Synthesis, Characterization and in Vitro Evaluation of New Composite Bisphosphonate Delivery Systems

Abstract
:1. Introduction
2. Results and Discussion
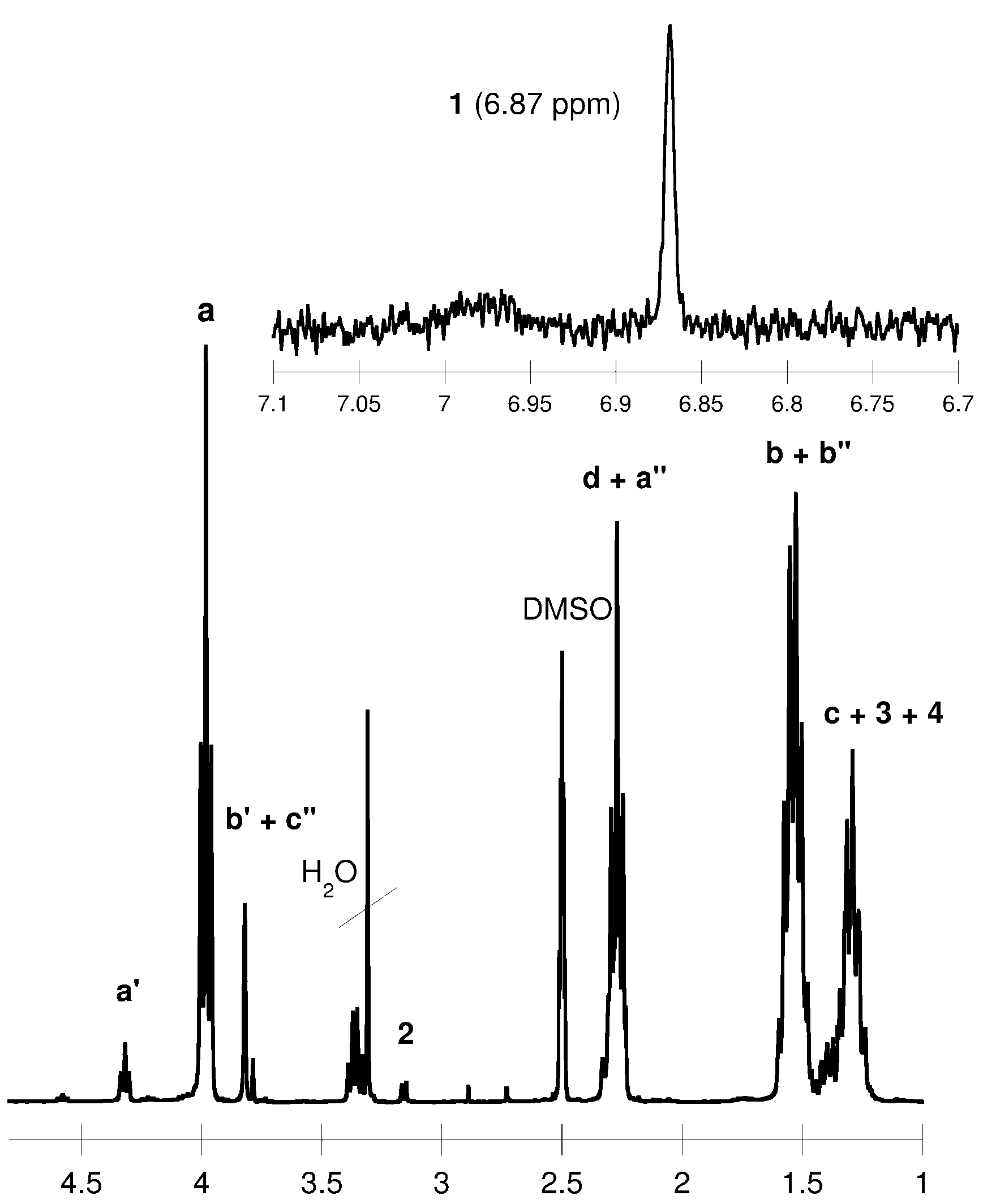
2.1. Synthesis of Polyols and Polyurethanes


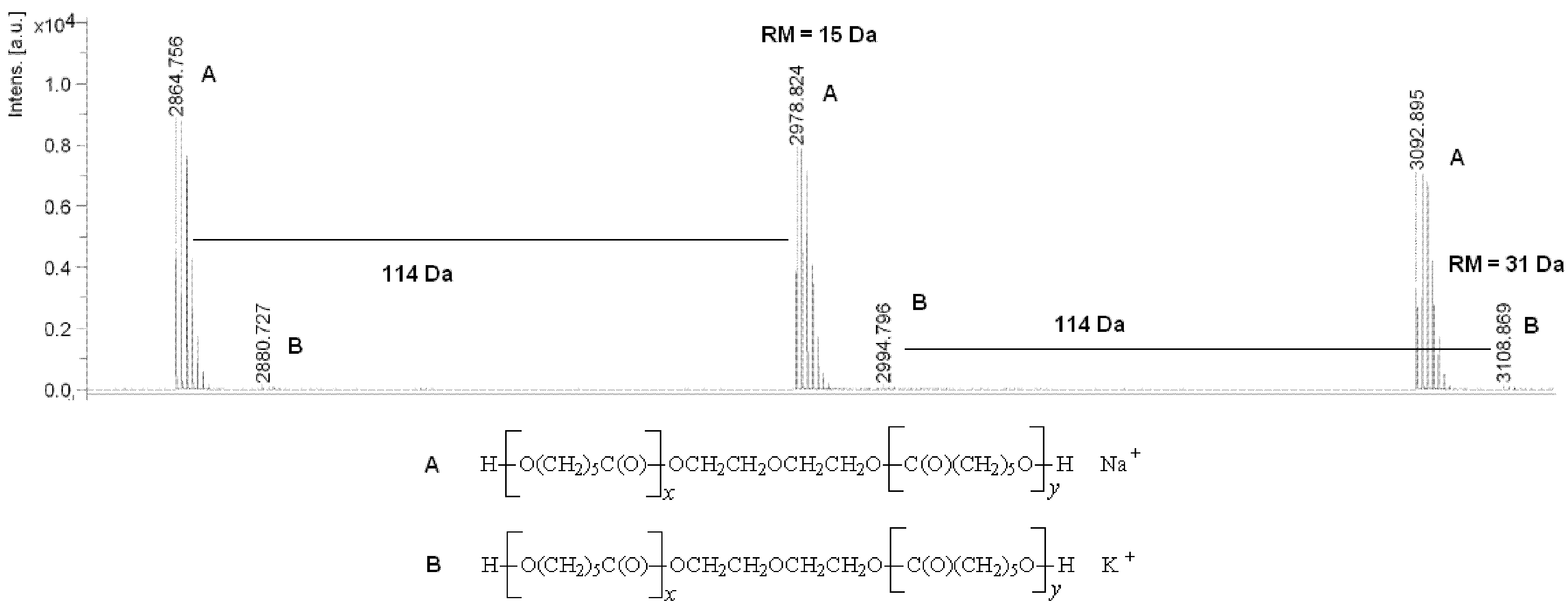{kind=link}
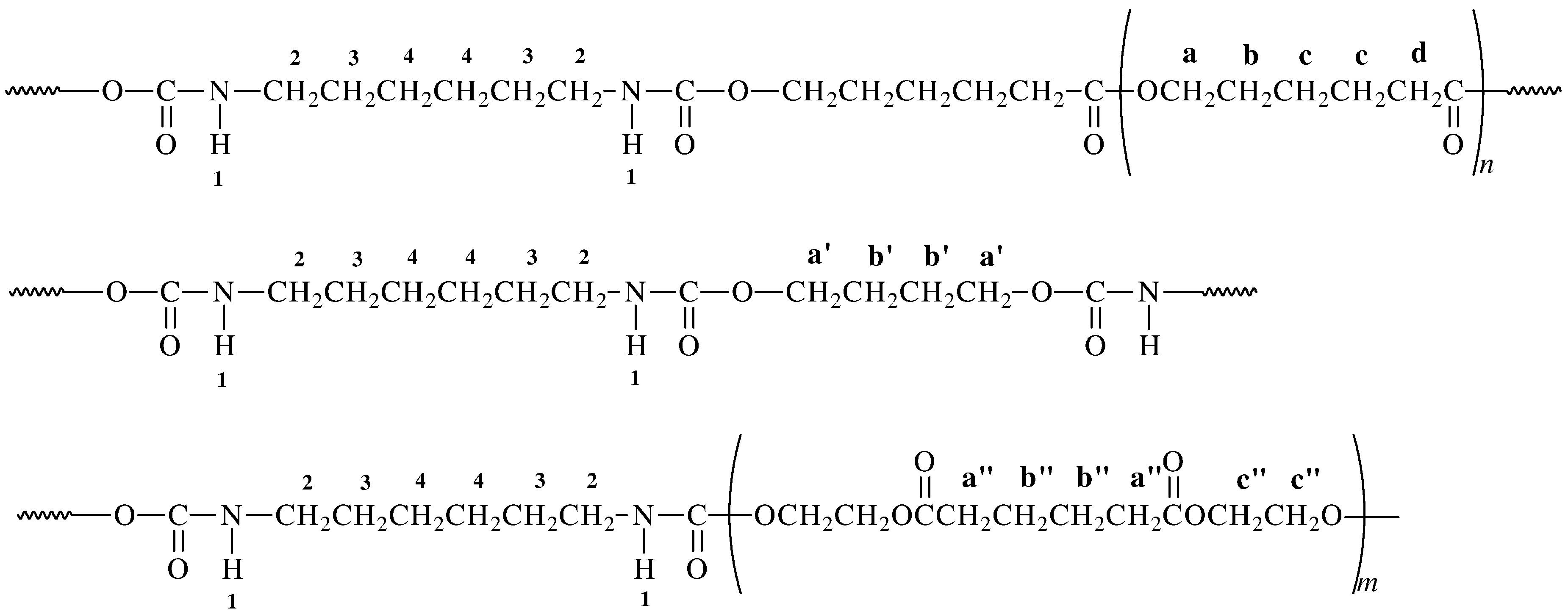{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | PU | Reagents | Mv (g/mol) | FS (MPa) | S100 (MPa) | ε (%) | ShH (Shore A) |
|---|---|---|---|---|---|---|---|
| 1. | PU-1 | HMDI/BD/PEAD/PCL-1 | 62,100 | 14.7 ± 0.8 | 5.2 ± 0.3 | 312 ± 12 | 44 ± 3 |
| 2. | PU-2 | HMDI/BD/PEAD/PCL-2 | 58,200 | 14.3 ± 0.9 | 5.4 ± 0.3 | 348 ± 13 | 42 ± 3 |
| 3. | PU-3 | HMDI/BD/PEAD/PCL-3 | 59,600 | 13.9 ± 0.7 | 5.5 ± 0.2 | 358 ± 11 | 41 ± 2 |


2.2. The Polyurethane/Hydroxyapatite Composites’ Fabrication
| No. | Code | Composition | d (g/cm3) | P (%) |
|---|---|---|---|---|
| 1. | PU-HA-1 | PU-1(PCL-1)/HA: 9/1 | 0.225 ± 0.003 | 66.5 ± 0.9 |
| 2. | PU-HA-2 | PU-1(PCL-1)/HA: 8/2 | 0.266 ± 0.003 | 53.6 ± 0.6 |
| 3. | PU-HA-3 | PU-2(PCL-2)/HA: 9/1 | 0.210 ± 0.002 | 59.4 ± 0.6 |
| 4. | PU-HA-4 | PU-2/HA PU(PCL-2): 8/2 | 0.259 ± 0.002 | 46.4 ± 0.4 |
| 5. | PU-HA-5 | PU-3(PCL-3)/HA: 9/1 | 0.207 ± 0.002 | 56.2 ± 0.5 |
| 6. | PU-HA-6 | PU-3(PCL-3)/HA: 8/2 | 0.244 ± 0.003 | 42.2 ± 0.5 |



2.3. Cytotoxic Tests
