Transcriptome-Wide Single Nucleotide Polymorphisms (SNPs) for Abalone (Haliotis midae): Validation and Application Using GoldenGate Medium-Throughput Genotyping Assays

Abstract
:1. Introduction
2. Results and Discussion
2.1. Transcriptome Data and SNP Discovery
2.2. SNP Performance
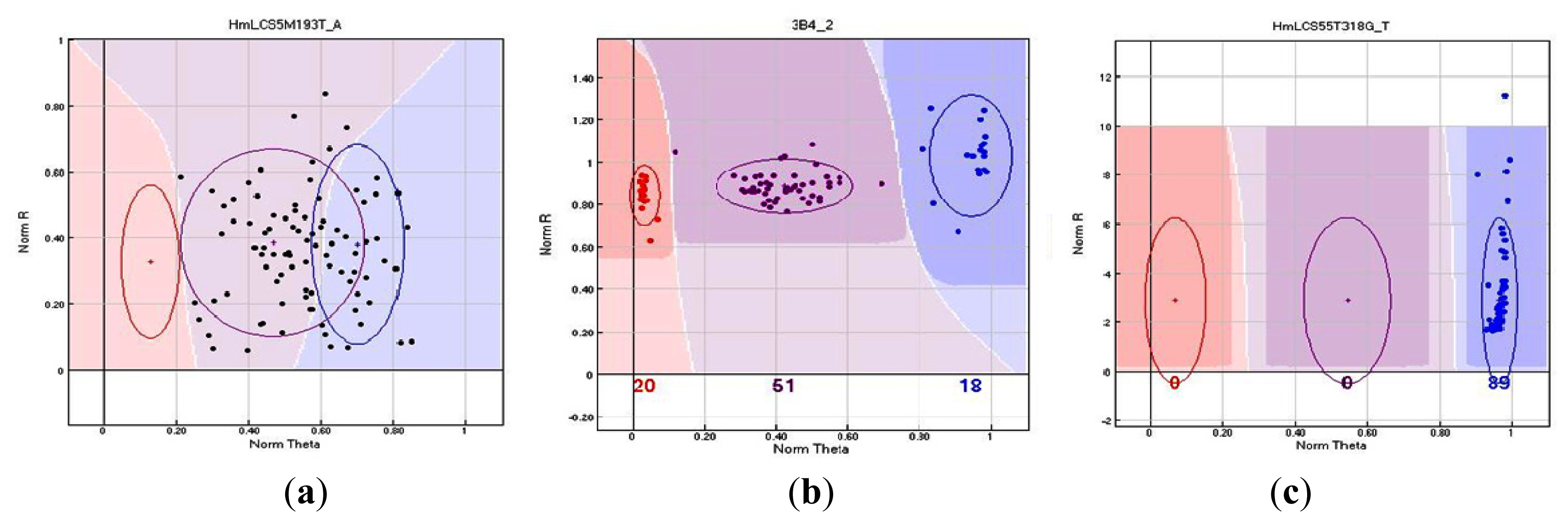
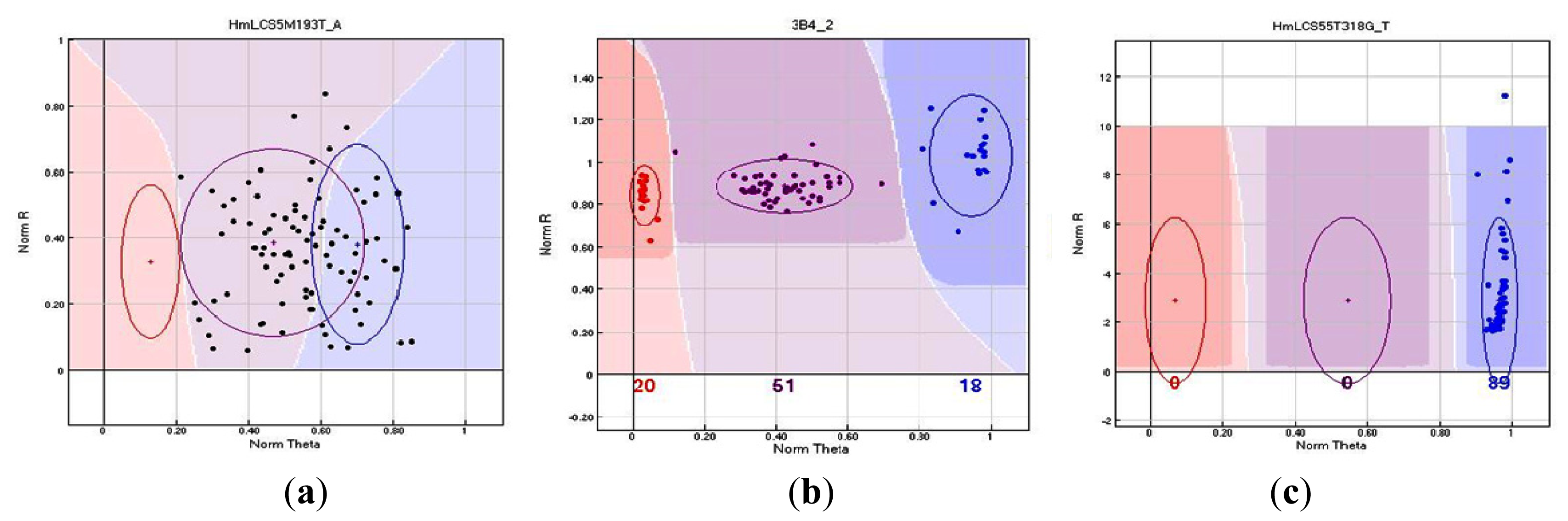
2.3. Genotyping Success of GoldenGate Assays
