
Cloud Computing-Based TagSNP Selection Algorithm for Human Genome Data


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
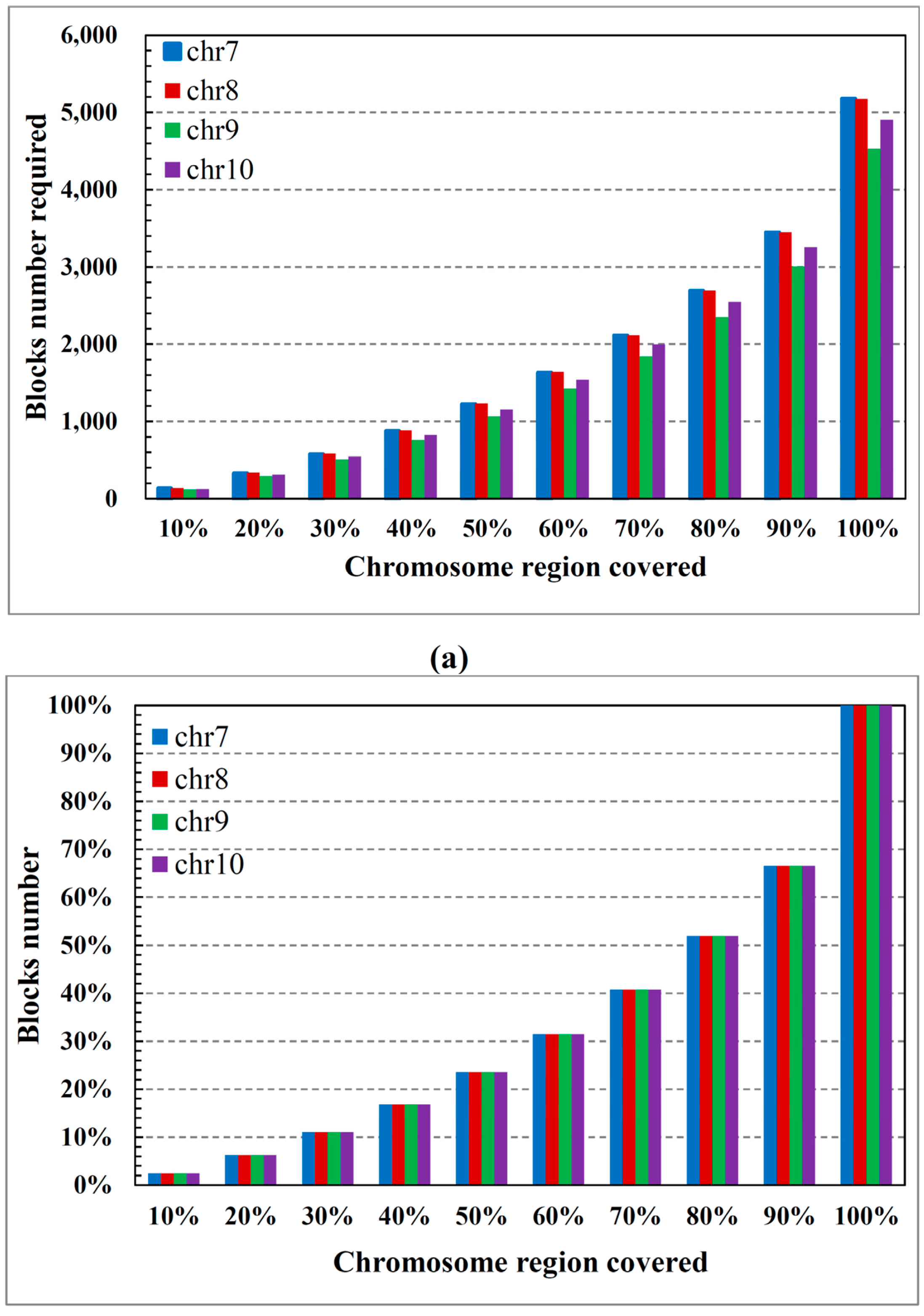
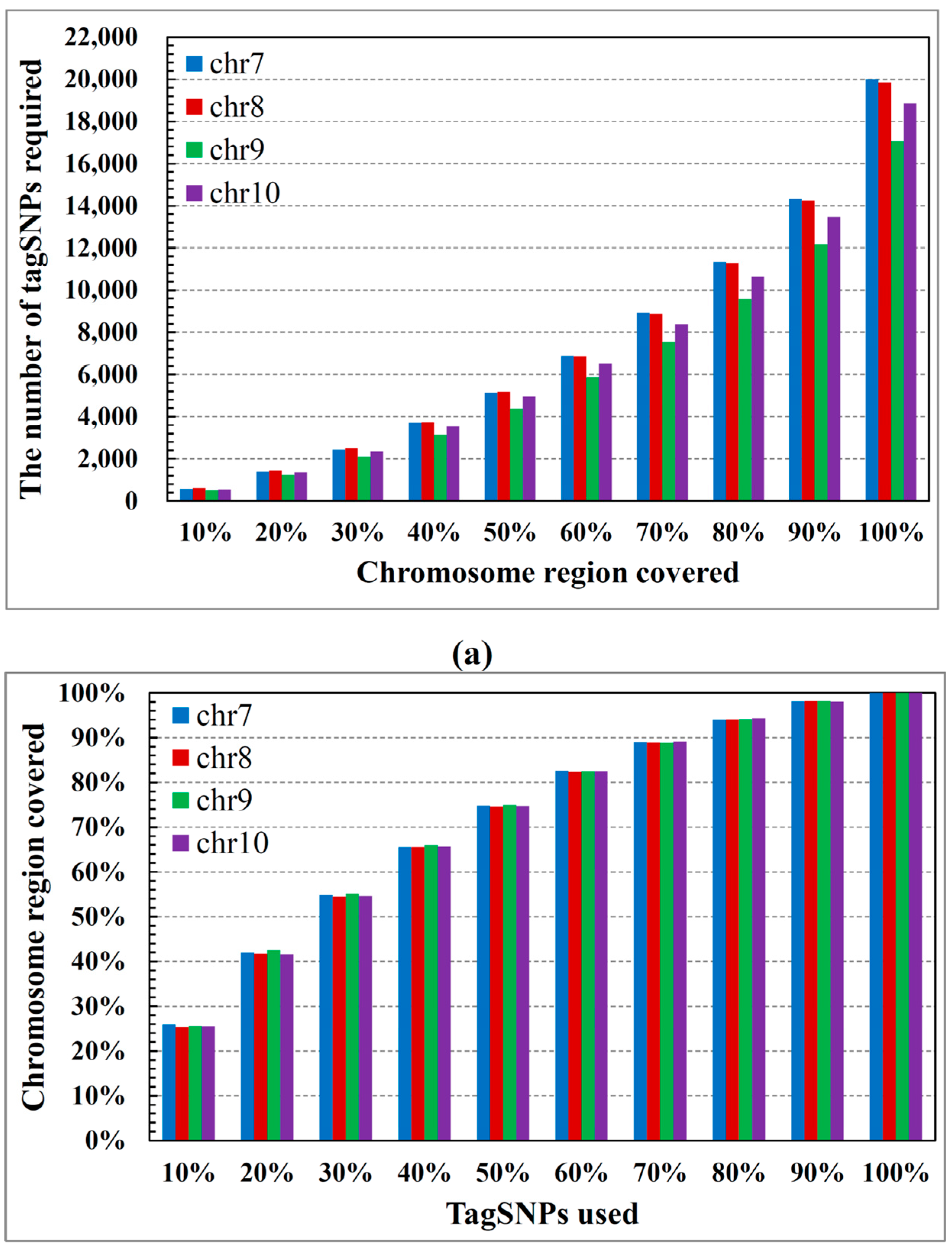
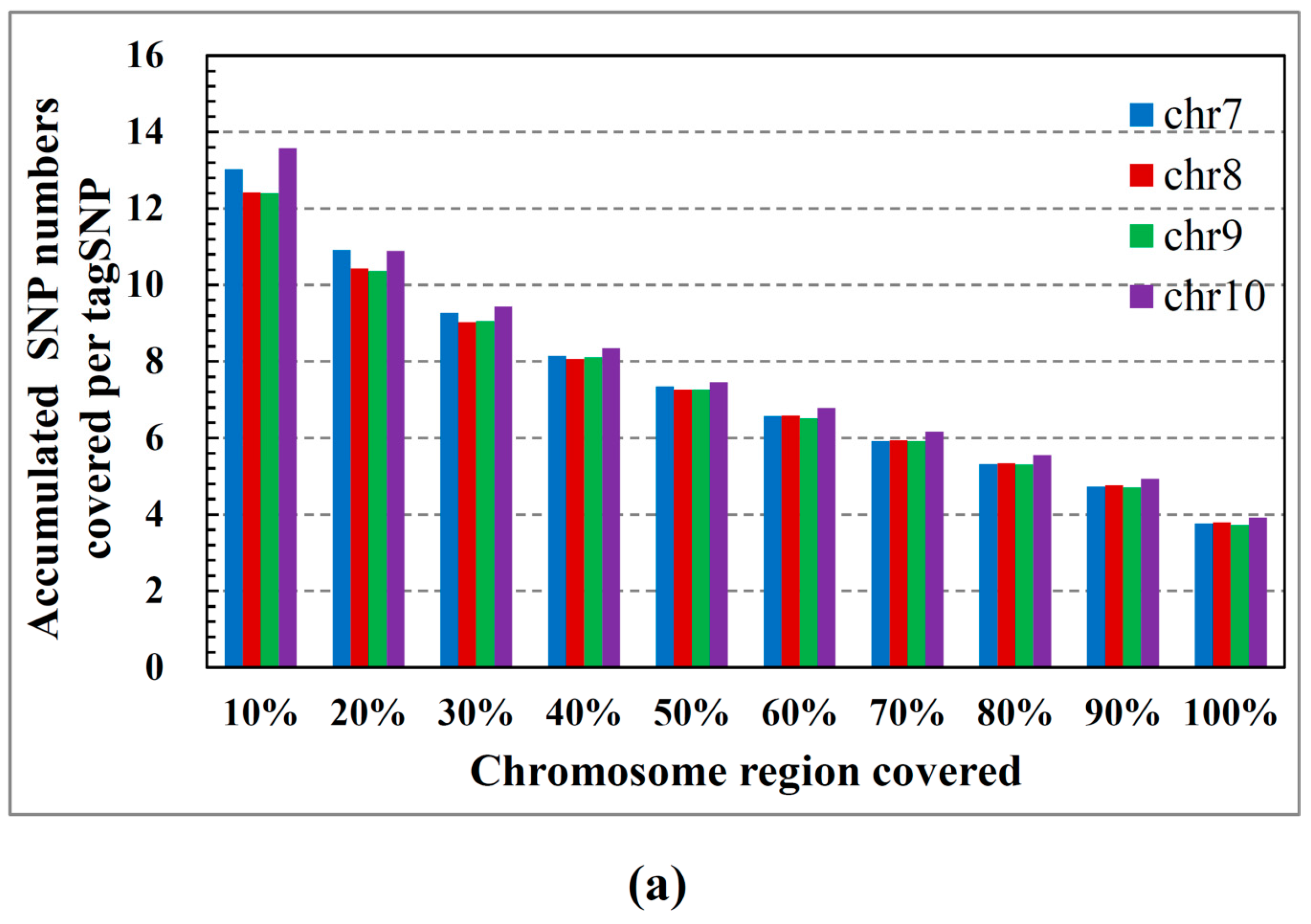
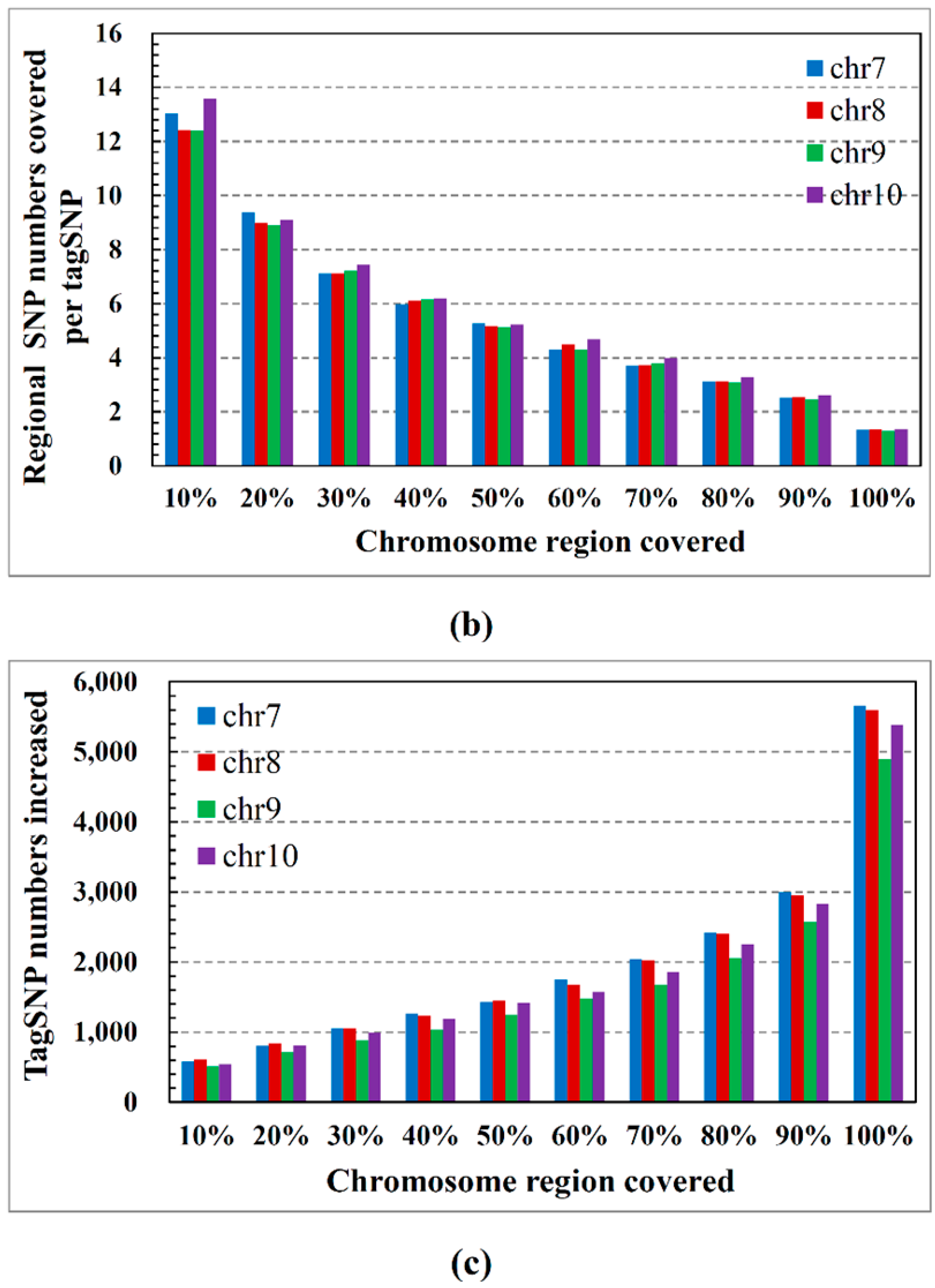
2.1. ASW Data Characteristics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common SNPs/Block | Zhang | FinKLB | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of Blocks | Length | Avg. Length | All Blocks (%) | Common SNPS (%) | No. of Blocks | Length | Avg. Length | All Blocks (%) | Common SNPs (%) | |
| ASW_chr7 | ||||||||||
| < 15 | 3525 | 28,090 | 7.97 | 64.29 | 37.29 | 3250 | 29,822 | 9.18 | 62.82 | 39.59 |
| 15 to 30 | 1604 | 32,524 | 20.28 | 29.25 | 43.18 | 1603 | 32,138 | 20.05 | 30.98 | 42.67 |
| > 30 | 354 | 14,706 | 41.54 | 6.46 | 19.53 | 321 | 13,360 | 41.62 | 6.20 | 17.74 |
| Total | 5483 | 75,320 | 13.74 | 100.00 | 100.00 | 5174 | 75,320 | 14.56 | 100.00 | 100.00 |
| Max. Blocks | 102 | 107 | ||||||||
| Tag SNP | 18,012 | 19,990 | ||||||||
| CPU Time | 409,834(s) ≡ 113.83(h) | 783(s) ≡ 0.22(h) | ||||||||
| ASW_chr8 | ||||||||||
