
New Therapies for Dedifferentiated Papillary Thyroid Cancer

Abstract
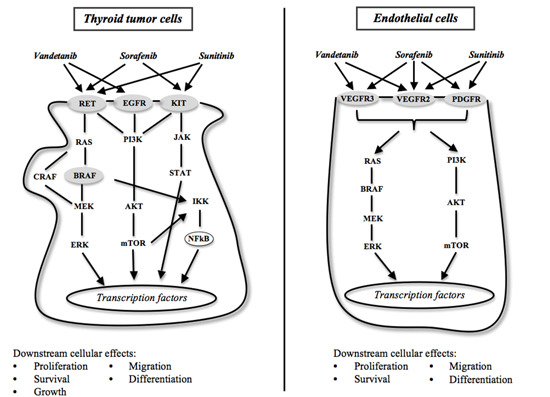
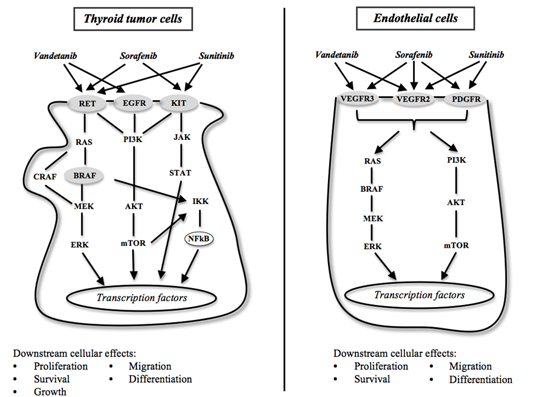
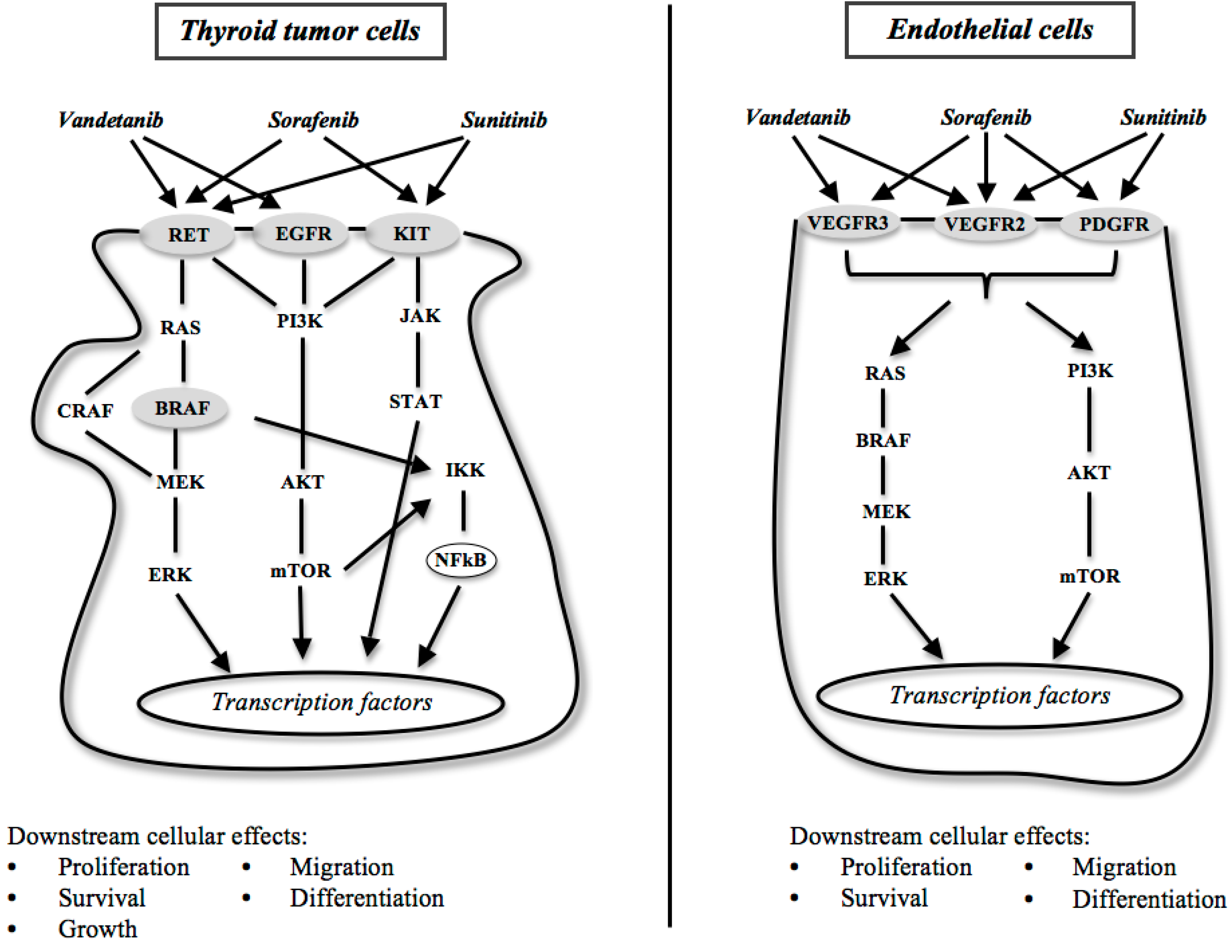
:
1. Introduction
2. Molecular Pathways Involved in DePTC
2.1. RET/PTC Rearrangements, BRAF, RAS, PAX8/PPARγ, Histone Acetylation
2.2. Factors Involved in Angiogenesis
2.2.1. Vascular Endothelial Growth Factor (VEGF)
2.2.2. EGF Receptor (EGFR)

2.3. Genomic Analysis
2.4. Tyrosine Kinase Inhibitors (TKIs)
3. Sorafenib

{kind=link}
{kind=link}
| Drug | Thyroid Cancer | Responses | Authors | |||
|---|---|---|---|---|---|---|
| PR | SD | PD | PFS (months) | |||
| Sorafenib | 30 DeTC | 23.3% | 53.3% | 7% | 21 | Gupta-Abramson et al. [55] |
| Sorafenib | 41 DeTC | 15% | 56% | – | 15 | Kloos et al. [56] |
| Sorafenib | 31 DeTC | 25% | 34% | 22% | 14.5 | Hoftijzer et al. [58] |
| Sorafenib | 13 DeTC | 20% | 60% | 20% | 19 | Cabanillas et al. [59] |
| Sorafenib | 19 DeTC 15 MTC | 18% DeTC 25% MTC | – | – | – | Ahmed et al. [60] |
| Sorafenib | 47 DeTC 5 ATC 3 MTC | 38% DeTC | 47% DeTC | – | 23.4 | Keefe et al. [61] |
| Sorafenib | 17 DeTC | 30% | 41% | 18% | 9 | Marotta et al. [62] |
| Sorafenib | 31 DeTC | 31% | 42% | – | 18 | Schneider et al. [63] |
| Sorafenib | 207 DeTC | – | – | – | 10.8 | Brose et al. [64] |
| Sorafenib | 8 DeTC | 12.5% | 62.5% | 25% | 14–24 | Pitoia [65] |
| Sorafenib Sunitinib Vandetanib | 32 DeTC 13 ATC 17 MTC | 15% vs. 8% DeTC 36% MTC | – | – | 6.7 vs. 7 DeTC | Massicotte et al. [66] |
4. Sunitinib
| Drug | Thyroid Cancer | Responses | Authors | |||
|---|---|---|---|---|---|---|
| PR | SD | PD | PFS (months) | |||
| Sunitinib | 37 DeTC 6 MTC | 13% DeTC | 68% DeTC 83% MTC | 10% DeTC 17% MTC | Cohen et al. [74] | |
| Sunitinib | 12 DeTC 1 ATC 4 MTC | 6% | 71% | Ravaud et al. [76] | ||
| Sunitinib | 7 MTC 28 DeTC | 28% PR + 3% CR | 46% | 17% | 12.8 | Carr et al. [77] |
| Sunitinib | 11 DeTC | 18% PR + 9% CR | 45% | 27% | 11.5 | Dìez et al. [78] |
5. Imatinib
6. Vandetanib
| Drug | Thyroid Cancer | Responses | Authors | |||
|---|---|---|---|---|---|---|
| PR | SD | PD | PFS (months) | |||
| Vandetanib | 30 MTC | 20% | 53% | 3% | 27.9 | Wells et al. [84] |
| Vandetanib | 19 MTC | 16% | 53% | 16% | 168 days | Robinson et al. [85] |
| Vandetanib | 231 MTC | 45% | 42% | – | – | Wells et al. [86] |
| Vandetanib | 145 DeTC | 8% | 57% | – | 11.1 | Lebolleux et al. [87] |
7. Motesanib Diphosphate
8. Axitinib
9. Cabozantinib
10. Gefitinib
11. Pazopanib