2.4. Functional Annotation and SNP Effect
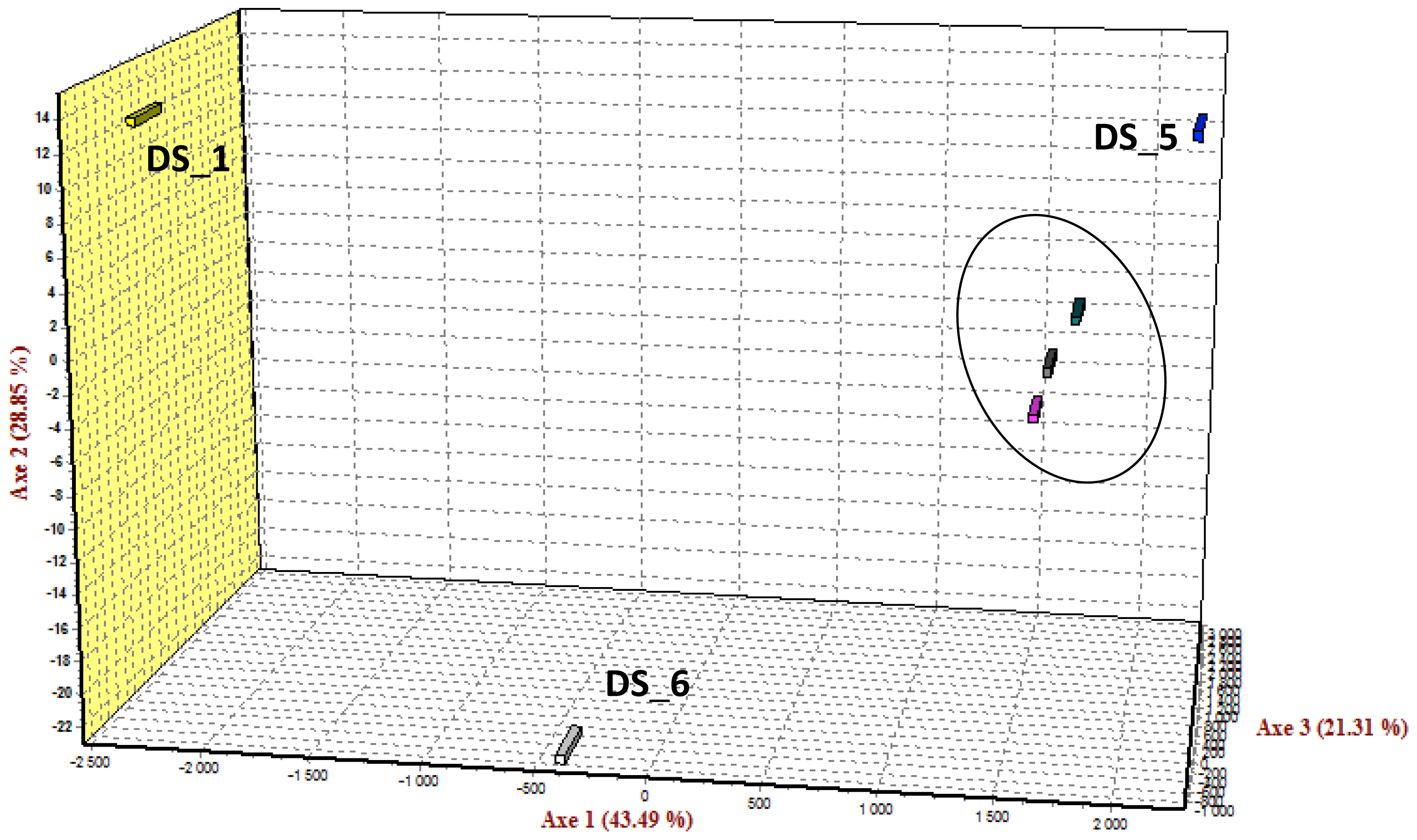
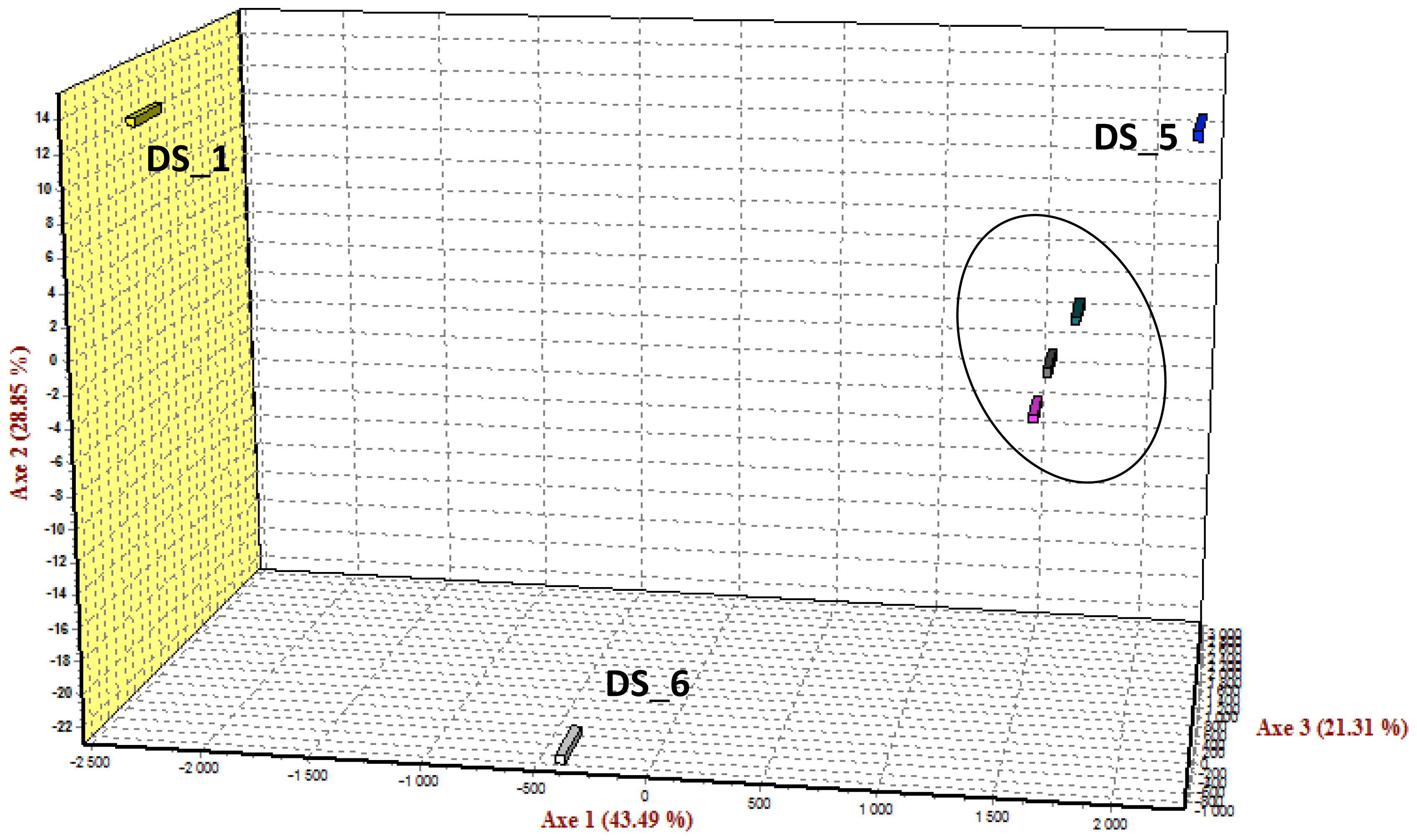
2.5. SNP Diversity, Population Differentiation and Family Informativeness
3. Experimental Section
3.1. Transcriptome Sequencing and SNP discovery
3.2. SNPs and Samples for GoldenGate Assays
3.3. SNP Genotyping and Functional Effect
3.4. Genetic Diversity, Population and Family Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- An, H.S.; Lee, J.W.; Kim, H.C.; Myeong, J.-I. Genetic characterization of five hatchery populations of the pacific abalone (Haliotis discus hannai) using microsatellite markers. Int. J. Mol. Sci 2011, 12, 4836–4849. [Google Scholar]
- Department of Agriculture Forestry and Fisheries. Aquaculture Annual Report 2011. March 2012, p. 15. Available online: http://www.nda.agric.za/doaDev/fisheries/ (accessed on 17 June 2013).
