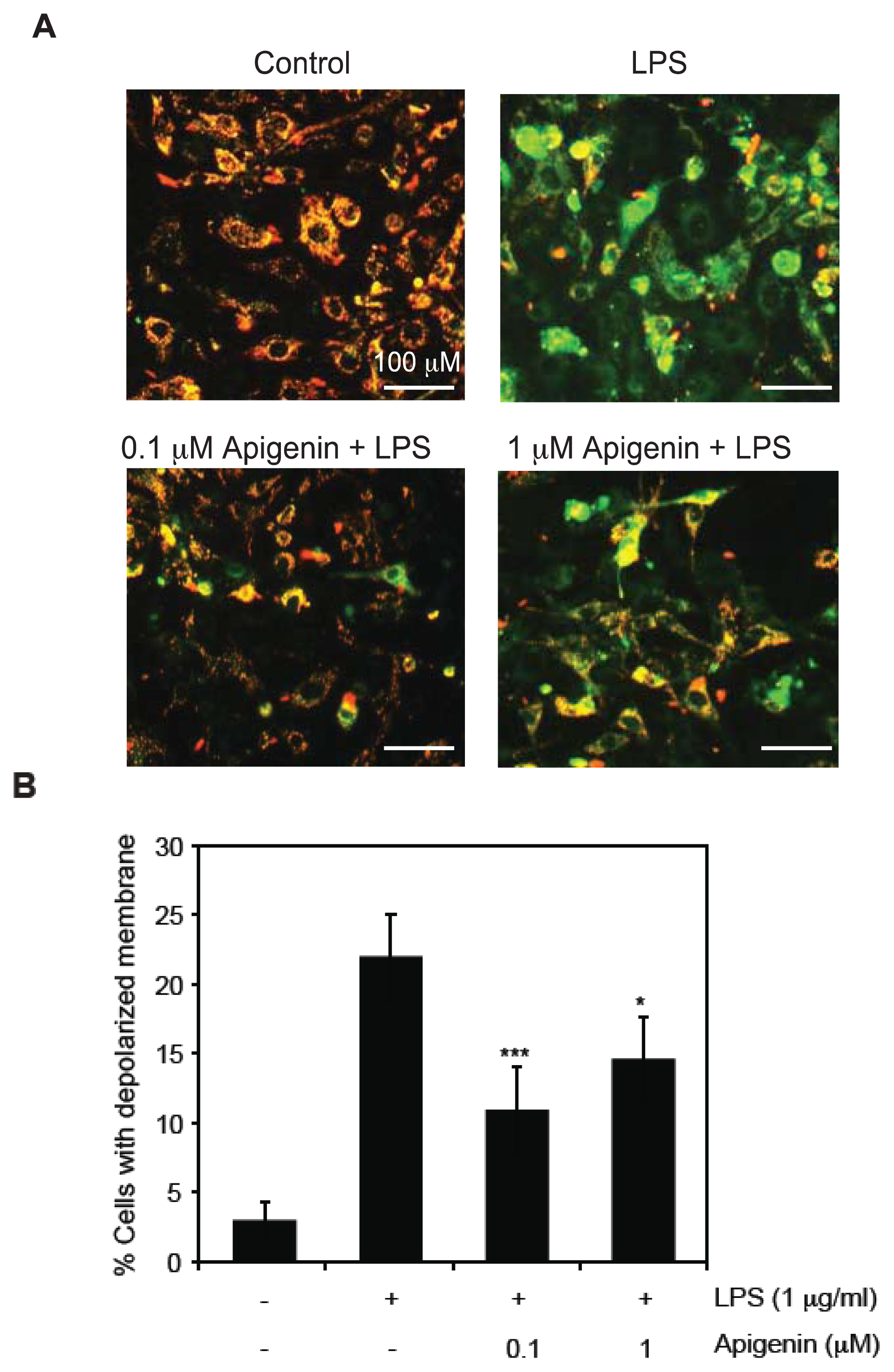
2.2. Apigenin Protects from LPS-Induced Mitochondrial Dysfunction
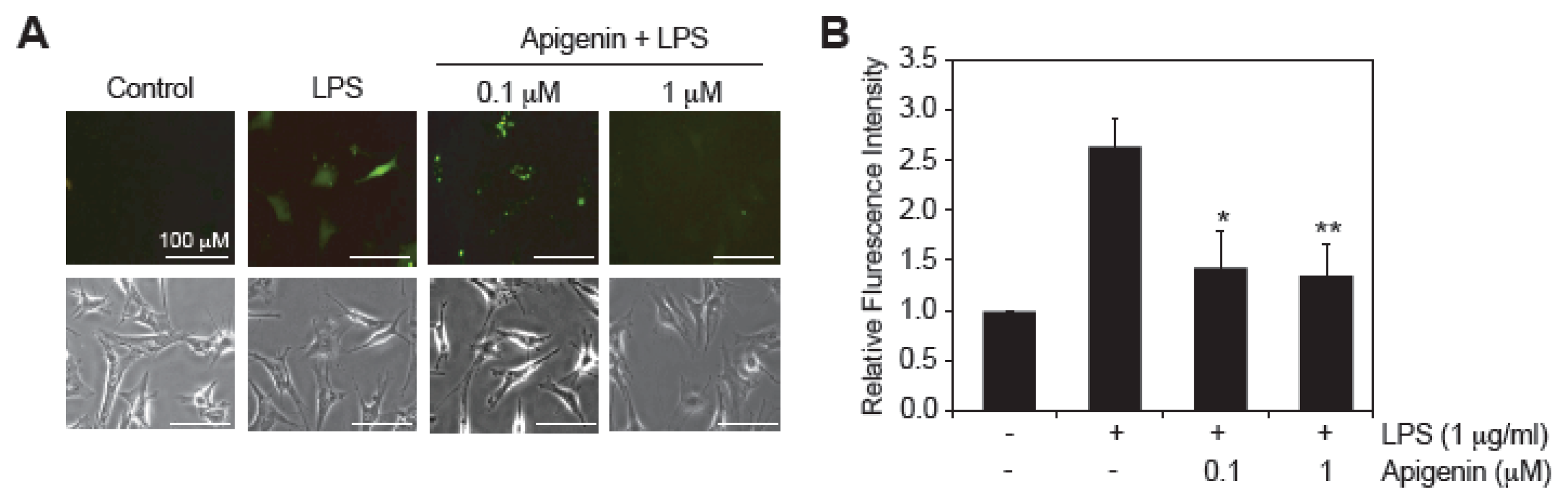
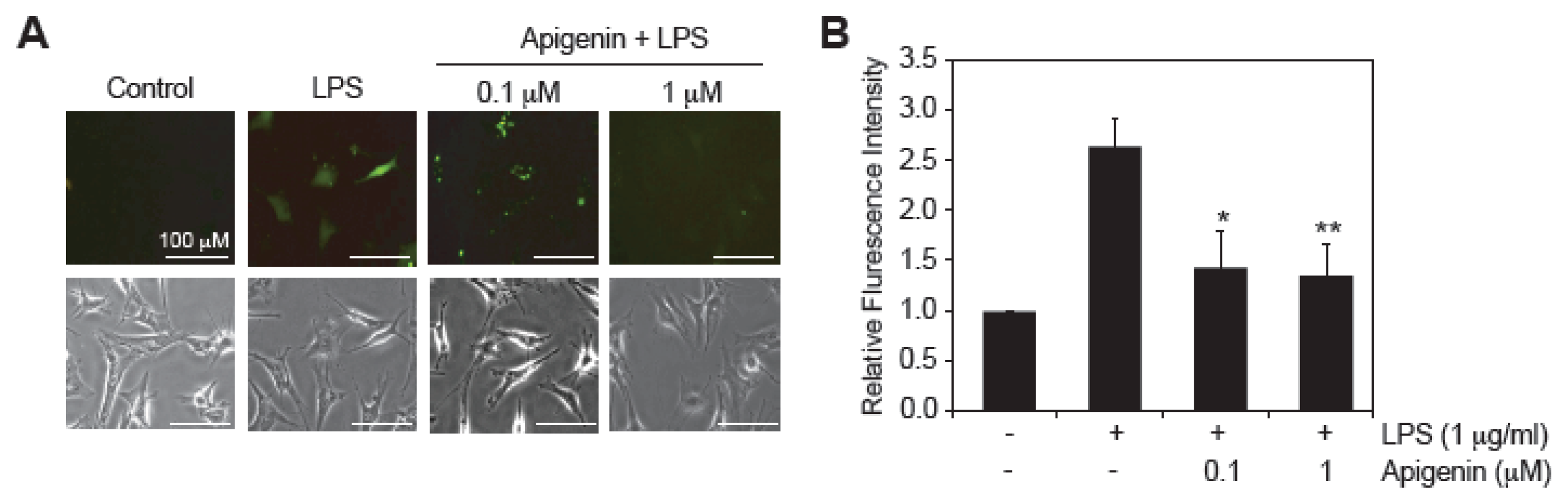
2.3. Apigenin Decreases LPS-Induced ROS Production
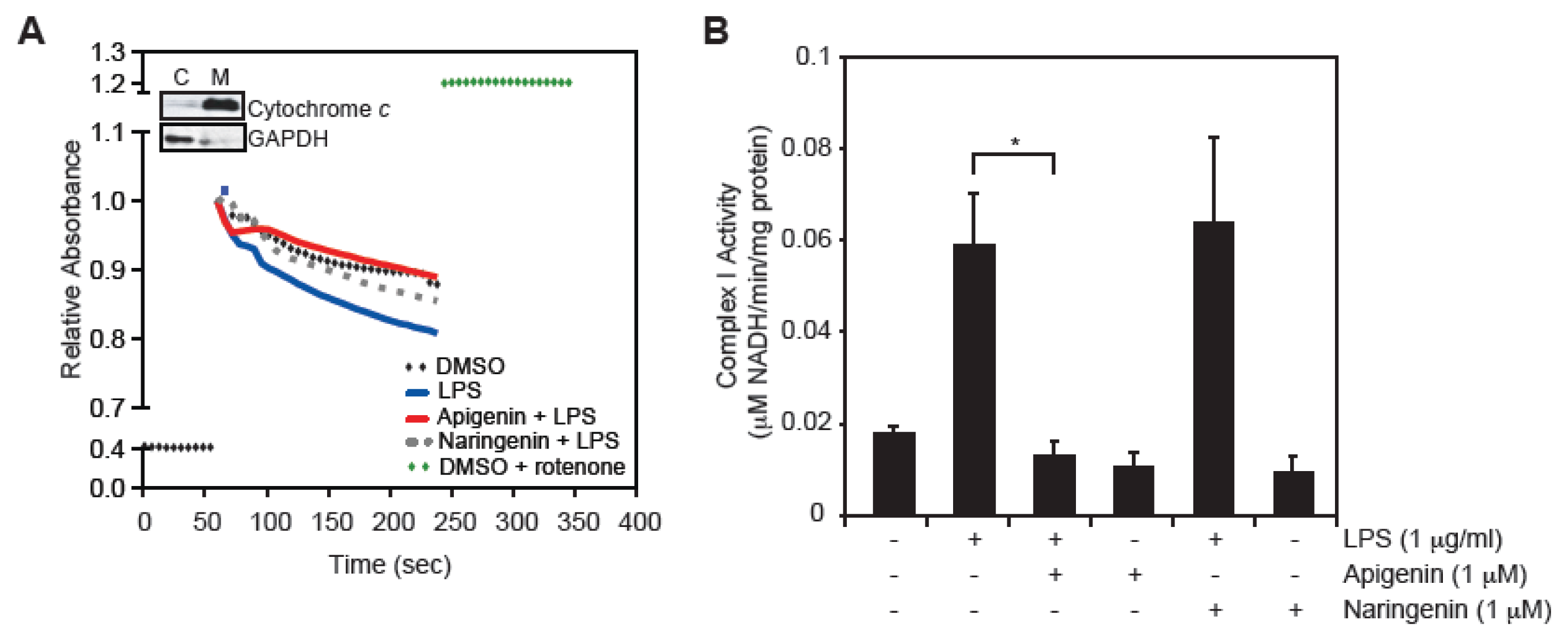
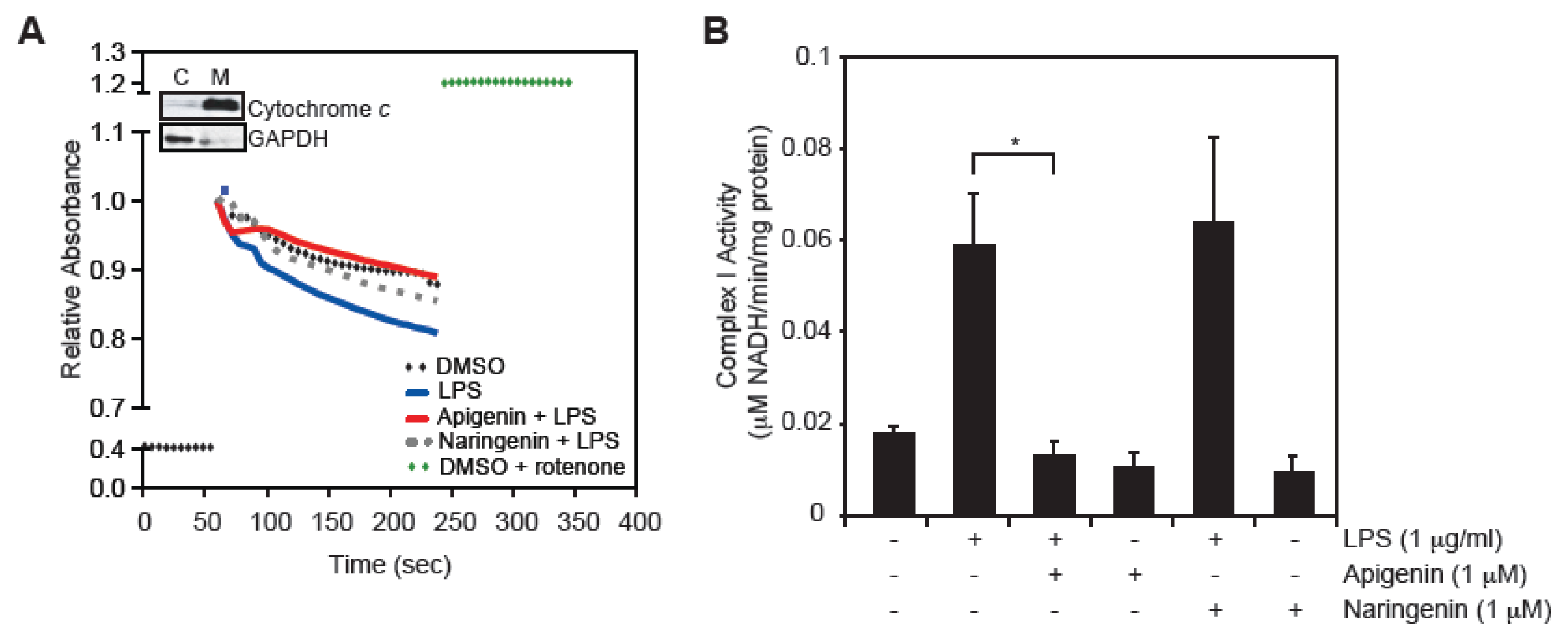
2.4. Apigenin Restores LPS-Induced Mitochondrial Complex I Activity
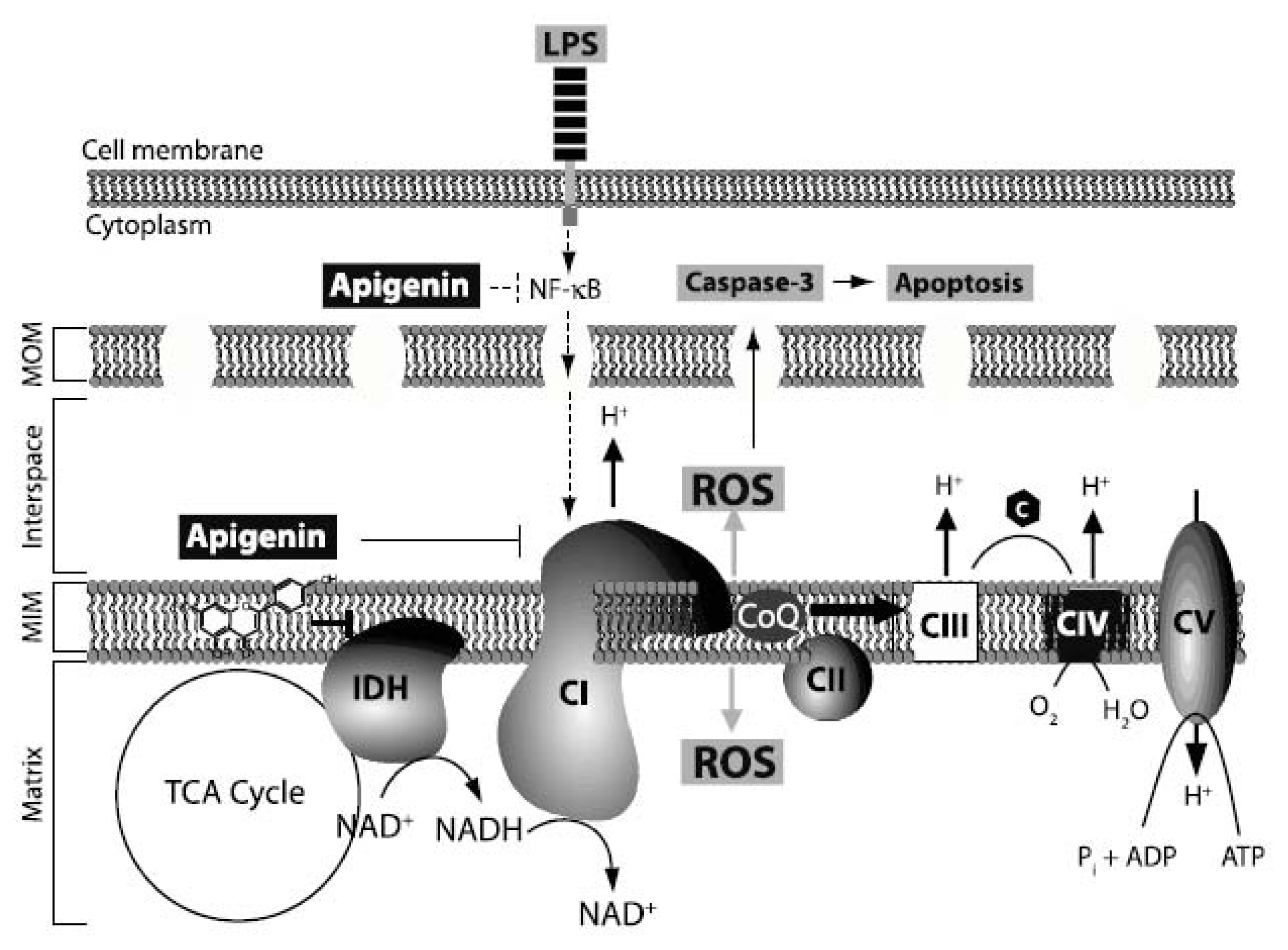
2.5. Discussion
3. Experimental Section
3.1. Chemicals, Cell Lines and Cultures
3.2. Cell Viability Assay (MTS Assay)
3.3. Determination of Caspase-3 Activity
3.4. Detection of Apoptosis
3.5. Determination of Mitochondrial Membrane Potential (MMP)
3.6. Detection of Reactive Oxygen Species (ROS)
3.7. Complex I Activity Assay
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med 2003, 348, 138–150. [Google Scholar]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis 2013, 13, 260–268. [Google Scholar]
- Lolis, E.; Bucala, R. Therapeutic approaches to innate immunity: Severe sepsis and septic shock. Nat. Rev. Drug Discov 2003, 2, 635–645. [Google Scholar]
- Fahy, R.J.; Doseff, A.I.; Wewers, M.D. Spontaneous human monocyte apoptosis utilizes a caspase-3-dependent pathway that is blocked by endotoxin and is independent of caspase-1. J. Immunol 1999, 163, 1755–1762. [Google Scholar]
- Henkels, K.M.; Frondorf, K.; Gonzalez-Mejia, M.E.; Doseff, A.L.; Gomez-Cambronero, J. IL-8-induced neutrophil chemotaxis is mediated by Janus kinase 3 (JAK3). FEBS Lett 2011, 585, 159–166. [Google Scholar]