12. Lenvatinib
13. BRAF Inhibitors
| Drug | Thyroid Cancer | Responses | Authors | |||
|---|---|---|---|---|---|---|
| PR | SD | PD | PFS (months) | |||
| Dabrafenib | 14 DeTC | 21% | – | – | – | Falchook et al. [123] |
| Selumetinib | 2 DeTC | – | 100% 19 cycles | – | – | Adjei et al. [127] |
| Selumetinib | 39 DeTC | 3% | 54% | 28% | 8 | Hayes et al. [128] |
14. mTOR Inhibitors
15. Histone Deacetylase Inhibitors
16. Limits and Drug Resistance
17. Alternative Therapeutic Strategies
18. Conclusions
Conflicts of Interest
References
- Kilfoy, B.A.; Zheng, T.; Holford, T.R.; Han, X.; Ward, M.H.; Sjodin, A.; Zhang, Y.; Bai, Y.; Zhu, C.; Guo, G.L.; et al. International patterns and trends in thyroid cancer incidence, 1973–2002. Cancer Causes Control 2009, 20, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.I. Thyroid carcinoma. Lancet 2003, 361, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferri, C.; Fallahi, P.; Pampana, A.; Ferrari, S.M.; Barani, L.; Marchi, S.; Ferrannini, E. Thyroid cancer in HCV-Related chronic hepatitis patients: A case-control study. Thyroid 2007, 17, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S.; Doherty, G.M.; Haugen, B.R.; Kloos, R.T.; Lee, S.L.; Mandel, S.J.; Mazzaferri, E.L.; McIver, B.; Pacini, F.; Schlumberger, M.; et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Miccoli, P.; Ferdeghini, M.; di Coscio, G.; Alberti, B.; Iacconi, P.; Baldi, V.; Fallahi, P.; Baschieri, L. Role of neck ultrasonography in the follow-up of patients operated on for thyroid cancer. Thyroid 1995, 5, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Miccoli, P.; Fallahi, P.; Grosso, M.; Nesti, C.; Spinelli, C.; Ferrannini, E. Role of neck ultrasonography in the follow-up of children operated on for thyroid papillary cancer. Thyroid 2003, 13, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Miccoli, P.; Fallahi, P.; Ferrari, S.M.; Grosso, M.; Boni, G.; Berti, P. Thyrotropin-stimulated serum thyroglobulin combined with neck ultrasonography has the highest sensitivity in monitoring differentiated thyroid carcinoma in children. Surgery 2006, 140, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Verburg, F.A.; Mäder, U.; Tanase, K.; Thies, E.D.; Diessl, S.; Buck, A.K.; Luster, M.; Reiners, C. Life expectancy is reduced in differentiated thyroid cancer patients ≥45 years old with extensive local tumor invasion, lateral lymph node, or distant metastases at diagnosis and normal in all other DTC patients. J. Clin. Endocrinol. Metab. 