- Rhode, C.; Hepple, J.; Jansen, S.; Davis, T.; Vervalle, J.; Bester-van der Merwe, A.E.; Roodt-Wilding, R. A Population genetic analysis of abalone domestication events in South Africa: Implications for the management of the abalone resource. Aquaculture 2012, 356, 235–242. [Google Scholar]
- Bester, A.E.; Slabbert, R.; D’Amato, M.E. Isolation and characterisation of microsatellite markers in the South African abalone (Haliotis midae). Mol. Ecol. Resour 2004, 4, 618–619. [Google Scholar]
- Slabbert, R.; Ruivo, N.R.; Van den Berg, N.C.; Lizamore, D.L.; Roodt-Wilding, R. Isolation and characterisation of 63 microsatellite loci for the abalone Haliotis midae. J. World Aquacult. Soc 2008, 39, 429–435. [Google Scholar]
- Slabbert, R.; Hepple, J.; Venter, A.; Nel, S.; Swart, L.; Van den Berg, N.C.; Roodt-Wilding, R. Isolation and inheritance of 44 microsatellite loci in the South African abalone Haliotis midae L. Anim. Genetics 2010, 41, 332–333. [Google Scholar]
- Slabbert, R.; Hepple, J.-A.; Rhode, C.; Bester-Van der Merwe, A.E.; Roodt-Wilding, R. New microsatellite markers for the abalone Haliotis midae developed by 454 pyrosequencing and in silico analyses. Gen. Mol. Res 2012, 11, 2769–2779. [Google Scholar]
- Bester, A.E.; Roodt-Wilding, R.; Whitaker, H.A. Discovery and evaluation of single nucleotide polymorphisms (SNPs) for Haliotis midae: A targeted EST approach. Anim. Genetics 2008, 39, 321–324. [Google Scholar]
- Rhode, C.; Slabbert, R.; Roodt-Wilding, R. Microsatellite flanking regions: A SNP mine in South African abalone (Haliotis midae). Anim. Genetics 2008, 39, 329. [Google Scholar]
- Lepoittevin, C.; Frigerio, J.M.; Garnier-Gere, P.; Salin, F.; Cervera, M.T.; Vornam, B.; Harvengt, L.; Plomion, C. In vitro vs. in silico detected SNPs for the development of a genotyping array: What can we learn from a non-model species? PLoS One 2010, 5, e11034. [Google Scholar]
- Hubert, S.; Bussey, J.T.; Higgins, B.; Curtis, B.A.; Bowman, S. Development of single nucleotide polymorphism markers for Atlantic cod (Gadus morhua) using expressed sequences. Aquaculture 2009, 296, 7–14. [Google Scholar]
- Moen, T.; Hayes, B.; Nilsen, F.; Delghandi, M.; Fjalestad, K.T.; Fevolden, S.E.; Berg, P.R.; Lien, S. Identification and characterisation of novel SNP markers in Atlantic cod: Evidence for directional selection. BMC Genetics 2008, 9, 18. [Google Scholar]
- Liu, W.; Li, H.; Bao, X.; He, C.; Li, W.; Shan, Z. The first set of EST-derived single nucleotide polymorphism markers for Japanese scallop Patinopecten yessoensis. J. World Aquacult. Soc 2011, 42, 456–461. [Google Scholar]
- Andreassen, R.; Lunner, S.; Hoyheim, B. Targeted SNP discovery in Atlantic salmon (Salmo salar) genes using a 3′UTR-primed SNP detection approach. BMC Genomics 2010, 11, 706. [Google Scholar]
- Hayes, B.; Laerdahl, J.K.; Lien, S.; Moen, T.; Berg, P.; Hindar, K.; Davidson, W.S.; Koop, B.F.; Adzhubei, A.; Hoyheim, B. An extensive resource of single nucleotide polymorphism markers associated with Atlantic salmon (Salmo salar) expressed sequences. Aquaculture 2007, 265, 82–90. [Google Scholar]
- Liu, S.; Zhou, Z.; Lu, J.; Sun, F.; Wang, S.; Liu, H.; Jiang, Y.; Kucuktas, H.; Kaltenboeck, L.; Peatman, E.; et al. Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array. BMC Genomics 2011, 12, 53. [Google Scholar]
- Qi, H.; Liu, X.; Zhang, G.; Wu, F. Mining expressed sequences for single nucleotide polymorphisms in Pacific abalone (Haliotis discus hannai). Aquac. Res 2009, 40, 1661–1667. [Google Scholar]
- Qi, H.; Liu, X.; Wu, F.; Zhang, G. Development of gene-targeted SNP markers for genomic mapping in Pacific abalone Haliotis discus hannai Ino. Mol. Biol. Rep 2010, 37, 3779–3784. [Google Scholar]
- Buetow, K.H.; Edmonson, M.N.; Cassidy, A.B. Reliable identification of large numbers of candidate SNPs from public EST data. Nat. Genetics 1999, 21, 323–325. [Google Scholar]
- Gurvey, V.; Berezikov, E.; Malik, R.; Plasterk, R.H.A.; Cuppen, E. Single nucleotide polymorphism associated with rat expressed sequences. Genome Res 2004, 14, 1438–1443. [Google Scholar]
- Hayes, B.J.; Nilsen, K.; Berg, P.R.; Grindflek, E.; Lien, S. SNP detection exploiting multiple sources of redundancy in large EST collections improves validation rates. Bioinformatics 2007, 23, 1692–1693. [Google Scholar]
- Bouck, A.; Vision, T. The molecular ecologist’s guide to expressed sequence tags. Mol. Ecol 2007, 16, 907–924. [Google Scholar]
