Trifluoroethanol Modulates Amyloid Formation by the All α-Helical URN1 FF Domain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
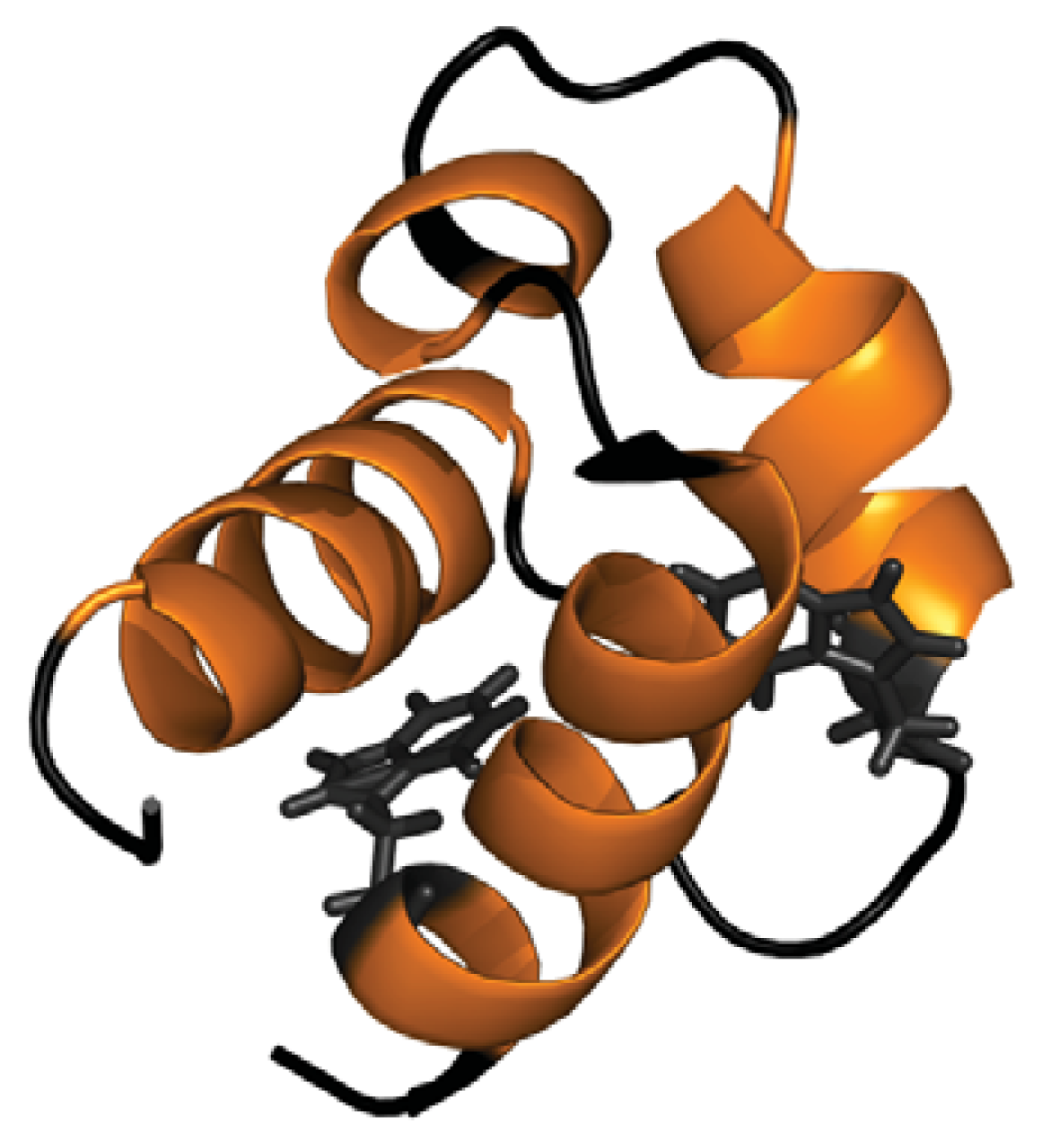
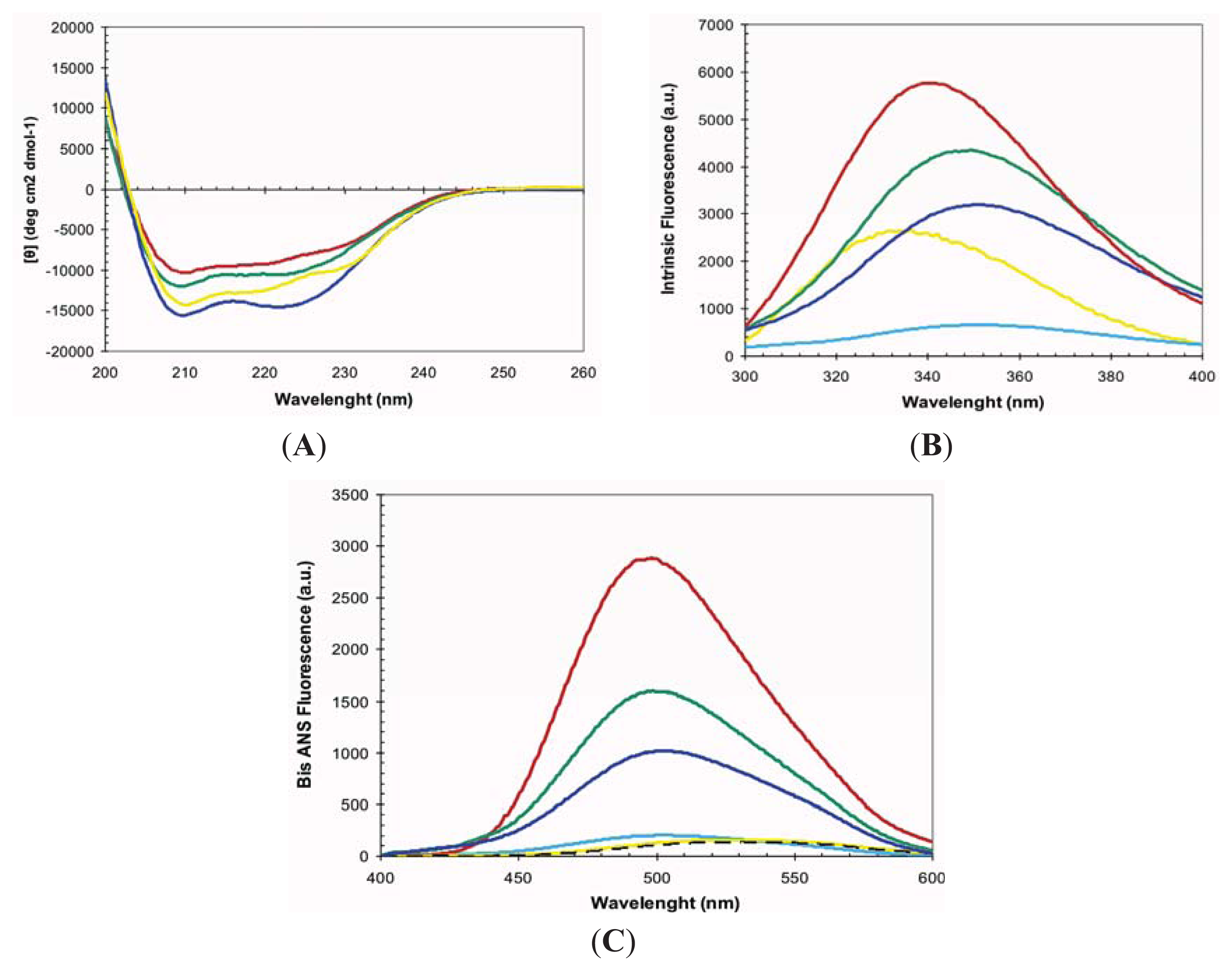
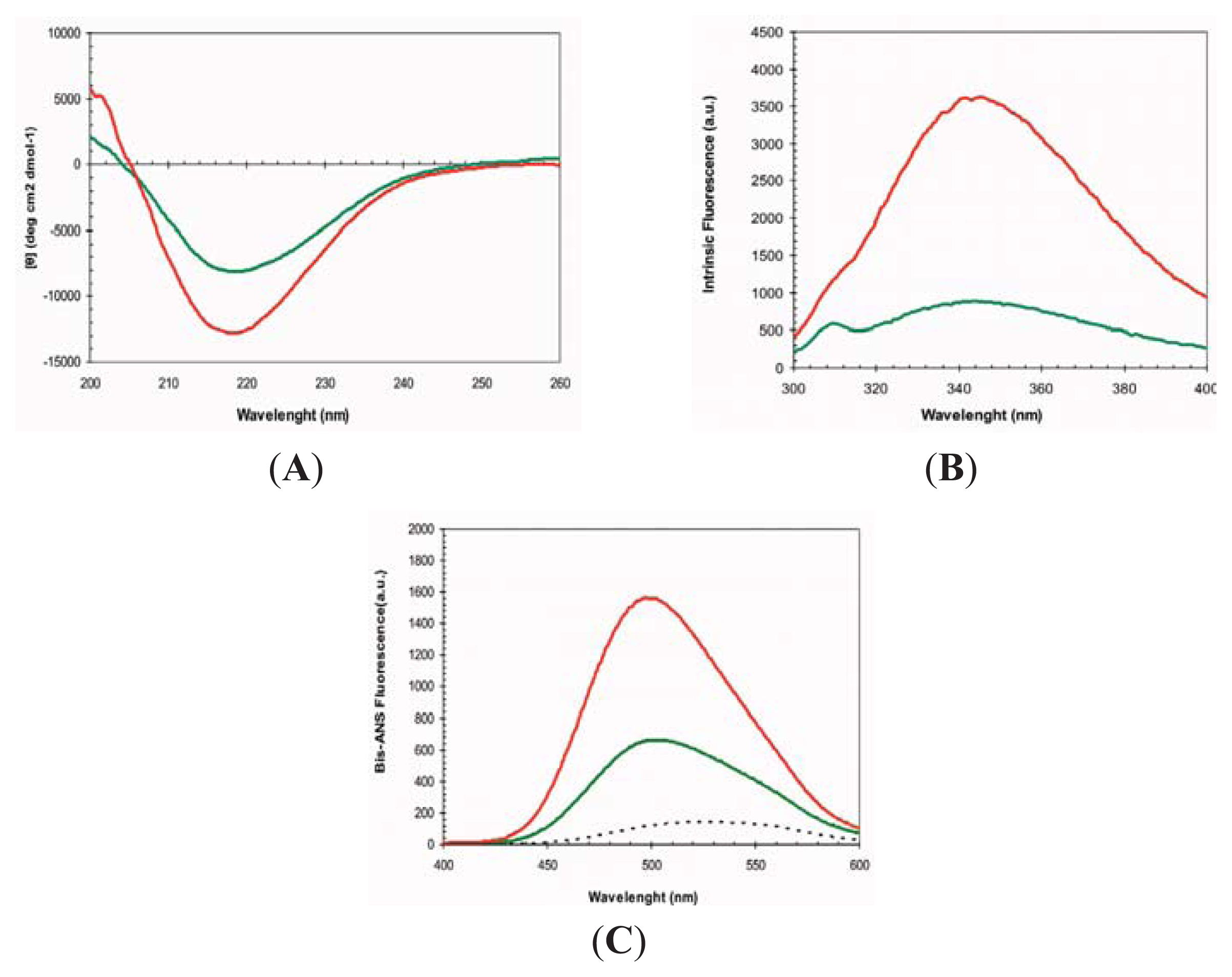
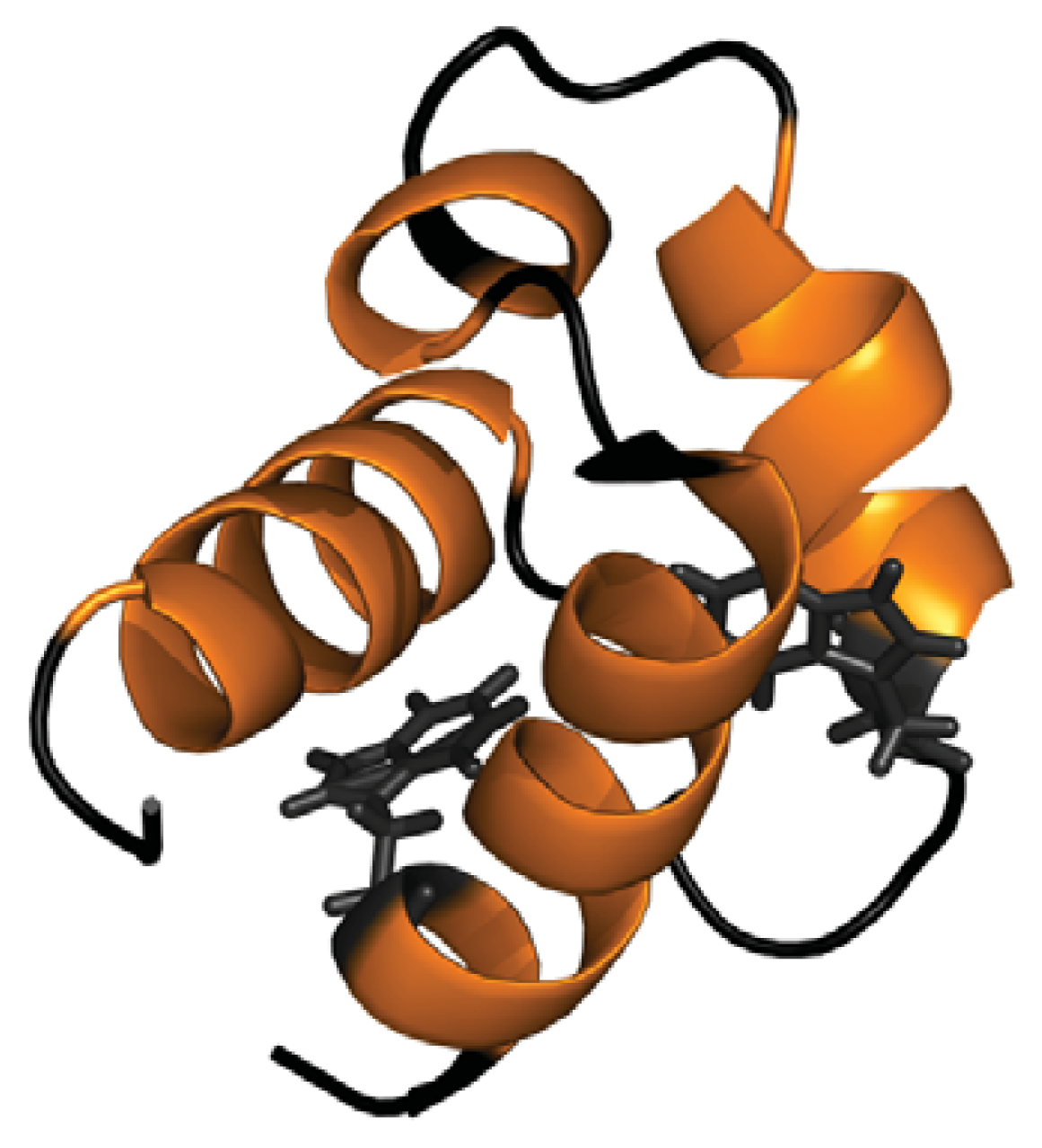
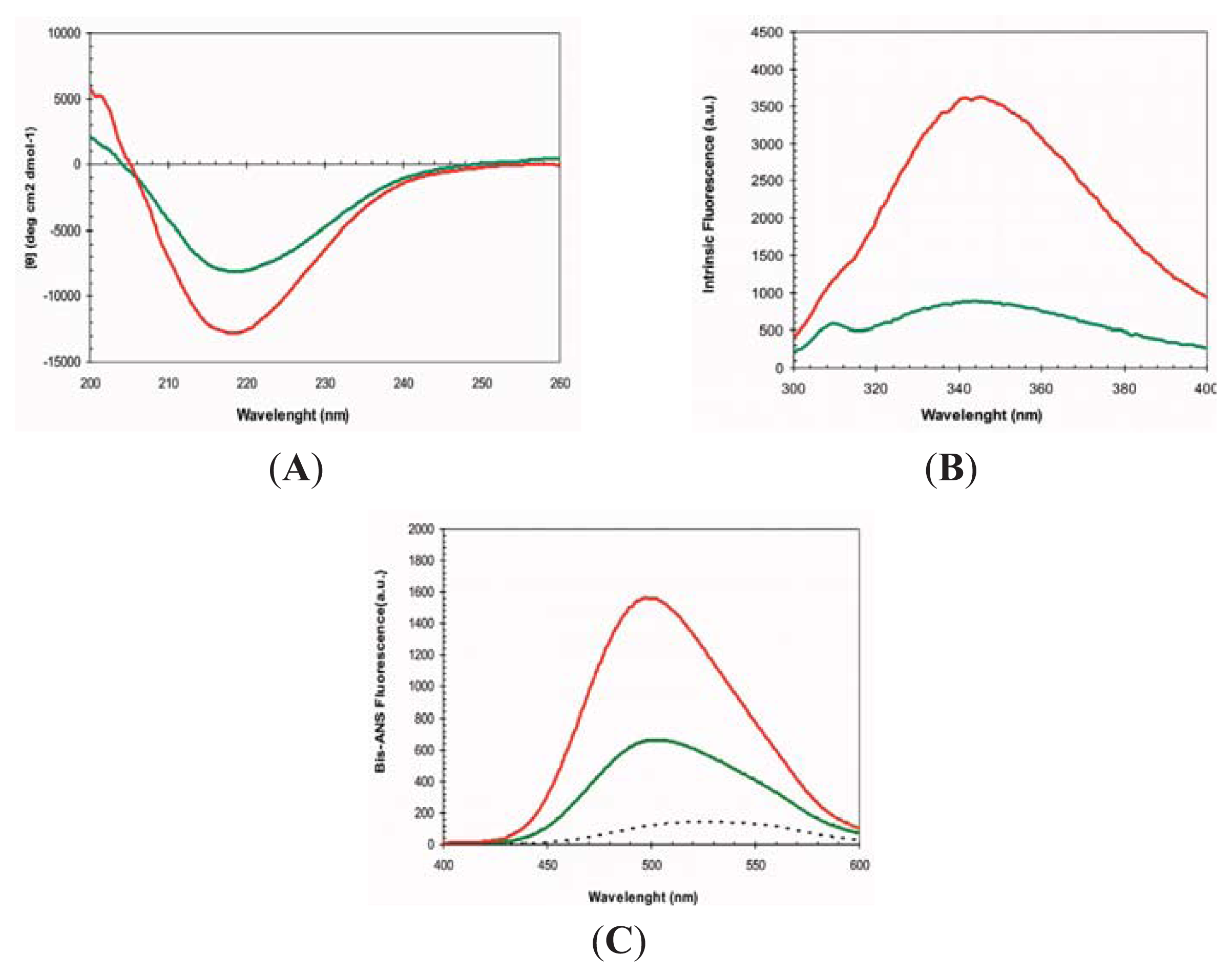
2.1. Conformational Properties of URN1-FF at Low pH in the Presence of TFE
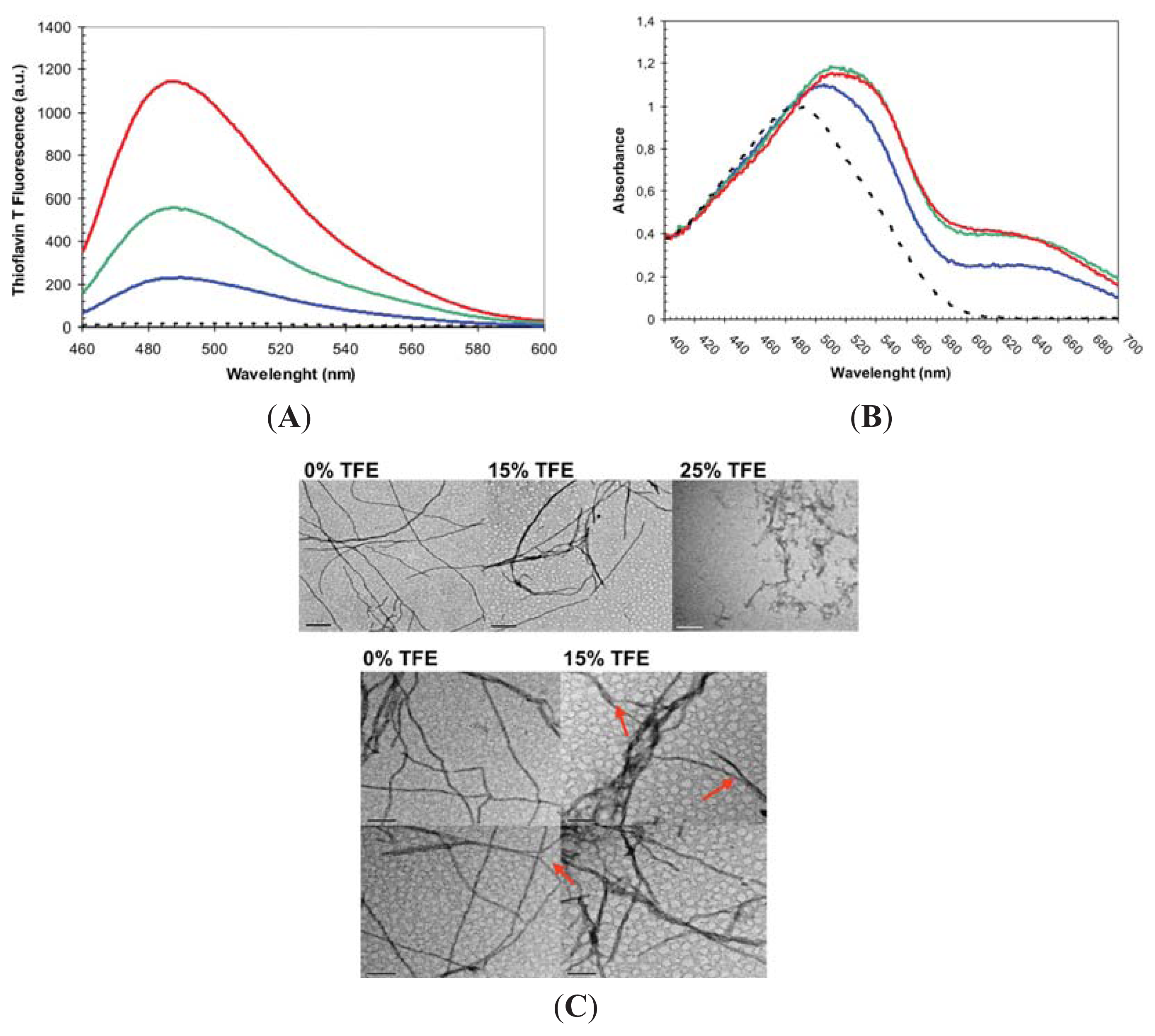
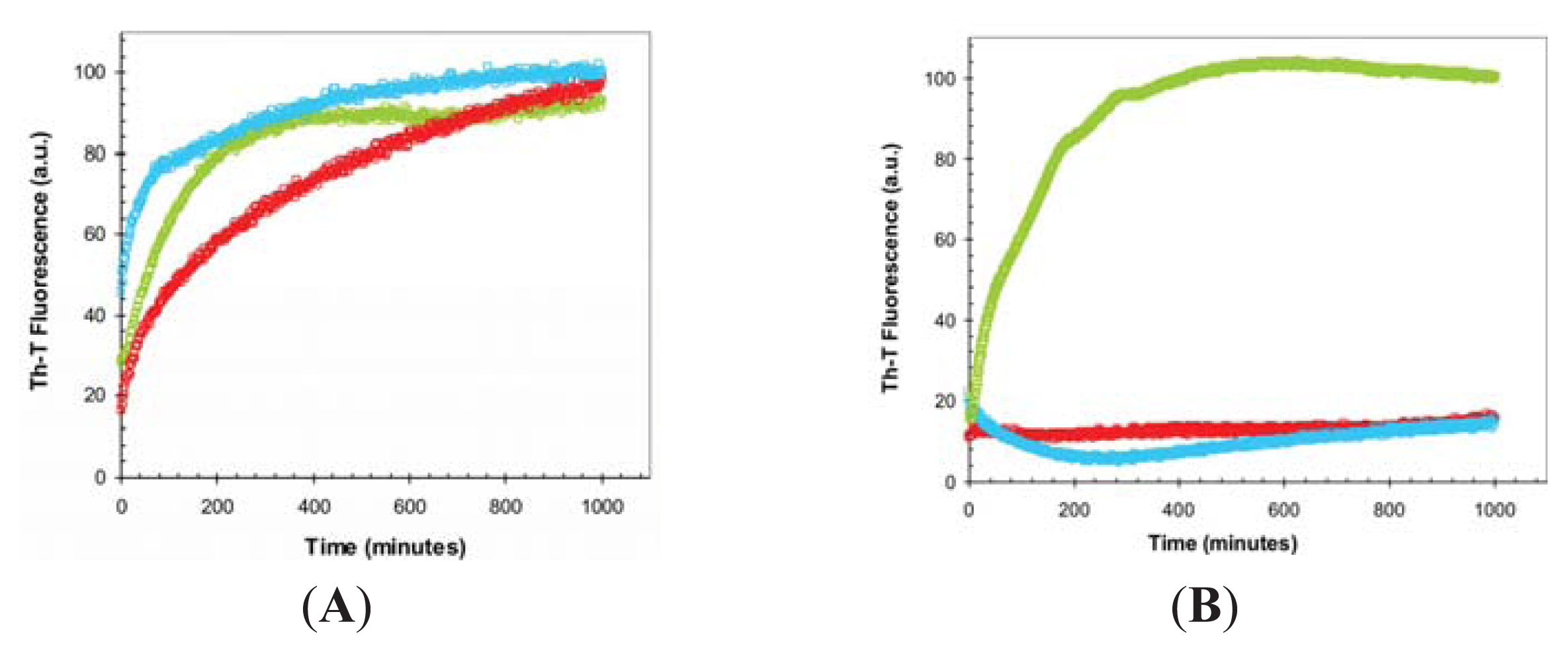
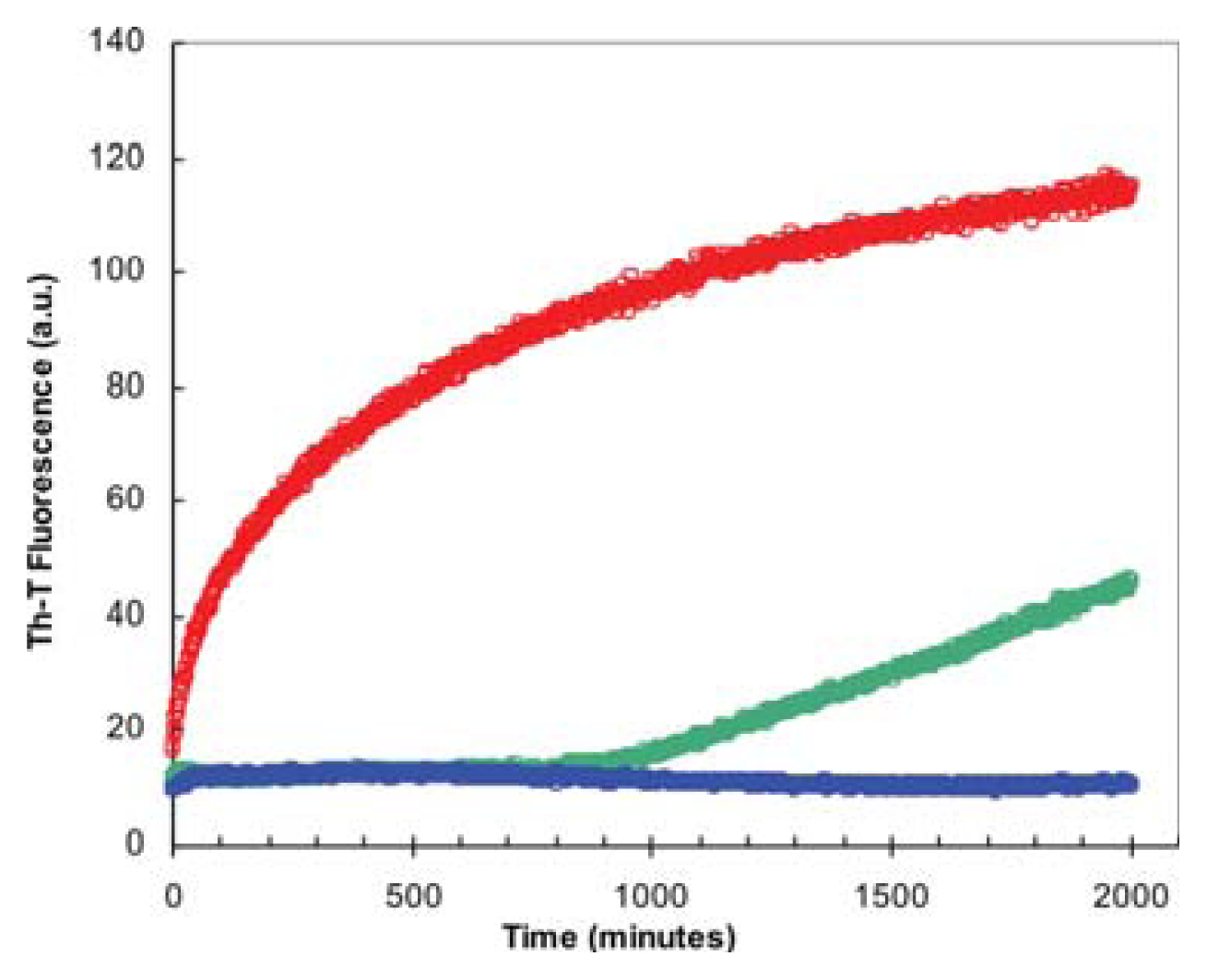
2.2. Effect of TFE on Amyloid Fibril Formation by the URN1-FF Domain
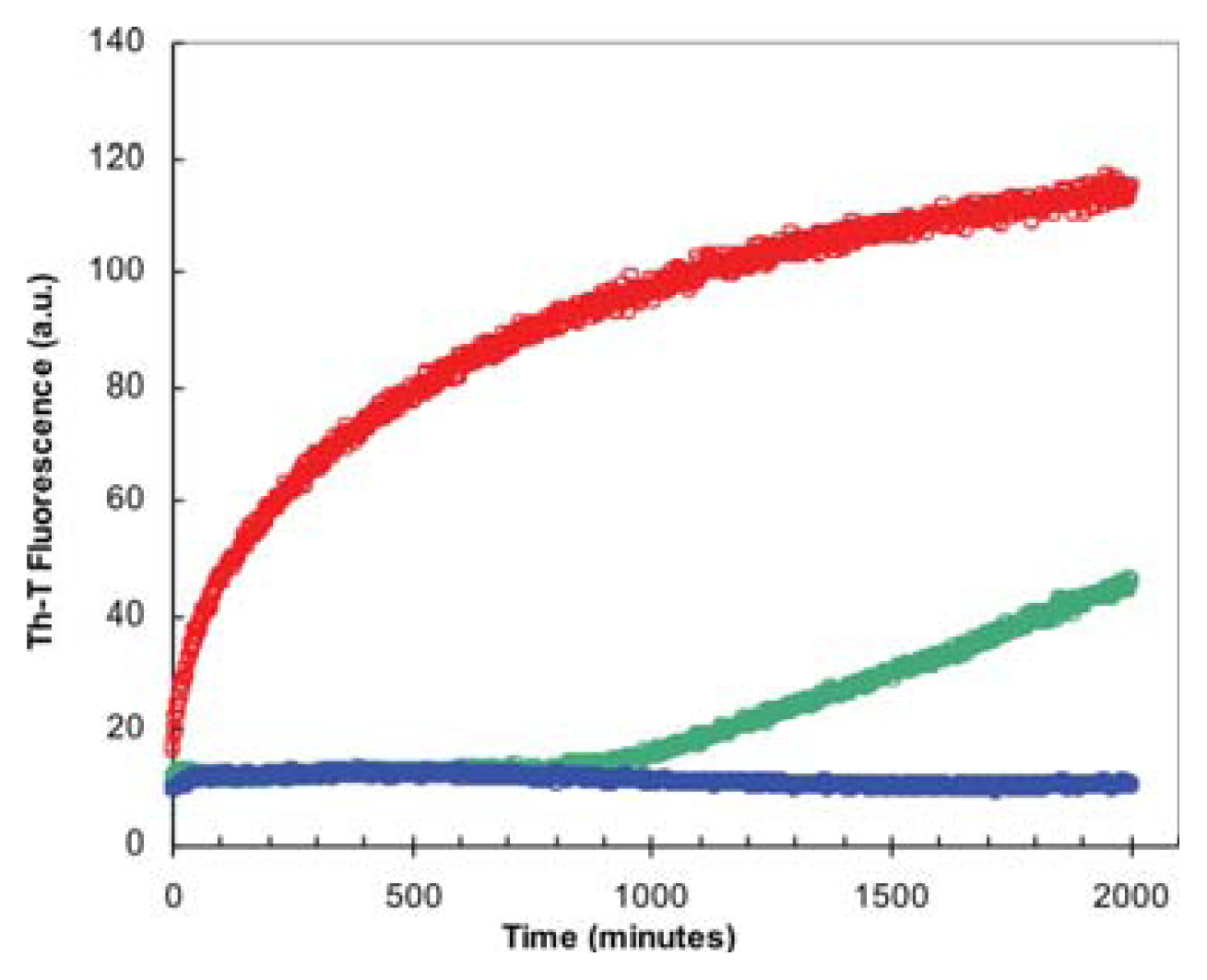
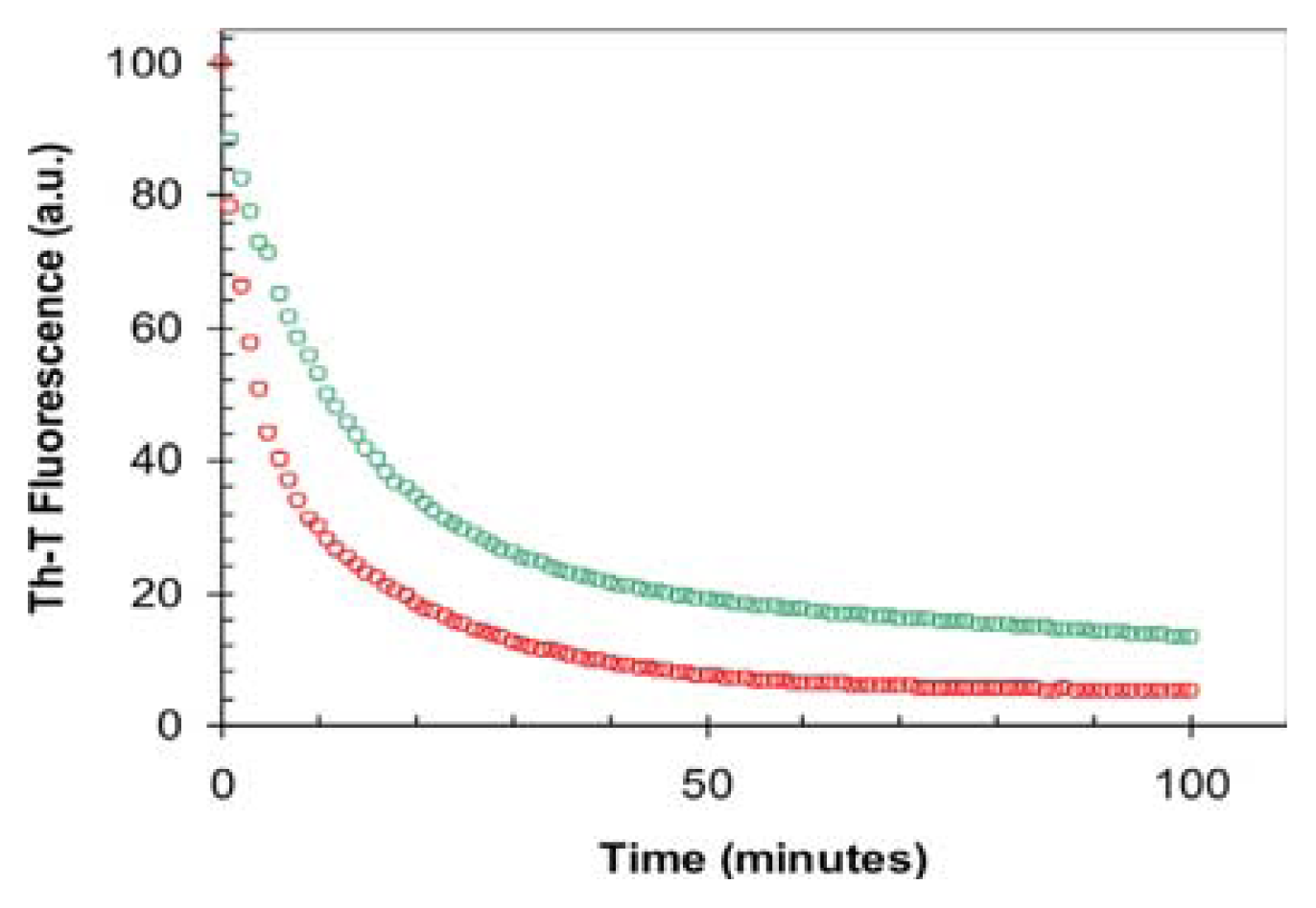
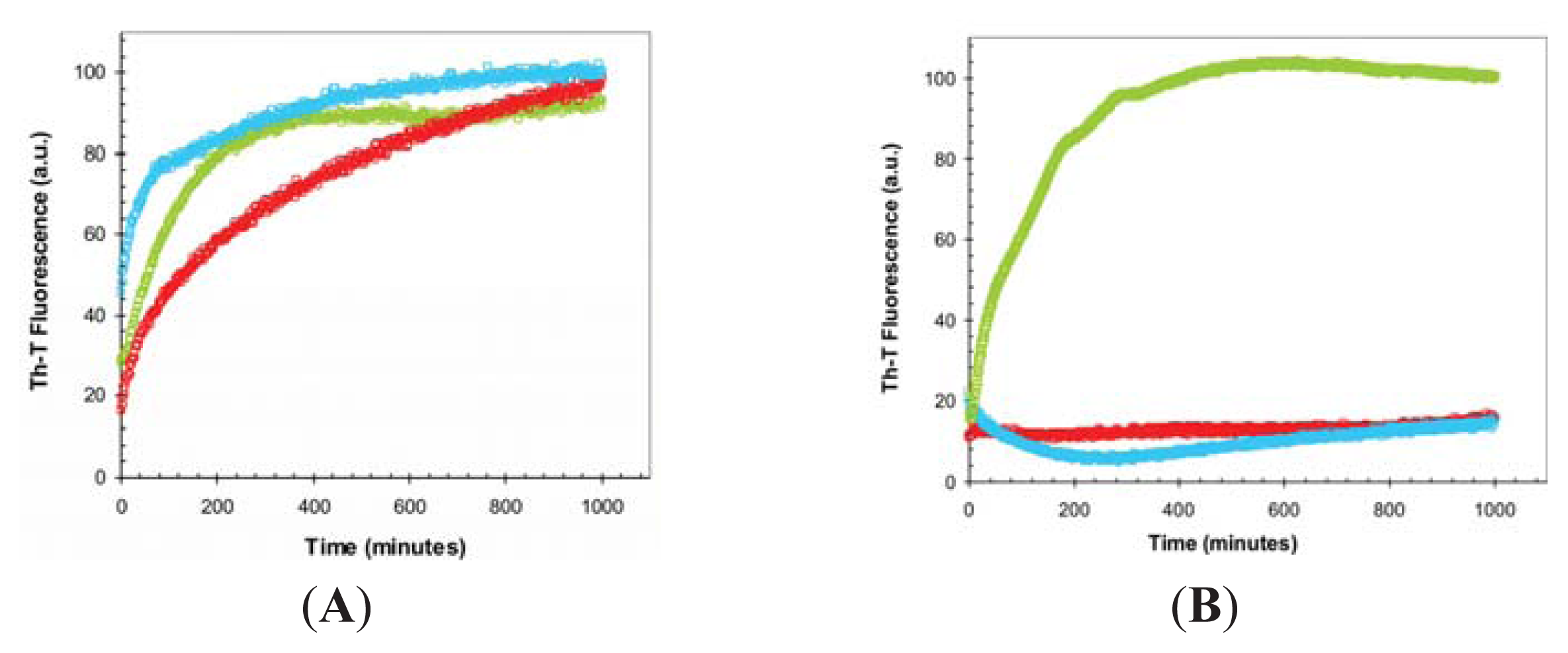
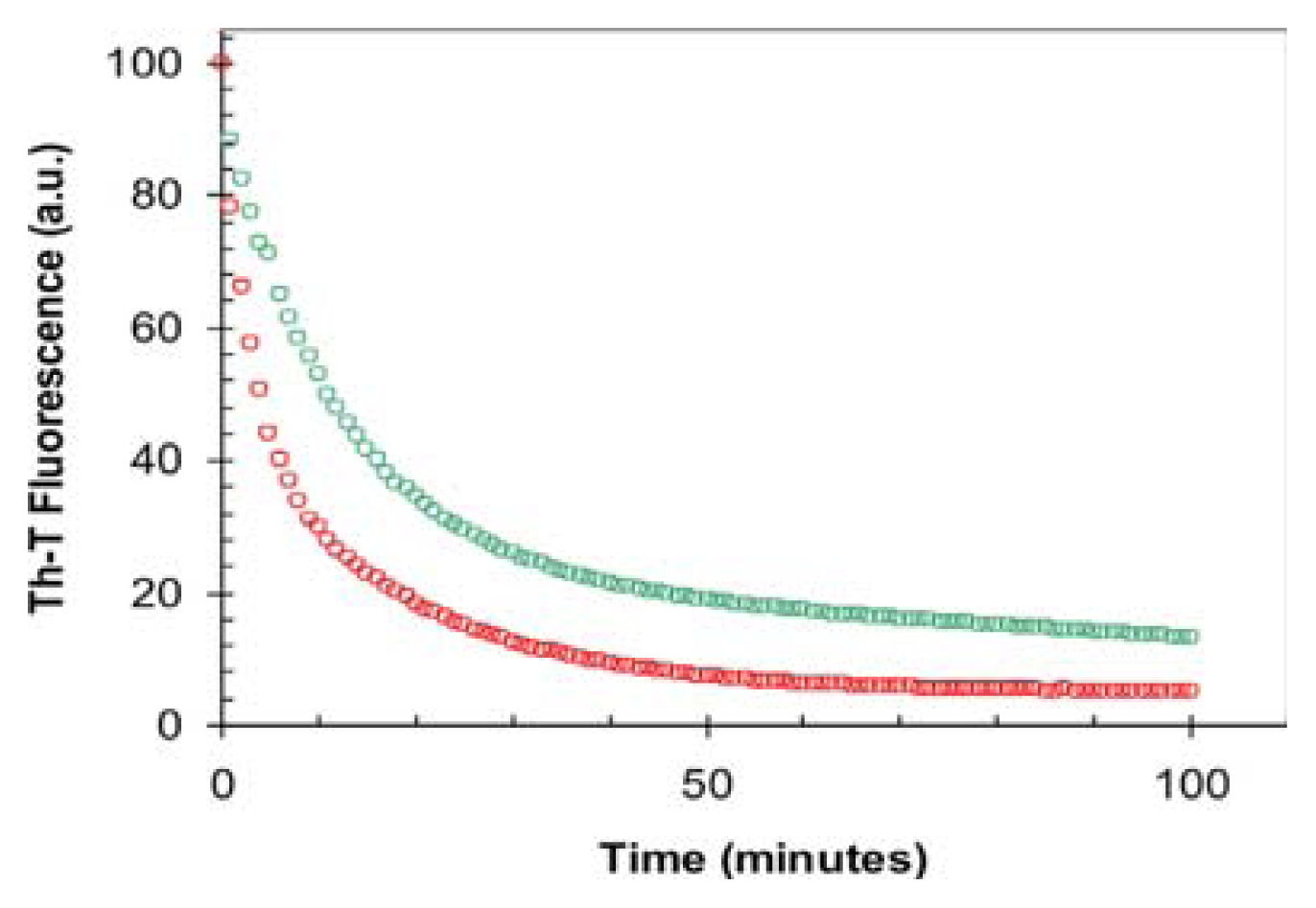
2.3. Seeding Properties of URN1-FF Domain Aggregates
3. Experimental Section
3.1. Protein Expression and Purification
3.2. Protein Preparation for URN1-FF Conformational Assays
3.3. Protein Preparation for URN1-FF Aggregation Assays
3.4. Circular Dichroism and Intrinsic Tryptophan Fluorescence
3.5. bis-ANS Binding Assay
3.6. Binding to ThT