| PU | Spirotox (24 h) | Microtox (15 min) | Protoxkit F (24 h) | |||
|---|---|---|---|---|---|---|
| Concentration (mg·mL−1) | 10 | 1 | 1 | 0.5 | 1 | 0.5 |
| PU-1 | 0 | 0 | 0 | 0 | 15 | 5 |
| PU-2 | 0 | 0 | 0 | 0 | 14 | 8 |
| PU-3 | 0 | 0 | 0 | 0 | 16 | 11 |
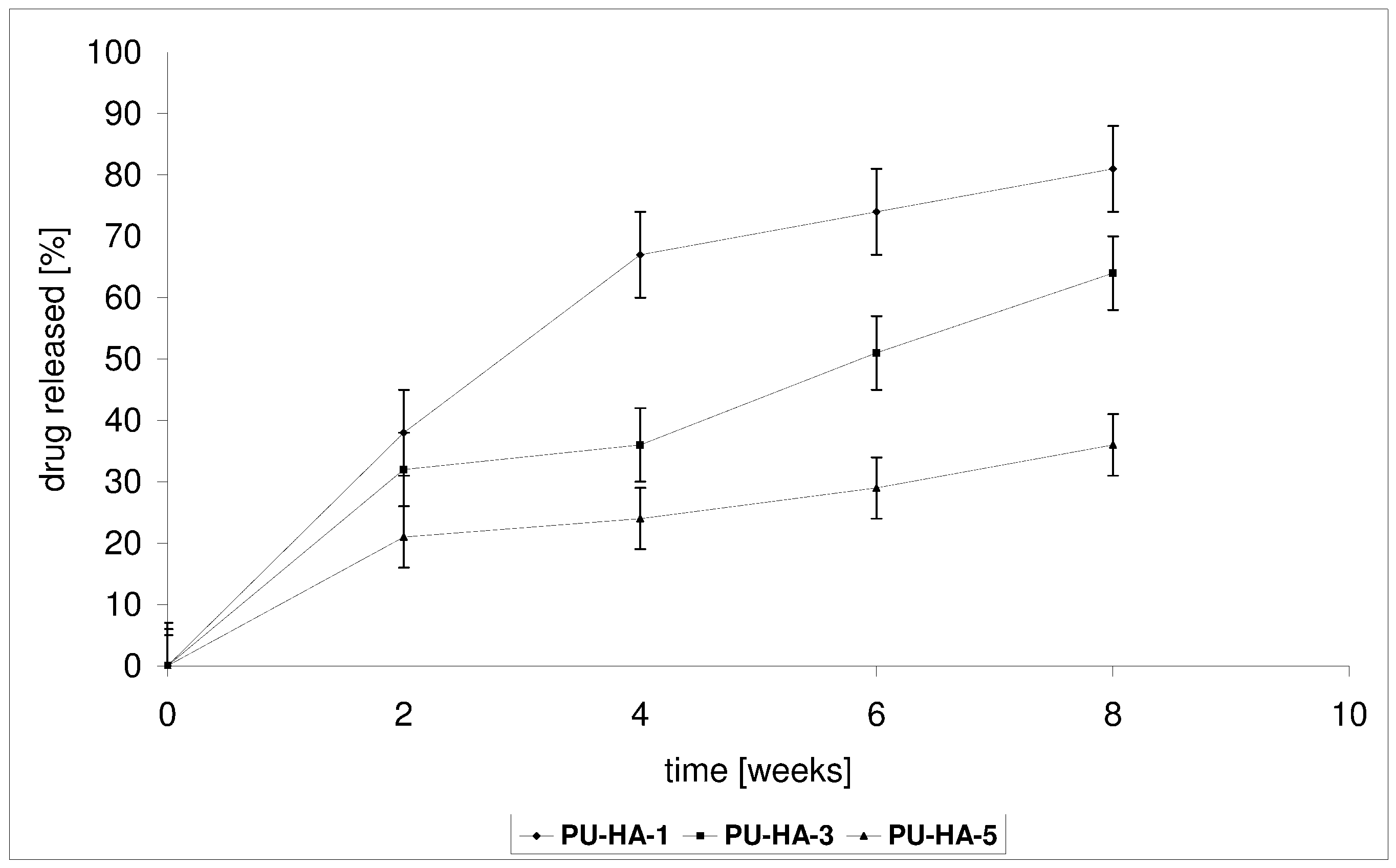
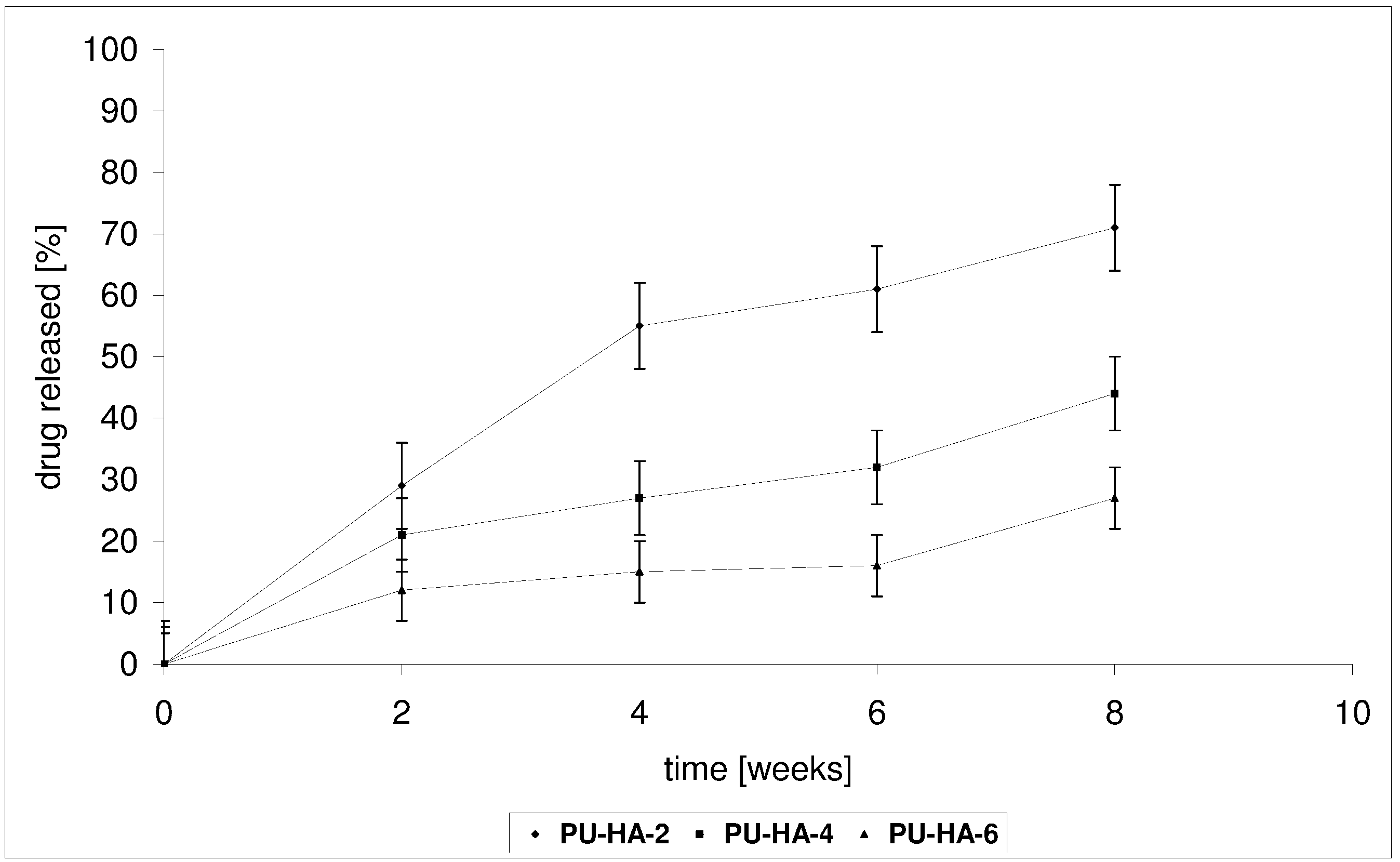
2.4. Drug Release from the Polyurethane/Hydroxyapatite Composites


| No. | PU | Reagents | Mv (g/mol) | Mv(4) (g/mol) | Mv(4) loss (%) | Mv(8) (g/mol) | Mv(8) loss (%) |
|---|---|---|---|---|---|---|---|
| 1 | PU-1 | HMDI/BD/PEAD/PCL-1 | 62,100 | 58,400 | 6.0 | 55,000 | 11.4 |
| 2 | PU-2 | HMDI/BD/PEAD/PCL-2 | 58,200 | 55,200 | 5.2 | 53,300 | 8.4 |
| 3 | PU-3 | HMDI/BD/PEAD/PCL-3 | 59,600 | 56,800 | 4.7 | 55,300 | 7.2 |


| No. | PU | Reagents | FS (MPa) | S100 (MPa) | ε (%) | ShH (Shore A) |
|---|---|---|---|---|---|---|
| 1. | PU-1 | HDI/BD/OEAD/PCL-1 | 11.2 ± 0.7 | 4.1 ± 0.2 | 252 ± 10 | 37 ± 3 |
| 2. | PU-2 | HDI/BD/OEAD/PCL-2 | 11.7 ± 0.8 | 4.5 ± 0.3 | 296 ± 9 | 37 ± 3 |
| 3. | PU-3 | HDI/BD/OEAD/PCL-3 | 12.2 ± 0.6 | 5.0 ± 0.2 | 326 ± 11 | 38 ± 2 |

3. Experimental Section
3.1. Materials
3.2. Synthesis of Poly(ε-caprolactone) Diols
FTIR and NMR Data
3.3. Synthesis of Polyurethanes
FTIR and NMR Data
3.4. Cytotoxicity Assays
3.5. Composite Production
3.6. Clodronate Impregnation of the Polyurethane Composites