- Wang, S.; Peatman, E.; Abernathy, J.; Waldbieser, G.; Lindquist, E.; Richardson, P.; Lucas, S.; Wang, M.; Li, P.; Thimmapuram, J.; et al. Assembly of 500,000 inter-specific catfish expressed sequence tags and large scale gene-associated marker development for whole genome association studies. Catfish Genome Consortium. Genome Biol 2010, 11, R8. [Google Scholar]
- Metzker, M.L. Applications of next-generation sequencing: Sequencing technologies—The next generation. Nat. Rev. Genetics 2010, 11, 31–46. [Google Scholar]
- Van Bers, N.E.M.; Van Oers, K.; Kerstens, H.H.D.; Dibbits, B.W.; Crooijmans, R.P.; Visser, M.E.; Groenen, M.A. SNP detection in the great tit Parus major using high throughput sequencing. Mol. Ecol 2010, 19, 89–99. [Google Scholar]
- Kerstens, H.H.D.; Crooijmans, R.P.M.A.; Veenendaal, A.; Dibbits, B.W.; Chin-A-Woenq, T.F.; Den Dunnen, J.T.; Groenen, M.A. Large scale single nucleotide polymorphism discovery in unsequenced genomes using second generation high throughput sequencing technology: Applied to turkey. BMC Genomics 2009, 10, 479. [Google Scholar]
- Renaut, S.; Nolte, A.W.; Bernatchez, L. Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs (Coregonus spp. Salmonidae). Mol. Ecol 2010, 19, 115–131. [Google Scholar]
- Stapley, J.; Reger, J.; Feulner, P.G.D.; Smadja, C.; Galindo, J.; Ekblom, R.; Bennison, C.; Ball, A.D.; Beckerman, A.P.; Slate, J. Adaptation genomics: The next generation. Trends Ecol. Evol 2010, 25, 705–712. [Google Scholar]
- Le Dantec, L.; Chagné, D.; Pot, D.; Cantin, O.; Garnier-Géré, P.; Bedon, F.; Frigerio, J.M.; Chaumeil, P.; Léger, P.; Garcia, V.; et al. Automated SNP detection in expressed sequence tags: Statistical considerations and application to maritime pine sequences. Plant Mol. Biol 2004, 54, 461–470. [Google Scholar]
- Diopere, E.; Hellemans, B.; Volckaert, F.A.M.; Maes, G.E. Identification and validation of single nucleotide polymorphisms in growth- and maturation-related candidate genes in sole (Solea solea L.). Mar. Genomics 2013, 9, 33–38. [Google Scholar]
- Useche, F.J.; Gao, G.; HanaFey, M.; Rafalski, A. High-Throughput Identification Database Storage and Analysis of SNPs in EST Sequences. Genome Inform 2001, 12, 194–203. [Google Scholar]
- Garvin, M.R.; Saitoh, K.; Gharrett, A.J. Application of single nucleotide polymorphisms to non-model species: A technical review. Mol. Ecol. Resour 2010, 10, 915–934. [Google Scholar]
- Fan, J.B.; Gunderson, K.L.; Bibikova, M.; Yeakley, J.M.; Chen, J.; Wickhamgarcia, E.; Lebruska, L.; Laurent, M.; Shen, R.; Barker, D. Illumina universal bead arrays. Methods Enzymol. 2006, 410, 57–73. [Google Scholar]
- Illumina. GoldenGate®Genotyping with VeraCode™ Technology: Custom 96-plex and 384-plex Assays. 2008. Available online: http://www.illumina.com/ (accessed on 28 June 2013).
- Illumina. VeraCode Technology. 2010. Avalaible online: http://www.illumina.com/ (accessed on 28 June 2013).
- Zhang, L.S.; Guo, X.M. Development and validation of single nucleotide polymorphism markers in the eastern oyster Crassostrea virginica Gmelin by mining ESTs and resequencing. Aquaculture 2010, 302, 124–129. [Google Scholar]
- Bai, Z.; Yin, Y.; Hu, S.; Wang, G.; Zhang, X.; Li, J. Identification of genes involved in immune response, microsatellite, and SNP markers from Expressed Sequence Tags generated from hemocytes of freshwater pearl mussel (Hyriopsis cumingii). Mar. Biotechnol 2009, 11, 520–530. [Google Scholar]
- Kang, J-H.; Appleyard, S.A.; Elliot, N.G.; Jee, Y-J.; Lee, J.B.; Kang, S.W.; Baek, M.K.; Han, Y.S.; Choi, T.J.; Lee, Y.S. Development of genetic markers in abalone through construction of a SNP database. Anim. Genetics 2011, 42, 309–315. [Google Scholar]
- Kim, W-J.; Jung, H.; Gaffney, P. Development of type I genetic markers from expressed sequence tags in highly polymorphic species. Mar. Biotechnol. 2010, 13, 1–6. [Google Scholar]
- Scofield, D.G.; Hong, X.; Lynch, M. Position of the final intron in full-length transcripts: Determined by NMD? Mol. Biol. Evol 2007, 24, 896–899. [Google Scholar]
- Wang, S.L.; Sha, Z.X.; Sonstegard, T.S.; Liu, H.; Xu, P.; Somridhivej, B.; Peatman, E.; Kucuktas, H.; Liu, Z.J. Quality assessment parameters for EST-derived SNPs from catfish. BMC Genomics 2008, 9, 450. [Google Scholar]