3.7. Binding to Congo Red
3.8. Electron Microscopy
3.9. Aggregation Kinetics and Seeding Assays
3.10. Depolymerization Assays
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| bis-ANS | 4,4′-Dianilino-1,1′-Binaphthyl-5,5′-Disulfonic Acid |
| CD | circular dichroism |
| CR | Congo red |
| MG | molten globule state |
| TFE | 2,2,2-trifluoroethanol |
| Th-T | Thioflavin-T |
| URN1-FF | FF domain of the URN1 splicing factor |
| a.u. | arbitrary units |
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [Google Scholar]
- Brender, J.R.; Salamekh, S.; Ramamoorthy, A. Membrane disruption and early events in the aggregation of the diabetes related peptide iapp from a molecular perspective. Acc. Chem. Res 2012, 45, 454–462. [Google Scholar]
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol 2012, 44, 1541–1554. [Google Scholar]
- Dobson, C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol 2004, 15, 3–16. [Google Scholar]
- Jahn, T.R.; Radford, S.E. The yin and yang of protein folding. FEBS J 2005, 272, 5962–5970. [Google Scholar]
- Linding, R.; Schymkowitz, J.; Rousseau, F.; Diella, F.; Serrano, L. A comparative study of the relationship between protein structure and beta-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol 2004, 342, 345–353. [Google Scholar]
- Seeliger, J.; Estel, K.; Erwin, N.; Winter, R. Cosolvent effects on the fibrillation reaction of human iapp. Phys. Chem. Chem. Phys 2013, 15, 8902–8907. [Google Scholar]
- Grudzielanek, S.; Jansen, R.; Winter, R. Solvational tuning of the unfolding, aggregation and amyloidogenesis of insulin. J. Mol. Biol 2005, 351, 879–894. [Google Scholar]
- Otzen, D.E. Amyloid formation in surfactants and alcohols: Membrane mimetics or structural switchers? Curr. Protein Pept. Sci 2010, 11, 355–371. [Google Scholar]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Trifluoroethanol effects on helix propensity and electrostatic interactions in the helical peptide from ribonuclease t1. Protein Sci 1998, 7, 383–388. [Google Scholar]
- Calamai, M.; Chiti, F.; Dobson, C.M. Amyloid fibril formation can proceed from different conformations of a partially unfolded protein. Biophys. J 2005, 89, 4201–4210. [Google Scholar]