- Bannerman, D.D.; Goldblum, S.E. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol 2003, 284, L899–L914. [Google Scholar]
- Aoki, M.; Nata, T.; Morishita, R.; Matsushita, H.; Nakagami, H.; Yamamoto, K.; Yamazaki, K.; Nakabayashi, M.; Ogihara, T.; Kaneda, Y. Endothelial apoptosis induced by oxidative stress through activation of NF-κB: Antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension 2001, 38, 48–55. [Google Scholar]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth 2011, 107, 57–64. [Google Scholar]
- Aird, W.C. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003, 101, 3765–3777. [Google Scholar]
- Crouser, E.D. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion 2004, 4, 729–741. [Google Scholar]
- Carre, J.E.; Orban, J.C.; Re, L.; Felsmann, K.; Iffert, W.; Bauer, M.; Suliman, H.B.; Piantadosi, C.A.; Mayhew, T.M.; Breen, P.; et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am. J. Respir. Crit. Care Med 2010, 182, 745–751. [Google Scholar]
- Zang, Q.; Maass, D.L.; Tsai, S.J.; Horton, J.W. Cardiac mitochondrial damage and inflammation responses in sepsis. Surg. Infect. (Larchmt. ) 2007, 8, 41–54. [Google Scholar]
- Zapelini, P.H.; Rezin, G.T.; Cardoso, M.R.; Ritter, C.; Klamt, F.; Moreira, J.C.; Streck, E.L.; Dal-Pizzol, F. Antioxidant treatment reverses mitochondrial dysfunction in a sepsis animal model. Mitochondrion 2008, 8, 211–218. [Google Scholar]
- Andrades, M.E.; Ritter, C.; Dal-Pizzol, F. The role of free radicals in sepsis development. Front. Biosci 2009, 1, 277–287. [Google Scholar]