- Jonker, R.M.; Zhang, Q.; Van Hooft, P.; Loonen, M.J.; Van der Jeugd, H.P.; Crooijmans, R.P.; Groenen, M.A.; Prins, H.H.; Kraus, R.H. The development of a genome wide SNP set for the Barnacle Goose Branta leucopsis. PLoS One 2012, 7, e38412. [Google Scholar]
- Eckert, A.J.; Pande, B.; Ersoz, E.S.; Wright, M.H.; Rashbrook, V.K.; Nicolet, C.M.; Neale, D.B. High-throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genet. Genomes 2009, 5, 225–234. [Google Scholar]
- Franchini, P.; Van der Merwe, M.; Roodt-Wilding, R. Transcriptome characterization of the South African abalone Haliotis midae using sequencing-by-synthesis. BMC Res. Notes 2011, 4, 59. [Google Scholar]
- Shen, R.; Fan, J.B.; Campbell, D.; Chang, W.; Chen, J.; Doucet, D.; Yeakley, J.; Bibikova, M.; Wickham Garcia, E.; McBride, C.; et al. High-throughput SNP genotyping on universal bead arrays. Mutat. Res 2005, 573, 70–82. [Google Scholar]
- Pavy, N.; Pelgas, B.; Beauseigle, S.; Blais, S.; Gagnon, F.; Gosselin, I.; Lamothe, M.; Isabel, N.; Bousquet, J. Enhancing genetic mapping of complex genomes through the design of highly-multiplexed SNP arrays: Application to the large and unsequenced genomes of white spruce and black spruce. BMC Genomics 2008, 9, 21. [Google Scholar]
- Fan, J.B.; Oliphant, A.; Shen, R.; Kermani, B.G.; Garcia, F.; Gunderson, K.L.; Hansen, M.; Steemers, F.; Butler, S.L.; Deloukas, P.; et al. Highly parallel SNP genotyping. Cold Spring Harb. Symp. Quant. Biol 2003, 68, 69–78. [Google Scholar]
- Montpetit, A.; Nelis, M.; Laflamme, P.; Magi, R.; Ke, X.; Remm, M.; Cardon, L.; Hudson, T.J.; Metspalu, A. An evaluation of the performance of tag SNPs derived from HapMap in a Caucasian population. PLoS Genet 2006, 2, 282–290. [Google Scholar]
- Clark, N.L.; Findlay, G.D.; Yi, X.; MaCoss, M.J.; Swanson, W.J. Duplication and selection on abalone sperm lysin in an allopatric population. Mol. Biol. Evol 2007, 24, 2081–2090. [Google Scholar]
- Vera, M.; Alvarez-Dios, J.A.; Millan, A.; Pardo, B.G.; Bouza, C.; Hermida, M.; Fernandez, C.; De la Herran, R.; Molina-Luzon, M.J.; Martinez, P. Validation of single nucleotide polymorphism (SNP) markers from an immune Expressed Sequence Tag (EST) turbot; Scophthalmus maximus; database. Aquaculture 2011, 313, 31–41. [Google Scholar]
- Vera, M.; Alvarez-Dios, J.-A.; Fernandez, C.; Bouza, C.; Vilas, R.; Martinez, P. Development and validation of single nucleotide polymorphisms (SNPs) markers from two transcriptome 454-runs of turbot (Scophthalmus maximus) using high-throughput genotyping. Int. J. Mol. Sci 2013, 14, 5694–5711. [Google Scholar]
- Bester-van der Merwe, A.E.; Roodt-Wilding, R.; Volckaert, F.A.M.; D’Amato, M.E. Historical isolation and hydrodynamically constrained gene flow in declining populations of the South African abalone Haliotis midae. Conserv. Genet 2011, 12, 543–555. [Google Scholar]
- Vervalle, J.; Hepple, J.; Jansen, S.; Du Plessis, J.; Wang, P.; Rhode, C.; Roodt-Wilding, R. Integrated linkage map of Haliotis midae Linnaeus based on microsatellites and SNPs. J. Shellfish Res 2013, 32, 89–103. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform 2003, 4, 41. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 2000, 25, 25–29. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res 2000, 28, 27–30. [Google Scholar]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucl. Acids Res 2006, 34, 354–357. [Google Scholar]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirikawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucl. Acids Res 2010, 38, 355–360. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer. BMC Bioinform 2008, 9, 253. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W improving the sensitivity of progressive multiple sequence alignment through sequence weighting; position-specific gap penalties and weight matrix choice. Nucl. Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program fro Windows 95/98/NT. Nucl. Acids Symp. Ser 1999, 41, 95–98. [Google Scholar]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour 2008, 8, 103–106. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimation F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Genetix, Logiciel Sous WindowsTM Pour la Génétique des Populations, version 4.04; Laboratoire Génome et populations, CNRS UPR 9060, Université de Montpellier II: Montpellier, France, 2002.
- JoinMap®, version 4; Software for the calculation of genetic linkage maps in experimental populations; Kyazma B.V. Wageningen: The Netherlands, 2006.