- Srisailam, S.; Kumar, T.K.; Rajalingam, D.; Kathir, K.M.; Sheu, H.S.; Jan, F.J.; Chao, P.C.; Yu, C. Amyloid-like fibril formation in an all beta-barrel protein. Partially structured intermediate state(s) is a precursor for fibril formation. J. Biol. Chem 2003, 278, 17701–17709. [Google Scholar]
- Gosal, W.S.; Clark, A.H.; Ross-Murphy, S.B. Fibrillar beta-lactoglobulin gels: Part 1. Fibril formation and structure. Biomacromolecules 2004, 5, 2408–2419. [Google Scholar]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational behavior and aggregation of alpha-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar]
- Pallares, I.; Vendrell, J.; Aviles, F.X.; Ventura, S. Amyloid fibril formation by a partially structured intermediate state of alpha-chymotrypsin. J. Mol. Biol 2004, 342, 321–331. [Google Scholar]
- Anderson, V.L.; Webb, W.W. A desolvation model for trifluoroethanol-induced aggregation of enhanced green fluorescent protein. Biophys. J 2012, 102, 897–906. [Google Scholar]
- Vivekanandan, S.; Brender, J.R.; Lee, S.Y.; Ramamoorthy, A. A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun 2011, 411, 312–316. [Google Scholar]