3.7. Clodronate Release from the Composites
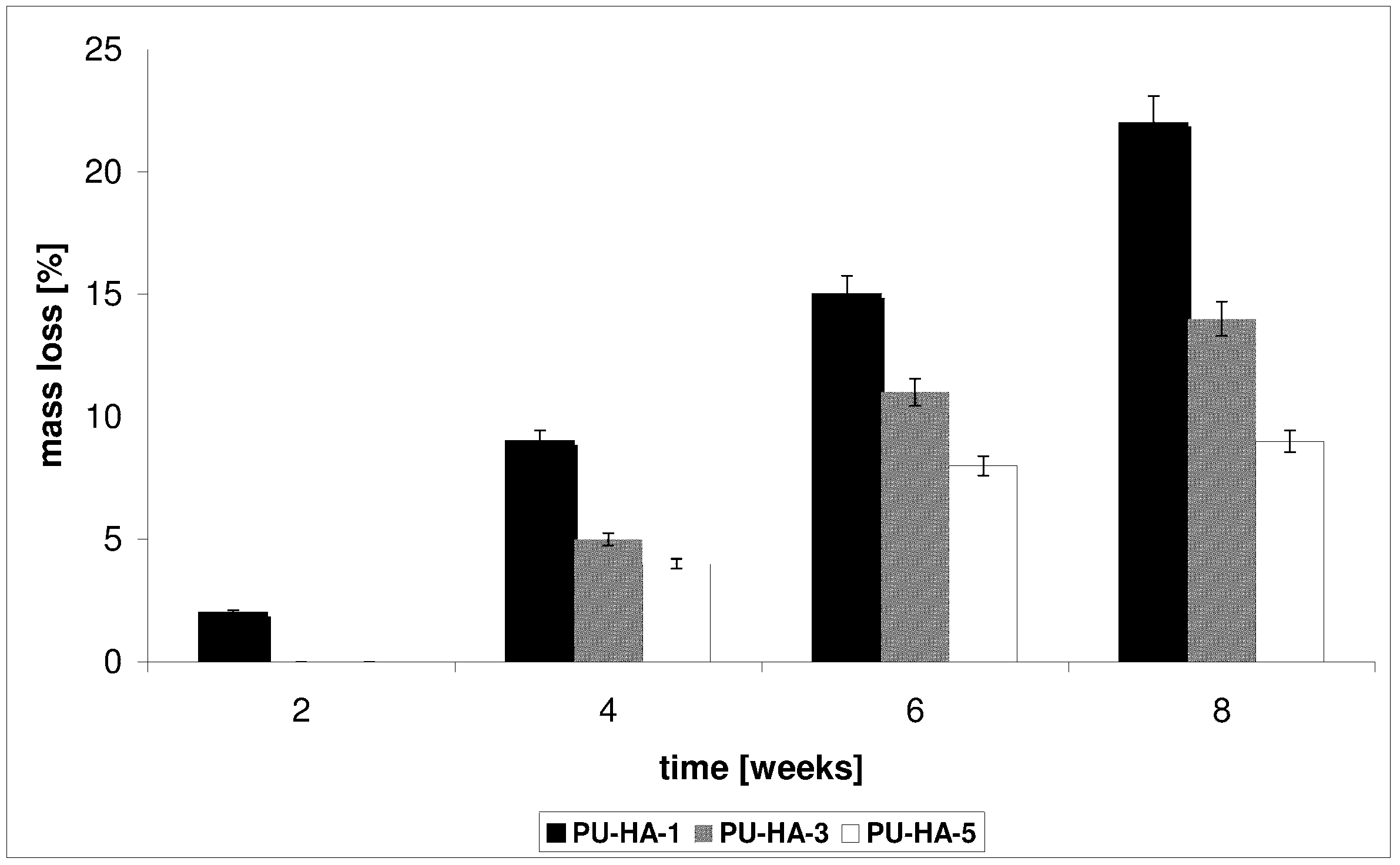
3.8. Degradation Test
3.9. Measurements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar]
- Holen, I.; Coleman, R.E. Bisphosphonates as treatment of bone metastases. Curr. Pharm. Des. 2010, 16, 1262–1271. [Google Scholar]
- Coleman, R.E. Bisphosphonates: Clinical experience. Oncologist 2004, 9, 14–27. [Google Scholar]
- Lipton, A. Pathophysiology of bone metastases: How this knowledge may lead to therapeutic intervention. J. Support. Oncol. 2004, 2, 205–220. [Google Scholar]
- Von Moos, R. Ibandronate provides efficacy and safety in the treatment of metastatic bone disease. Eur. J. Cancer Suppl. 2006, 4, 13–18. [Google Scholar]
- Russell, R.G.; Watts, N.B.; Ebetino, F.H.; Rogers, M.J. Mechanisms of action of bisphosphonates: Similarities and differences and their potential influence on clinical efficacy. Osteoporos. Int. 2008, 19, 733–759. [Google Scholar]
- Russell, R.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–11. [Google Scholar]
- Padalecki, S.S.; Carreon, M.; Grubbs, B.; Cui, Y.; Guise, T.A. Androgen deprivation enhances bone loss and prostate cancer metastases to bone: Prevention by zoledronic acid. Oncology 2003, 17, (Suppl.). 32–54. [Google Scholar]