2013, 98, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Baudin, E.; Schlumberger, M. New therapeutic approaches for metastatic thyroid carcinoma. Lancet Oncol. 2007, 8, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Fallahi, P.; Ferrari, S.M.; Carpi, A.; Berti, P.; Materazzi, G.; Minuto, M.; Guastalli, M.; Miccoli, P. Dedifferentiated thyroid cancer: A therapeutic challenge. Biomed. Pharmacother. 2008, 62, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Burman, K.D. Is poorly differentiated thyroid cancer poorly characterized? J. Clin. Endocrinol. Metab. 2014, 99, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.N.; Shaha, A.R. Poorly differentiated thyroid cancer. Curr. Opin. Otolaryngol. Head Neck Surg. 2014, 22, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferri, C.; Ferrari, S.M.; Sebastiani, M.; Colaci, M.; Ruffilli, I.; Fallahi, P. New targeted molecular therapies for dedifferentiated thyroid cancer. J. Oncol. 2010, 2010, 921682. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Lima, J.; Preto, A.; Castro, P.; Vinagre, J.; Celestino, R.; Couto, J.P.; Prazeres, H.; Eloy, C.; Máximo, V.; et al. Genetic alterations in poorly differentiated and undifferentiated thyroid carcinomas. Curr. Genomics 2011, 12, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, P.; Ferrari, S.M.; Mazzi, V.; Vita, R.; Benvenga, S.; Antonelli, A. Personalization of targeted therapy in advanced thyroid cancer. Curr. Genomics 2014, 15, 190–202. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.W.; Links, T.P.; Plukker, J.T.; Lips, C.J.; Hofstra, R.M. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr. Rev. 2006, 5, 535–560. [Google Scholar] [CrossRef]
- Antonelli, A.; Fallahi, P.; Ferrari, S.M.; Mancusi, C.; Colaci, M.; Santarpia, L.; Ferri, C. RET TKI: Potential role in thyroid cancers. Curr. Oncol. Rep. 2012, 14, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E. Thyroid carcinoma: Molecular pathways and therapeutic targets. Mod. Pathol. 2008, 21, S37–S43. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Dathan, N.A.; Berlingieri, M.T.; Bongarzone, I.; Paulin, C.; Grieco, M.; Pierotti, M.A.; Vecchio, G.; Fusco, A. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene 1994, 9, 509–516. [Google Scholar] [PubMed]
- Vitale, M. Rethinking the role of oncogenes in papillary thyroid cancer initiation. Front. Endocrinol. (Lausanne) 2012, 3, 83. [Google Scholar]
- Romei, C.; Elisei, R. RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front. Endocrinol. (Lausanne) 2012, 3, 54. [Google Scholar]