| < 15 | 3514 | 27,976 | 7.96 | 63.90 | 37.17 | 3225 | 29,837 | 9.25 | 62.32 | 39.64 |
| 15 to 30 | 1640 | 33,156 | 20.22 | 29.82 | 44.05 | 1638 | 32,798 | 20.02 | 31.65 | 43.57 |
| > 30 | 345 | 14,140 | 40.99 | 6.28 | 18.78 | 312 | 12,637 | 40.50 | 6.03 | 16.79 |
| Total | 5499 | 75,272 | 13.69 | 100 | 100.00 | 5175 | 75,272 | 14.55 | 100.00 | 100.00 |
| Max. Blocks | 105 | 105 | ||||||||
| Tag SNP | 17,957 | 19,844 | ||||||||
| CPU Time | 299,970(s) ≡ 83.32(h) | 924(s) ≡ 0.25(h) | ||||||||
| ASW_chr9 | ||||||||||
| < 15 | 3175 | 24,607 | 7.75 | 65.98 | 38.68 | 2945 | 26,714 | 9.07 | 65.02 | 42.00 |
| 15 to 30 | 1343 | 27,093 | 20.17 | 27.91 | 42.59 | 1330 | 26,664 | 20.05 | 29.37 | 41.92 |
| > 30 | 294 | 11,912 | 40.52 | 6.11 | 18.73 | 254 | 10,234 | 40.29 | 5.61 | 16.09 |
| Total | 4812 | 63,612 | 13.22 | 100.00 | 100.00 | 4529 | 63,612 | 14.05 | 100 | 100.00 |
| Max. Blocks | 83 | 83 | ||||||||
| Tag SNP | 15,308 | 17,064 | ||||||||
| CPU Time | 47,786(s) ≡ 13.27(h) | 645(s) ≡ 0.17(h) | ||||||||
| ASW_chr10 | ||||||||||
| < 15 | 3261 | 25,850 | 7.93 | 62.52 | 35.01 | 2973 | 27,623 | 9.29 | 60.62 | 37.41 |
| 15 to 30 | 1556 | 31,559 | 20.28 | 29.83 | 42.75 | 1585 | 31,855 | 20.10 | 32.31 | 43.15 |