- Rosen, L.S.; Gordon, D.; Kaminski, M.; Howell, A.; Belch, A.; Mackey, J.; Apffelstaedt, J.; Hussein, M.A. Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast cancer: A randomized, double-blind, multicenter, comparative trial. Cancer 2003, 98, 1735–1744. [Google Scholar]
- Barret-Lee, P.; Casbard, A.; Abraham, J.; Hood, K.; Coleman, R.; Simmonds, P.; Timmins, H.; Wheatley, D.; Grieve, R.; Griffiths, G.; et al. Oral ibandronic acid versus intravenous zoledronic acid in treatment of bone metastases from breast cancer: A randomised, open label, non-inferiority phase 3 trial. Lancet Oncol. 2014, 15, 114–122. [Google Scholar]
- Tanvetyanon, T.; Stiff, P.J. Management of the adverse effects associated with intravenous bisphosphonates. Ann. Oncol. 2006, 17, 897–907. [Google Scholar]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar]
- Ouchi, T.; Ohya, Y. Macromolecular prodrugs. Prog. Polym. Sci. 1995, 20, 211–257. [Google Scholar]
- Sobczak, M.; Olędzka, E.; Kołodziejski, W.; Kuźmicz, R. Pharmaceutical application of polymers. Polimery 2007, 52, 411–420. [Google Scholar]
- Giger, E.V.; Castagner, B.; Leroux, J.-C. Biomedical applications of bisphosphonates. J. Control. Release 2013, 167, 175–188. [Google Scholar]
- Karrholm, J.; Borssen, B.; Lowenhielm, G.; Snorrason, F. Does early micromotion of femoral stem prostheses matter? 4-7-Year stereoradiographic follow-up of 84 cemented prostheses. J. Bone Jt. Surg. Br. 1994, 76, 912–917. [Google Scholar]