- Surget-Groba, Y.; Montoya-Burgos, J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res 2010, 20, 1432–1440. [Google Scholar]
- Quinn, N.L.; Levenkova, N.; Chow, W.; Bouffard, P.; Boroevich, K.A.; Knight, J.R.; Jarvie, T.P.; Lunieniecki, K.P.; Desany, B.A.; Koop, B.F.; et al. Assessing the feasibility of GS FLX Pyrosequencing for sequencing the Atlantic salmon genome. BMC Genomics 2008, 9, 404. [Google Scholar]
- Slabbert, R.; Bester, A.E.; D’Amato, M.E. Analyses of genetic diversity and parentage within a South African hatchery of the abalone Haliotis midae Linnaeus using microsatellite markers. J. Shellfish Res 2009, 28, 369–375. [Google Scholar]

{kind=link}
{kind=link}
| In vitro (Velvet assembly) | In silico (CLC assembly) | |
|---|---|---|
| Number of contigs | 58 | 256 |
| Number of putative SNPs | 105 | 400 |
| Transversions: | ||
| A/T | 15 (14.3%) | 52 (13.0%) |
| A/C | 7 (6.7%) | 35 (8.8%) |
| C/G | 10 (9.5%) | 15 (3.8%) |
| T/G | 6 (5.7%) | 43 (10.8%) |
| Transitions: | ||
| A/G | 35 (33.3%) | 124 (31.0%) |
| T/C | 30 (28.6%) | 129 (32.3%) |
| Other | 2 (1.9%) | 2 (0.5%) |
| Plex-48 | Plex-192 | |
|---|---|---|
| Functionality score | 0.75 | 0.8 |
| ESTs/Contigs | 35 | 139 |
| Feasible SNPs | 48 | 186 |
| GenTrain score | 0.25 | 0.45 |
| Total | 48 | 186 |
| Failures | 7 (14.58%) | 44 (23.7%) |
| Monomorphic | 10 (20.83%) | 14 (7.5%) |
| Polymorphic | 31 (64.58%) | 128 (68.8%) |
| SNP Name | EST/Contig | Functional annotation | Variant | SNP effect |
|---|---|---|---|---|
| 3B4_2 | 3B4 | 60s Ribosomal protein L8 | T/C | UTR |
| 3B4_7 | A/T | UTR | ||
| 3D10_1 | 3D10 | Hemocyanin | A/G | UTR |
| 2H9_2 | 2H9 | Ribosomal protein L22 | A/T | UTR |
| HdSNPc148_820T_C | HdSNPc148 | Actin | T/C | Synonymous |
| HdSNPc106_688C_T | HdSNPc106 | Tubulin alpha-1a chain isoform 2 | C/T | Synonymous |
| HmSNPc4_815C_T | HmSNPc4 | Microsatellite sequence | C/T | Non-synonymous |
| HaSNPdw500_207C_T | HaSNPdw | Microsatellite sequence | C/T | UTR |
| HmLCS5M193T_A | HmLCS5M | - | T/A | - |
| HmLCS5M479C_T | Opacity protein | C/T | Synonymous | |
| HmLCS55T318G_T | HmLCS55T | Microsatellite sequence | G/T | Non-synonymous |
| HmRS36T262T_C | HmRS36T | 5-Formyltetrahydrofolate cyclo-ligase | T/C | Synonymous |
| SNP101_113 | Contig 101 | Myosin heavy chain | A/C | UTR |
| SNP101_201 | C/G | UTR | ||
| SNP146.2_132 | Contig 146 | ADP/ATP carrier protein | A/G | Synonymous |
| SNP146.3_123 | T/G | Non-synonymous | ||
| SNP149.1_106 | Contig 149 | Heat shock protein 70 | A/C | UTR |
| SNP149.1_374 | C/G | Non-synonymous | ||
| SNP149.2_165 | A/G | Synonymous | ||
| SNP149.4_75 | A/G | UTR | ||
| SNP149.4_341 | T/C | UTR | ||
| SNP210_266 | Contig 210 | - | T/G | - |
| SNP214_86 | Contig 214 | Ribosomal protein L10 | T/C | Non-synonymous |
| SNP214_434 | T/C | UTR | ||
| SNP342.2_537 | Contig 342 | Heat shock protein | T/C | UTR |
| SNP449.2_110 | Contig 449 | s-Adenosylmethionine synthetase isoform type-1 | A/G | Non-synonymous |
| SNP449.2_443 | T/C | Synonymous | ||
| SNP1718_109 | Contig 1718 | NADH dehydrogenase subunit 1 | A/T | UTR |
| SNP1833_160 | Contig 1833 | Alpha tubulin | A/G | UTR |
| SNP1834_76 | Contig 1834 | Tubulin alpha-1a chain- partial | A/G | UTR |
| SNP1834_464 | A/G | Non-synonymous | ||
| SNP1949_235 | Contig 1949 | Ribosomal protein L3 | A/C | UTR |
| SNP4691_183 | Contig 4691 | Heat shock protein 70 | A/G | UTR |
| SNP17550.1_463 | Contig 17550 | Clathrin heavy chain 1 | A/G | Non-synonymous |
| SNP17550.3_221 | A/T | UTR | ||
| SNP17550.3_555 | A/T | UTR | ||
| SNP48_322 | Contig 48 | Collagen alpha-4 chain | T/G | UTR |
| SNP67_164 | Contig 67 | Collagen alpha-6 partial | A/G | UTR |
| SNP140_2421 | Contig 140 | Na+ K+-ATPase alpha subunit | T/C | Synonymous |
| SNP229_2772 | Contig 229 | 14-3-3 Zeta | T/C | UTR |
| SNP300_1828 | Contig 300 | - | A/G | - |
| SNP972_1055 | Contig 972 | Myosin heavy chain | T/C | Synonymous |