| > 30 | 399 | 16,423 | 41.16 | 7.65 | 22.24 | 347 | 14,354 | 41.37 | 7.07 | 19.44 |
| Total | 5216 | 73,832 | 14.15 | 100 | 100.00 | 4905 | 73,832 | 15.05 | 100 | 100.00 |
| Max. Blocks | 112 | 112 | ||||||||
| Tag SNP | 17,012 | 18,862 | ||||||||
| CPU Time | 46,580(s) ≡ 12.93(h) | 919(s) ≡ 0.25(h) | ||||||||




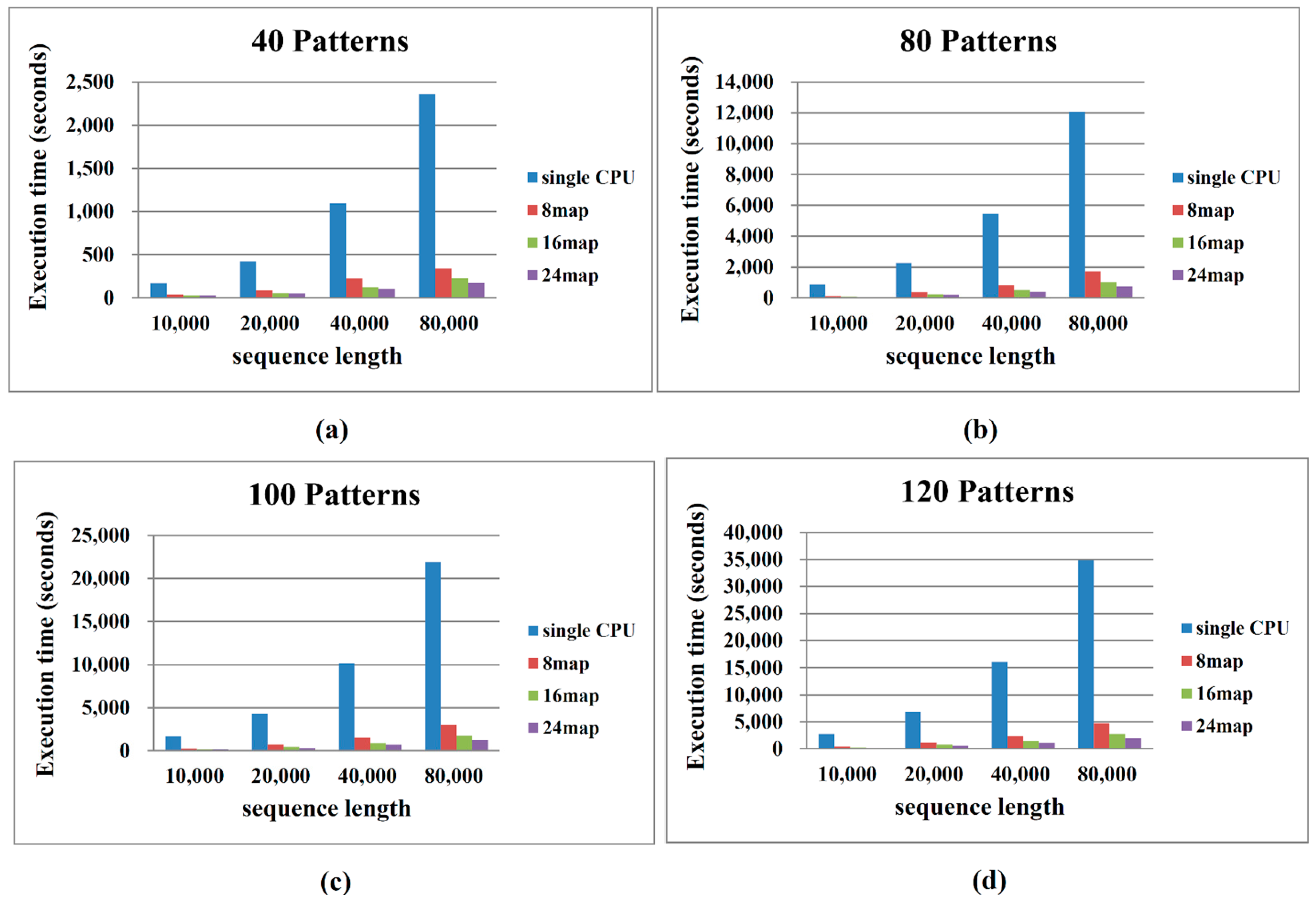
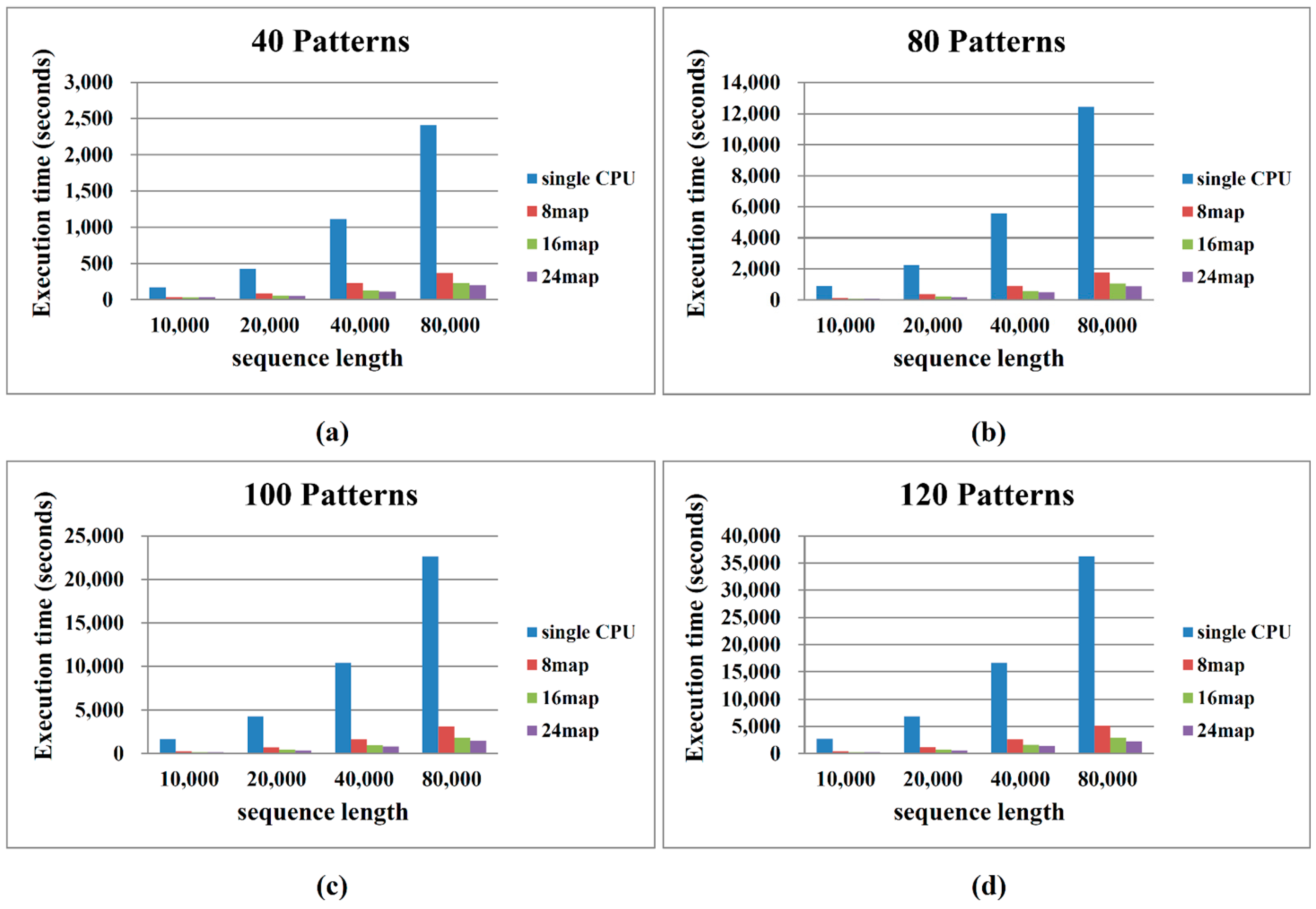
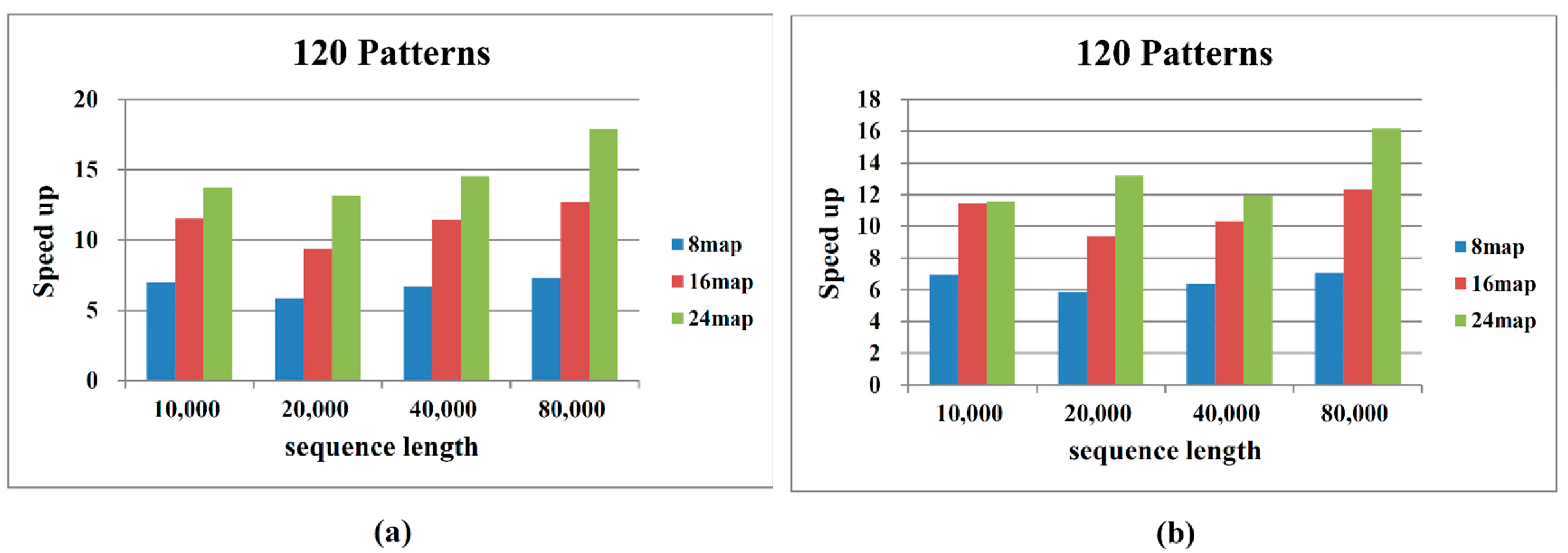
2.2. Performance on Cloud Computing



3. Methods
3.1. Diversity Function

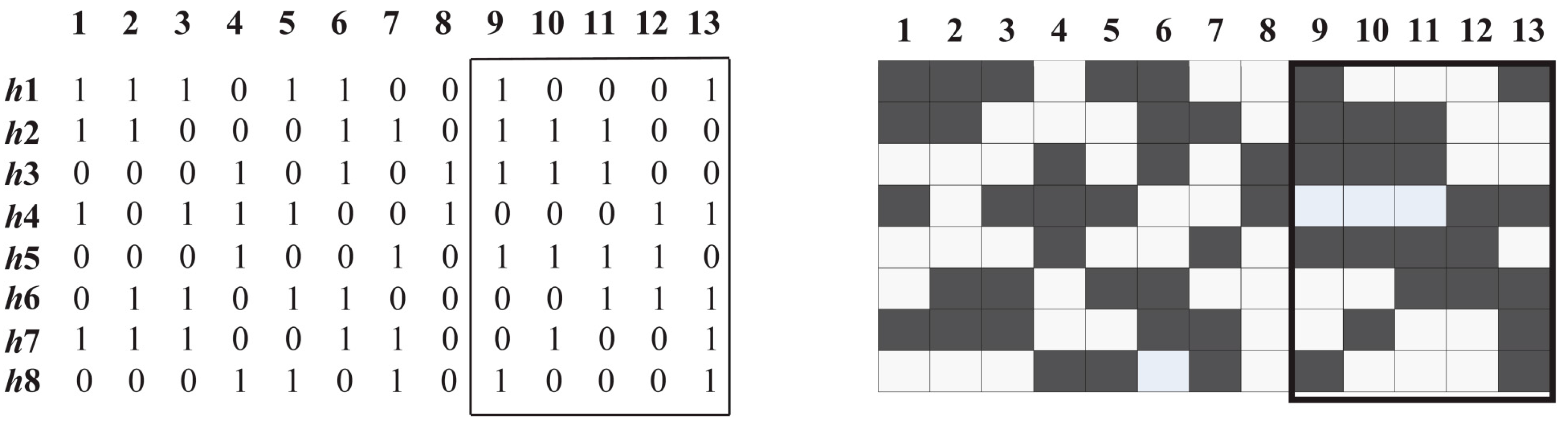
3.2. Common Haplotypes
3.3. Haplotype Block Partitioning
3.4. TagSNPs Selection
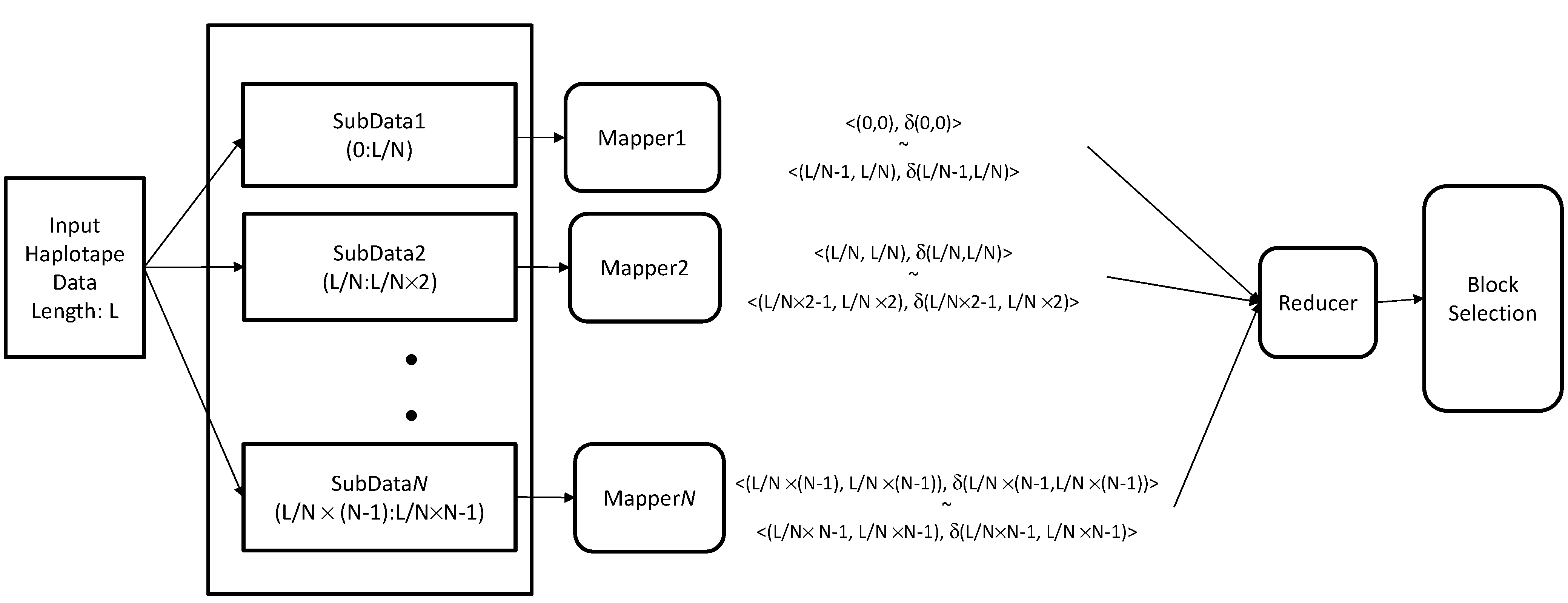
3.5. Hadoop Framework
3.6. Hadoop-Based Block Partitioning and Selection Scheme

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bonnen, P.E.; Wang, P.J.; Kimmel, M.; Chakraborty, R.; Nelson, D.L. Haplotype and linkage disequilibrium architecture for human cancer-associated genes. Genome Res. 2002, 12, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Gray, I.C.; Campbell, D.A.; Spurr, N.K. Single nucleotide polymorphisms as tools in human genetics. Hum. Mol. Genet. 