- Bemporad, F.; Calloni, G.; Campioni, S.; Plakoutsi, G.; Taddei, N.; Chiti, F. Sequence and structural determinants of amyloid fibril formation. Acc. Chem. Res 2006, 39, 620–627. [Google Scholar]
- Sabate, R.; Espargaro, A.; Grana-Montes, R.; Reverter, D.; Ventura, S. Native structure protects sumo proteins from aggregation into amyloid fibrils. Biomacromolecules 2012, 13, 1916–1926. [Google Scholar]
- Bedford, M.T.; Leder, P. The FF domain: A novel motif that often accompanies WW domains. Trends Biochem. Sci 1999, 24, 264–265. [Google Scholar]
- Bonet, R.; Ramirez-Espain, X.; Macias, M.J. Solution structure of the yeast urn1 splicing factor FF domain: Comparative analysis of charge distributions in FF domain structures-ffs and surps, two domains with a similar fold. Proteins 2008, 73, 1001–1009. [Google Scholar]
- Allen, M.; Friedler, A.; Schon, O.; Bycroft, M. The structure of an FF domain from human hypa/fbp11. J. Mol. Biol 2002, 323, 411–416. [Google Scholar]
- Gasch, A.; Wiesner, S.; Martin-Malpartida, P.; Ramirez-Espain, X.; Ruiz, L.; Macias, M.J. The structure of prp40 ff1 domain and its interaction with the crn-tpr1 motif of clf1 gives a new insight into the binding mode of FF domains. J. Biol. Chem 2006, 281, 356–364. [Google Scholar]