- Galley, H.F. Bench-to-bedside review: Targeting antioxidants to mitochondria in sepsis. Crit. Care 2010, 14, 230. [Google Scholar]
- Lowes, D.A.; Thottakam, B.M.; Webster, N.R.; Murphy, M.P.; Galley, H.F. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic. Biol. Med 2008, 45, 1559–1565. [Google Scholar]
- Havsteen, B.H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther 2002, 96, 67–202. [Google Scholar]
- Estruch, R.; Ros, E.; Salas-Salvado, J.; Covas, M.I.; Corella, D.; Aros, F.; Gomez-Gracia, E.; Ruiz-Gutierrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N. Engl. J. Med 2013, 368, 1279–1290. [Google Scholar]
- Nicholas, C.; Batra, S.; Vargo, M.A.; Voss, O.H.; Gavrilin, M.A.; Wewers, M.D.; Guttridge, D.C.; Grotewold, E.; Doseff, A.I. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-κB through the suppression of p65 phosphorylation. J. Immunol 2007, 179, 7121–7127. [Google Scholar]
- Hostetler, G.; Riedl, K.; Cardenas, H.; Diosa-Toro, M.; Arango, D.; Schwartz, S.; Doseff, A.I. Flavone deglycosylation increases their anti-inflammatory activity and absorption. Mol. Nutr. Food Res 2012, 56, 558–569. [Google Scholar]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Remesy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr 2005, 81, 230S–242S. [Google Scholar]
- Day, A.J.; Canada, F.J.; Diaz, J.C.; Kroon, P.A.; McLauchlan, R.; Faulds, C.B.; Plumb, G.W.; Morgan, M.R.; Williamson, G. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett 2000, 468, 166–170. [Google Scholar]