| SNP1001_388 | Contig 1001 | Cre-sca-1 protein | T/C | UTR |
| SNP2091_264 | Contig 2091 | - | A/C | - |
| SNP3129_923 | Contig 3129 | Arginine kinase | A/G | Synonymous |
| SNP5837_204 | Contig 5837 | Mucus-associated protein partial | T/C | Non-synonymous |
| SNP13865_165 | Contig 13865 | Cathepsin l | T/C | UTR |
| SNP20648_3041 | Contig 20648 | Filamin-c isoform 4 | A/G | Synonymous |
| SNP Name | Minor allele frequency | Heterozygosity | Inbreeding coefficient | Probability HW | |
|---|---|---|---|---|---|
| Observed | Expected | ||||
| 3B4_2 | 0.4549 | 0.592 | 0.465 | −0.274 | 0.01 |
| 3B4_7 | 0.1901 | 0.537 | 0.473 | −0.134 | <0.01 |
| 3D10_1 | 0.0391 | 0.281 | 0.472 | 0.406 | 0.001 |
| HdSNPc148_820T_C | 0.4564 | 0.183 | 0.172 | −0.066 | 0.000 |
| HdSNPc106_688C_T | 0.0015 | 0.721 | 0.462 | −0.563 | <0.01 |
| HmRS36T262T_C | 0.0420 | 0.009 | 0.331 | 0.972 | 0.002 |
| SNP101_113 | 0.0557 | 0.069 | 0.073 | 0.049 | 0.530 |
| SNP101_201 | 0.0015 | 0.091 | 0.126 | 0.278 | 0.786 |
| SNP146.2_132 | 0.1352 | 0.285 | 0.245 | −0.165 | 0.011 |
| SNP149.1_374 | 0.0651 | 0.025 | 0.031 | 0.189 | 0.799 |
| SNP149.2_165 | 0.4079 | 0.483 | 0.478 | −0.009 | 0.001 |
| SNP149.4_75 | 0.2464 | 0.003 | 0.003 | 0.000 | - |
| SNP210_266 | 0.0669 | 0.044 | 0.043 | −0.021 | 1.000 |
| SNP214_86 | 0.0000 | 0.041 | 0.040 | −0.019 | 0.875 |
| SNP342.2_537 | 0.1023 | 0.098 | 0.111 | 0.116 | 0.793 |
| SNP449.2_110 | 0.4035 | 0.32 | 0.497 | 0.357 | 0.000 |
| SNP1834_76 | 0.4781 | 0.013 | 0.012 | −0.005 | - |
| SNP1834_464 | 0.0014 | 0.788 | 0.489 | −0.614 | <0.01 |
| SNP1949_235 | 0.1764 | 0.309 | 0.266 | −0.162 | 0.000 |
| SNP4691_183 | 0.0667 | 0.138 | 0.353 | 0.610 | 0.025 |
| SNP17550.3_221 | 0.0189 | 0.013 | 0.013 | −0.005 | - |
| SNP17550.3_555 | 0.0841 | 0.119 | 0.112 | −0.062 | 0.081 |
| SNP67_164 | 0.0696 | 0.182 | 0.176 | −0.033 | 0.181 |
| SNP140_2421 | 0.1272 | 0.178 | 0.162 | −0.096 | 0.310 |
| SNP229_2772 | 0.0338 | 0.099 | 0.128 | 0.228 | 0.880 |
| SNP300_1828 | 0.0678 | 0.195 | 0.181 | −0.075 | 0.058 |
| SNP972_1055 | 0.2267 | 0.082 | 0.394 | 0.794 | <0.01 |
| SNP1001_388 | 0.1893 | 0.280 | 0.250 | −0.119 | 0.000 |
| SNP2091_264 | 0.2580 | 0.314 | 0.486 | 0.354 | 0.001 |
| SNP3129_923 | 0.2246 | 0.318 | 0.462 | 0.312 | <0.01 |
| SNP20648_3041 | 0.0889 | 0.164 | 0.166 | 0.015 | 0.063 |
| Sample origin | Number of individuals | |
|---|---|---|
| Plex-48 | Plex-192 | |
| Family DS 1 | 103 | 70 |
| Family DS_2 | 94 | 87 |
| Family DS_5 | 90 | - |
| Family DS_6 | 94 | - |
| Family D | - | 72 |
| Family H | - | 71 |
| Family I | - | 81 |
| Family J | - | 72 |
| Saldanha Bay | 23 | - |
| Witsand | 26 | - |
| Riet Point | 26 | - |
| Positive controls | 2 per plate | 3 per plate |
| Negative controls | - | 1 per plate |
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bester-Van Der Merwe, A.; Blaauw, S.; Du Plessis, J.; Roodt-Wilding, R. Transcriptome-Wide Single Nucleotide Polymorphisms (SNPs) for Abalone (Haliotis midae): Validation and Application Using GoldenGate Medium-Throughput Genotyping Assays. Int. J. Mol. Sci. 2013, 14, 19341-19360. https://doi.org/10.3390/ijms140919341
Bester-Van Der Merwe A, Blaauw S, Du Plessis J, Roodt-Wilding R. Transcriptome-Wide Single Nucleotide Polymorphisms (SNPs) for Abalone (Haliotis midae): Validation and Application Using GoldenGate Medium-Throughput Genotyping Assays. International Journal of Molecular Sciences. 2013; 14(9):19341-19360. https://doi.org/10.3390/ijms140919341
Chicago/Turabian StyleBester-Van Der Merwe, Aletta, Sonja Blaauw, Jana Du Plessis, and Rouvay Roodt-Wilding. 2013. "Transcriptome-Wide Single Nucleotide Polymorphisms (SNPs) for Abalone (Haliotis midae): Validation and Application Using GoldenGate Medium-Throughput Genotyping Assays" International Journal of Molecular Sciences 14, no. 9: 19341-19360. https://doi.org/10.3390/ijms140919341