- Cremers, S.; Papapoulos, S. Pharmacology of bisphosphonates. Bone 2011, 49, 42–49. [Google Scholar]
- Katsumi, H.; Takashima, M.; Sano, J.-I.; Nishiyama, K.; Kitamura, N.; Sakane, T.; Hibi, T.; Yamamoto, A. Development of polyethylene glycol-conjugated alendronate, a novel nitrogen-containing bisphosphonate derivative: Evaluation of absorption, safety, and effects after intrapulmonary administration in rats. J. Pharm. Sci. 2011, 100, 3783–3792. [Google Scholar]
- Bellido, T.; Plotkin, L.I. Novel actions of bisphosphonates in bone: Preservation of osteoblast and osteocyte viability. Bone 2011, 49, 50–55. [Google Scholar]
- Gutman, D.; Golomb, G. Liposomal alendronate for the treatment of restenosis. J. Control. Release 2012, 161, 619–627. [Google Scholar]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjallman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar]
- Salzano, G.; Marra, M.; Porru, M.; Zappavigna, S.; Abbruzzese, A.; la Rotonda, M.I.; Leonetti, C.; Caraglia, M.; de Rosa, G. Self-assembly nanoparticles for the delivery of bisphosphonates into tumors. Int. J. Pharm. 2011, 403, 292–297. [Google Scholar]
- Wang, G.; Mostafa, N.Z.; Incani, V.; Kucharski, C.; Uludag, H. Bisphosphonate decorated lipid nanoparticles designed as drug carriers for bone diseases. J. Biomed. Mater. Res. A 2012, 100, 684–693. [Google Scholar]