- Nielsen, I.L.; Chee, W.S.; Poulsen, L.; Offord-Cavin, E.; Rasmussen, S.E.; Frederiksen, H.; Enslen, M.; Barron, D.; Horcajada, M.N.; Williamson, G. Bioavailability is improved by enzymatic modification of the citrus flavonoid hesperidin in humans: A randomized, double-blind, crossover trial. J. Nutr 2006, 136, 404–408. [Google Scholar]
- Richelle, M.; Pridmore-Merten, S.; Bodenstab, S.; Enslen, M.; Offord, E.A. Hydrolysis of isoflavone glycosides to aglycones by beta-glycosidase does not alter plasma and urine isoflavone pharmacokinetics in postmenopausal women. J. Nutr 2002, 132, 2587–2592. [Google Scholar]
- Vargo, M.A.; Voss, O.H.; Poustka, F.; Cardounel, A.J.; Grotewold, E.; Doseff, A.I. Apigenin-induced-apoptosis is mediated by the activation of PKCδ and caspases in leukemia cells. Biochem. Pharmacol 2006, 72, 681–692. [Google Scholar]
- Arango, D.; Morohashi, K.; Yilmaz, A.; Kuramochi, K.; Parihar, A.; Brahimaj, B.; Grotewold, E.; Doseff, A.I. Molecular basis for the action of a dietary flavonoid revealed by the comprehensive identification of apigenin human targets. Proc. Natl. Acad. Sci. USA 2013, 1303726110. [Google Scholar] [CrossRef]
- Tseng, H.W.; Juan, H.F.; Huang, H.C.; Lin, J.Y.; Sinchaikul, S.; Lai, T.C.; Chen, C.F.; Chen, S.T.; Wang, G.J. Lipopolysaccharide-stimulated responses in rat aortic endothelial cells by a systems biology approach. Proteomics 2006, 6, 5915–5928. [Google Scholar]
- Doseff, A.I.; Baker, J.H., Jr; Bourgeois, T.A.; Wewers, M.D. Interleukin-4-induced apoptosis entails caspase activation and suppression of extracellular signal-regulated kinase phosphorylation. Am. J. Respir. Cell. Mol. Biol. 2003, 29, 367–374. [Google Scholar]