- Korzhnev, D.M.; Religa, T.L.; Banachewicz, W.; Fersht, A.R.; Kay, L.E. A transient and low-populated protein-folding intermediate at atomic resolution. Science 2010, 329, 1312–1316. [Google Scholar]
- Korzhnev, D.M.; Religa, T.L.; Kay, L.E. Transiently populated intermediate functions as a branching point of the FF domain folding pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 17777–17782. [Google Scholar]
- Castillo, V.; Chiti, F.; Ventura, S. The N-terminal helix controls the transition between the soluble and amyloid states of an FF domain. PLoS One 2013, 8, e58297. [Google Scholar]
- Fandrich, M.; Forge, V.; Buder, K.; Kittler, M.; Dobson, C.M.; Diekmann, S. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc. Natl. Acad. Sci. USA 2003, 100, 15463–15468. [Google Scholar]
- Brender, J.R.; Nanga, R.P.; Popovych, N.; Soong, R.; Macdonald, P.M.; Ramamoorthy, A. The amyloidogenic sevi precursor, pap248–286, is highly unfolded in solution despite an underlying helical tendency. Biochim. Biophys. Acta 2011, 1808, 1161–1169. [Google Scholar]
- Merz, P.A.; Wisniewski, H.M.; Somerville, R.A.; Bobin, S.A.; Masters, C.L.; Iqbal, K. Ultrastructural morphology of amyloid fibrils from neuritic and amyloid plaques. Acta Neuropathol 1983, 60, 113–124. [Google Scholar]
- Taddei, N.; Capanni, C.; Chiti, F.; Stefani, M.; Dobson, C.M.; Ramponi, G. Folding and aggregation are selectively influenced by the conformational preferences of the alpha-helices of muscle acylphosphatase. J. Biol. Chem 2001, 276, 37149–37154. [Google Scholar]
- Hartman, K.; Brender, J.R.; Monde, K.; Ono, A.; Evans, M.L.; Popovych, N.; Chapman, M.R.; Ramamoorthy, A. Bacterial curli protein promotes the conversion of pap248–286 into the amyloid sevi: Cross-seeding of dissimilar amyloid sequences. Peer J 2013, 1, e5. [Google Scholar]
- Jha, A.; Narayan, S.; Udgaonkar, J.B.; Krishnamoorthy, G. Solvent-induced tuning of internal structure in a protein amyloid protofibril. Biophys. J 2012, 103, 797–806. [Google Scholar]