- Cherng, J.Y.; Houa, T.Y.; Shih, M.F.; Talsma, H.; Hennink, W.E. Polyurethane-based drug delivery systems. Int. J. Pharm. 2013, 450, 145–162. [Google Scholar]
- Errassifi, F.; Sarda, S.; Barroug, A.; Legrouri, A.; Sfihi, H.; Rey, C. Infrared, Raman and NMR investigations of risedronate adsorption on nanocrystalline apatites. J. Colloid Interface Sci. 2014, 420, 101–111. [Google Scholar]
- Iafisco, M.; Palazzo, B.; Falini, G.; di Foggia, M.; Bonora, S.; Nicolis, S.; Casella, L.; Roveri, N. Adsorption and conformational change of myoglobin on biomimetic hydroxyapatite nanocrystals functionalized with alendronate. Langmuir 2008, 24, 4924–4930. [Google Scholar]
- Josse, S.; Faucheux, C.; Soueidan, A.; Grimandi, G.; Massiot, D.; Alonso, B.; Janvier, P.; Laïb, S.; Pilet, P.; Gauthier, O.; et al. Novel materials for bisphosphonates delivery. Biomaterials 2005, 26, 2073–2080. [Google Scholar]
- Cukrowski, I.; Popović, L.; Barnard, W.; Paul, S.O.; van Royen, P.H.; Liles, D.C. Modeling and spectroscopic studies of bisphosphonate-bone interactions. The Raman, NMR and crystallographic investigations of Ca-HEDP complexes. Bone 2007, 41, 668–678. [Google Scholar]