- Ceaser, E.K.; Ramachandran, A.; Levonen, A.L.; Darley-Usmar, V.M. Oxidized low-density lipoprotein and 15-deoxy-delta 12,14-PGJ2 increase mitochondrial complex I activity in endothelial cells. Am. J. Physiol. Heart Circ. Physiol 2003, 285, H2298–H2308. [Google Scholar]
- Cannino, G.; El-Khoury, R.; Pirinen, M.; Hutz, B.; Rustin, P.; Jacobs, H.T.; Dufour, E. Glucose modulates respiratory complex I activity in response to acute mitochondrial dysfunction. J. Biol. Chem 2012, 287, 38729–38740. [Google Scholar]
- Tatsuta, T.; Model, K.; Langer, T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell 2005, 16, 248–259. [Google Scholar]
- Choi, K.B.; Wong, F.; Harlan, J.M.; Chaudhary, P.M.; Hood, L.; Karsan, A. Lipopolysaccharide mediates endothelial apoptosis by a FADD-dependent pathway. J. Biol. Chem 1998, 273, 20185–20188. [Google Scholar]
- Hull, C.; McLean, G.; Wong, F.; Duriez, P.J.; Karsan, A. Lipopolysaccharide signals an endothelial apoptosis pathway through TNF receptor-associated factor 6-mediated activation of c-Jun NH2-terminal kinase. J. Immunol 2002, 169, 2611–2618. [Google Scholar]
- Scott, S.V.; Cassidy-Stone, A.; Meeusen, S.L.; Nunnari, J. Staying in aerobic shape: how the structural integrity of mitochondria and mitochondrial DNA is maintained. Curr. Opin. Cell Biol 2003, 15, 482–488. [Google Scholar]
- Donovan, M.; Cotter, T.G. Control of mitochondrial integrity by Bcl-2 family members and caspase-independent cell death. Biochim. Biophys 2004, 1644, 133–147. [Google Scholar]