2000, 9, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Indap, A.R.; Marth, G.T.; Struble, C.A.; Tonellato, P.J.; Olivier, M. Analysis of concordance of different haplotype block partitioning algorithms haplotype tagging for the identification of common disease genes. BMC Bioinformatics 2005, 6, 303. [Google Scholar] [CrossRef] [PubMed]
- Mas, A.; Blanco, E.; Monux, G.; Urcelay, E.; Serrano, F.J.; de la Concha, E.G.; Martinez, A. DRB1-TNF-alpha-TNF-beta haplotype is strongly associated with severe aortoiliac occlusive disease, a clinical form of atherosclerosis. Hum. Immunol. 2005, 66, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, P.; Kwon, J.M.; Goate, A.M. SNP analysis to dissect human traits. Curr. Opin. Neurobiol. 2001, 11, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Reif, A.; Herterich, S.; Strobel, A.; Ehlis, A.C.; Saur, D.; Jacob, C.P.; Wienker, T.; Topner, T.; Fritzen, S.; Walter, U.; et al. A neuronal nitri coxide synthase (NOS-I) haplotype associated with schizo-phrenia modifies prefront alcortex function. Mol. Psychiatry 2006, 11, 286–300. [Google Scholar]
- Daly, M.J.; Rioux, J.D.; Schaffner, S.F.; Hudson, T.J.; Lander, E.S. High-resolution haplotype structure in the human genome. Nat. Genet. 2001, 29, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar]
- Patil, N.; Berno, A.J.; Hinds, D.A.; Barrett, W.A.; Doshi, J.M.; Hacker, C.R.; Kautzer, C.R.; Lee, D.H.; Marjoribanks, C.; McDonough, D.P.; et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 2001, 294, 1719–1723. [Google Scholar]
- Dawson, E.; Abecasis, G.R.; Bumpstead, S.; Chen, Y.; Hunt, S.; Beare, D.M.; Pabial, J.; Dibling, T.; Tinsley, E.; Kirby, S. First-generation linkage disequilibrium map of human chromosome 22. Nature 2002, 418, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Mahdevar, G.; Zahiri, J.; Sadeghi, M.; Nowzari-Dalini, A.; Ahrabian, H. Tag SNP selection via a genetic algorithm. J. Biomed. Inform. 2010. [Google Scholar] [CrossRef]
- Zhang, K.; Calabrese, P.; Nordborg, M.; Sun, F. Haplotype block structure and its applications to association studies: Power and study designs. Am. J. Hum. Genet. 2002, 71, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.D.; Pritchard, J.K. Assessing the performance of the haplotype block model of linkage disequilibrium. Am. J. Hum. Genet. 