- Alghamdi, H.S.; Bosco, R.; Both, S.K.; Iafisco, M.; Leeuwenburgh, S.C.G.; Jansen, J.A.; van den Beucken, J.J.J.P. Synergistic effect of bisphosphonates and calcium phosphate nanoparticles on peri-implant bone responses in osteoporotic rats. Biomaterials 2014, 35, 5482–5490. [Google Scholar]
- Pascaud, P.; Errassifi, F.; Brouillet, F.; Sarda, S.; Barroug, A.; Legrouri, A.; Rey, C. Adsorption on apatitic calcium phosphates for drug delivery: Interaction with bisphosphonates molecules. J. Mater. Sci. Mater. Med. 2014. [Google Scholar] [CrossRef]
- Sobczak, M. Enzyme-catalyzed ring-opening polymerization of cyclic esters in the presence of poly(ethylene glycol). J. Appl. Polym. Sci. 2012, 125, 3602–3609. [Google Scholar]
- Sobczak, M.; Dębek, C.; Olędzka, E.; Nałęcz-Jawecki, G.; Kołodziejski, W.L.; Rajkiewicz, M. Segmented polyurethane elastomers derived from aliphatic polycarbonate and poly(ester-carbonate) soft segments for biomedical applications. J. Polym. Sci. A Polym. Chem. 2012, 50, 3904–3913. [Google Scholar]
- Sobczak, M.; Nałęcz-Jawecki, G.; Kołodziejski, W.L.; Goś, P.; Żółtowska, K. Synthesis and study of controlled release of ofloxacin from polyester conjugates. Int. J. Pharm. 2010, 402, 37–43. [Google Scholar]
- Gorna, K.; Gogolewski, S. In vitro degradation of novel medical biodegradable aliphatic polyurethanes based on ε-caprolactone and Pluronics® with various hydrophilicities. Polym. Degrad. Stab. 2002, 75, 113–122. [Google Scholar]