- Lee, W.L.; Slutsky, A.S. Sepsis and endothelial permeability. N. Engl. J. Med 2010, 363, 689–691. [Google Scholar]
- Larche, J.; Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Marchetti, P.; Chopin, C.; Neviere, R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J. Am. College Cardiol 2006, 48, 377–385. [Google Scholar]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar]
- Gellerich, F.N.; Trumbeckaite, S.; Opalka, J.R.; Gellerich, J.F.; Chen, Y.; Neuhof, C.; Redl, H.; Werdan, K.; Zierz, S. Mitochondrial dysfunction in sepsis: Evidence from bacteraemic baboons and endotoxaemic rabbits. Biosci. Rep 2002, 22, 99–113. [Google Scholar]
- Vanasco, V.; Cimolai, M.C.; Evelson, P.; Alvarez, S. The oxidative stress and the mitochondrial dysfunction caused by endotoxemia are prevented by alpha-lipoic acid. Free Radic. Res 2008, 42, 815–823. [Google Scholar]
- Vanasco, V.; Magnani, N.D.; Cimolai, M.C.; Valdez, L.B.; Evelson, P.; Boveris, A.; Alvarez, S. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and ATP content without affecting membrane potential. J. Bioenerg. Biomembr 2012, 44, 243–252. [Google Scholar]
- Tanaka, J.; Kono, Y.; Shimahara, Y.; Sato, T.; Jones, R.T.; Cowley, R.A.; Trump, B.F. A study of oxidative phosphorylative activity and calcium-induced respiration of rat liver mitochondria following living Escherichia coli injection. Adv. Shock Res 1982, 7, 77–90. [Google Scholar]
- Taylor, D.E.; Kantrow, S.P.; Piantadosi, C.A. Mitochondrial respiration after sepsis and prolonged hypoxia. Am. J. Physiol 1998, 275, L139–L144. [Google Scholar]