2003, 73, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.C.L.; Esposito, L.; Barratt, B.J.; Smith, A.N.; Heward, J.; Di Genova, G.; Ueda, H.; Cordell, H.J.; Eaves, I.A.; Dudbridge, F.; et al. Haplotype tagging for the identification of common disease genes. Nat. Genet. 2001, 29, 233–237. [Google Scholar]
- Zahirib, J.; Mahdevar, G.; Nowzari-dalini, A.; Ahrabian, H.; Sadeghic, M. A novel efficient dynamic programming algorithm for haplotype block partitioning. J. Theor. Biol. 2010, 267, 164–170. [Google Scholar]
- Greenspan, G.; Geiger, D. High density linkage disequilibrium mapping using models of haplotype block variation. Bioinformatics 2004, 20, 137. [Google Scholar] [CrossRef]
- Wang, N.; Akey, J.M.; Zhang, K.; Chakraborty, R.; Jin, L. Distribution of recombination crossovers and the origin of haplotype blocks: The interplay of population history, recombination, and mutation. Am. J. Hum. Genet. 2002, 71, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [PubMed]
- Anderson, E.C.; Novembre, J. Finding haplotype block boundaries by using the minimum-description-length principle. Am. J. Hum. Genet. 2003, 73, 336–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Deng, M.; Chen, T.; Waterman, M.S.; Sun, F. A dynamic programming algorithm for haplotype block partitioning. Proc. Natl. Acad. Sci. USA 2002, 99, 7335–7339. [Google Scholar] [CrossRef] [PubMed]
- Hadoop-Apache Software Foundation project home page. Available online: http://hadoop.apache.org/ (accessed on 10 September 2014).
- Taylor, R.C. An overview of the Hadoop/MapReduce/HBase framework and its current applications in bioinformatics. BMC Bioinformatics 2010, 11, S1. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.; Ghemawat, S. MapReduce: A Flexible Data Processing Tool. Commun. ACM 2010, 53, 72–77. [Google Scholar] [CrossRef]
- Schatz, M. Cloudburst: Highly sensitive read mapping with MapReduce. Bioinformatics 2009, 25, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Hua, G.J. Cloud Computing for Protein-Ligand Binding Site Comparison. Biomed. Res. Int. 2013. [Google Scholar] [CrossRef]
- Hung, C.L.; Lin, Y.L. Implementation of a Parallel Protein Structure Alignment Service on Cloud. Int. J. Genomics 2013. [Google Scholar] [CrossRef]
- International HapMap Project. Available online: http://hapmap.ncbi.nlm.nih.gov/ (accessed on 10 August 2014).