- Sobczak, M.; Dębek, C.; Goś, P. Preparation and mechanical properties of PCL-based polyurethanes as potential biomaterials for short-term applications. E-Polymers 2010, 10, 1661–1669. [Google Scholar]
- Asefnejad, A.; Khorasani, M.T.; Behnamghader, A.; Farsadzadeh, B.; Bonakdar, S. Manufacturing of biodegradable polyurethane scaffolds based on polycaprolactone using a phase separation method: Physical properties and in vitro assay. Int. J. Nanomed. 2011, 6, 2375–2384. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kolmas, J.; Sobczak, M.; Olędzka, E.; Nałęcz-Jawecki, G.; Dębek, C. Synthesis, Characterization and in Vitro Evaluation of New Composite Bisphosphonate Delivery Systems. Int. J. Mol. Sci. 2014, 15, 16831-16847. https://doi.org/10.3390/ijms150916831
Kolmas J, Sobczak M, Olędzka E, Nałęcz-Jawecki G, Dębek C. Synthesis, Characterization and in Vitro Evaluation of New Composite Bisphosphonate Delivery Systems. International Journal of Molecular Sciences. 2014; 15(9):16831-16847. https://doi.org/10.3390/ijms150916831
Chicago/Turabian StyleKolmas, Joanna, Marcin Sobczak, Ewa Olędzka, Grzegorz Nałęcz-Jawecki, and Cezary Dębek. 2014. "Synthesis, Characterization and in Vitro Evaluation of New Composite Bisphosphonate Delivery Systems" International Journal of Molecular Sciences 15, no. 9: 16831-16847. https://doi.org/10.3390/ijms150916831