- Kozlov, A.V.; Gille, L.; Miller, I.; Piskernik, C.; Haindl, S.; Staniek, K.; Nohl, H.; Bahrami, S.; Ohlinger, W.; Gemeiner, M.; et al. Opposite effects of endotoxin on mitochondrial and endoplasmic reticulum functions. Biochem. Biophys. Res. Commun 2007, 352, 91–96. [Google Scholar]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol 2011, 13, 1272–1279. [Google Scholar]
- Munoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis 2012, 3, e248. [Google Scholar]
- Lagoa, R.; Graziani, I.; Lopez-Sanchez, C.; Garcia-Martinez, V.; Gutierrez-Merino, C. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. Biochim. Biophys 2011, 1807, 1562–1572. [Google Scholar]
- Santos, A.C.; Uyemura, S.A.; Lopes, J.L.; Bazon, J.N.; Mingatto, F.E.; Curti, C. Effect of naturally occurring flavonoids on lipid peroxidation and membrane permeability transition in mitochondria. Free Radic. Biol. Med 1998, 24, 1455–1461. [Google Scholar]
- Das, A.; Gopalakrishnan, B.; Voss, O.H.; Doseff, A.I.; Villamena, F.A. Inhibition of ROS-induced apoptosis in endothelial cells by nitrone spin traps via induction of phase II enzymes and suppression of mitochondria-dependent pro-apoptotic signaling. Biochem. Pharmacol 2012, 84, 486–497. [Google Scholar]
- Voss, O.H.; Kim, S.; Wewers, M.D.; Doseff, A.I. Regulation of monocyte apoptosis by the protein kinase Cδ-dependent phosphorylation of caspase-3. J. Biol. Chem 2005, 280, 17371–17379. [Google Scholar]
- Voss, O.H.; Batra, S.; Kolattukudy, S.J.; Gonzalez-Mejia, M.E.; Smith, J.B.; Doseff, A.I. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J. Biol. Chem 2007, 282, 25088–25099. [Google Scholar]