- Chen, W.P.; Hung, C.L.; Lin, Y.L. Efficient Haplotype Block Partitioning and Tag SNP Selection Algorithms under Various Constraints. Biomed Res. Int. 2013. [Google Scholar] [CrossRef]
- Chen, W.P.; Hung, C.L.; Tsai, S.J.J.; Lin, Y.L. Novel and efficient tag SNPs selection algorithms. Biomed. Mater. Eng. 2014, 24, 1383–1389. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, C.-L.; Chen, W.-P.; Hua, G.-J.; Zheng, H.; Tsai, S.-J.J.; Lin, Y.-L. Cloud Computing-Based TagSNP Selection Algorithm for Human Genome Data. Int. J. Mol. Sci. 2015, 16, 1096-1110. https://doi.org/10.3390/ijms16011096
Hung C-L, Chen W-P, Hua G-J, Zheng H, Tsai S-JJ, Lin Y-L. Cloud Computing-Based TagSNP Selection Algorithm for Human Genome Data. International Journal of Molecular Sciences. 2015; 16(1):1096-1110. https://doi.org/10.3390/ijms16011096
Chicago/Turabian StyleHung, Che-Lun, Wen-Pei Chen, Guan-Jie Hua, Huiru Zheng, Suh-Jen Jane Tsai, and Yaw-Ling Lin. 2015. "Cloud Computing-Based TagSNP Selection Algorithm for Human Genome Data" International Journal of Molecular Sciences 16, no. 1: 1096-1110. https://doi.org/10.3390/ijms16011096