- Chatani, E.; Yagi, H.; Naiki, H.; Goto, Y. Polymorphism of beta2-microglobulin amyloid fibrils manifested by ultrasonication-enhanced fibril formation in trifluoroethanol. J. Biol. Chem 2012, 287, 22827–22837. [Google Scholar]
- Nerelius, C.; Sandegren, A.; Sargsyan, H.; Raunak, R.; Leijonmarck, H.; Chatterjee, U.; Fisahn, A.; Imarisio, S.; Lomas, D.A.; Crowther, D.C.; et al. Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9191–9196. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marinelli, P.; Castillo, V.; Ventura, S. Trifluoroethanol Modulates Amyloid Formation by the All α-Helical URN1 FF Domain. Int. J. Mol. Sci. 2013, 14, 17830-17844. https://doi.org/10.3390/ijms140917830
Marinelli P, Castillo V, Ventura S. Trifluoroethanol Modulates Amyloid Formation by the All α-Helical URN1 FF Domain. International Journal of Molecular Sciences. 2013; 14(9):17830-17844. https://doi.org/10.3390/ijms140917830
Chicago/Turabian StyleMarinelli, Patrizia, Virginia Castillo, and Salvador Ventura. 2013. "Trifluoroethanol Modulates Amyloid Formation by the All α-Helical URN1 FF Domain" International Journal of Molecular Sciences 14, no. 9: 17830-17844. https://doi.org/10.3390/ijms140917830