- Arango, D.; Parihar, A.; Villamena, F.A.; Wang, L.; Freitas, M.A.; Grotewold, E.; Doseff, A.I. Apigenin induces DNA damage through the PKCδ-dependent activation of ATM and H2AX causing down-regulation of genes involved in cell cycle control and DNA repair. Biochem. Pharmacol 2012, 84, 1571–1580. [Google Scholar]
- Reisch, A.S.; Elpeleg, O. Biochemical assays for mitochondrial activity: assays of TCA cycle enzymes and PDHc. Method Cell Biol 2007, 80, 199–222. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Duarte, S.; Arango, D.; Parihar, A.; Hamel, P.; Yasmeen, R.; Doseff, A.I. Apigenin Protects Endothelial Cells from Lipopolysaccharide (LPS)-Induced Inflammation by Decreasing Caspase-3 Activation and Modulating Mitochondrial Function. Int. J. Mol. Sci. 2013, 14, 17664-17679. https://doi.org/10.3390/ijms140917664
Duarte S, Arango D, Parihar A, Hamel P, Yasmeen R, Doseff AI. Apigenin Protects Endothelial Cells from Lipopolysaccharide (LPS)-Induced Inflammation by Decreasing Caspase-3 Activation and Modulating Mitochondrial Function. International Journal of Molecular Sciences. 2013; 14(9):17664-17679. https://doi.org/10.3390/ijms140917664
Chicago/Turabian StyleDuarte, Silvia, Daniel Arango, Arti Parihar, Patrice Hamel, Rumana Yasmeen, and Andrea I. Doseff. 2013. "Apigenin Protects Endothelial Cells from Lipopolysaccharide (LPS)-Induced Inflammation by Decreasing Caspase-3 Activation and Modulating Mitochondrial Function" International Journal of Molecular Sciences 14, no. 9: 17664-17679. https://doi.org/10.3390/ijms140917664