- Powell, D.J., Jr.; Russell, J.; Nibu, K.; Li, G.; Rhee, E.; Liao, M.; Goldstein, M.; Keane, W.M.; Santoro, M.; Fusco, A.; et al. The RET/PTC3 oncogene: Metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res. 1998, 58, 5523–5528. [Google Scholar] [PubMed]
- Corvi, R.; Martinez-Alfaro, M.; Harach, H.R.; Zini, M.; Papotti, M.; Romeo, G. Frequent RET rearrangements in thyroid papillary microcarcinoma detected by interphase fluorescence in situ hybridization. Lab. Investig. 2001, 81, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, P.; Giannini, R.; Miccoli, P.; Antonelli, A.; Basolo, F. Molecular diagnostics of fine needle aspiration for the presurgical screening of thyroid nodules. Curr. Genomics 2014, 15, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Handkiewicz-Junak, D.; Czarniecka, A.; Jarzab, B. Molecular prognostic markers in papillary and follicular thyroid cancer: Current status and future directions. Mol. Cell. Endocrinol. 2010, 322, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., II; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J. Clin. Endocrinol. Metab. 2002, 87, 3947–3952. [Google Scholar] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Turner, H.E.; Harris, A.L.; Melmed, S.; Wass, J.A. Angiogenesis in endocrine tumors. Endocr. Rev. 2003, 24, 600–632. [Google Scholar] [CrossRef] [PubMed]
- Bunone, G.; Vigneri, P.; Mariani, L.; Butó, S.; Collini, P.; Pilotti, S.; Pierotti, M.A.; Bongarzone, I. Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical pathological features. Am. J. Pathol. 1999, 155, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Santarpia, L.; Gagel, R.F. The evolving field of tyrosine kinase inhibitors in the treatment of endocrine tumors. Endocr. Rev. 2010, 31, 578–599. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Rixie, O. Axitinib—A selective inhibitor of the vascular endothelial growth factor (VEGF) receptor. Target Oncol. 2009, 4, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Picard, E.; Vignaud, J.M.; Marie, B.; Bresler, L.; Toussaint, B.; Weryha, G.; Duprez, A.; Leclère, J. Vascular endothelial growth factor gene and protein: Strong expression in thyroiditis and thyroid carcinoma. J. Endocrinol. 1999, 161, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. VEGF-targeted cancer therapeutics-paradoxical effects in endocrine organs. Nat. Rev. Endocrinol. 2014, 10, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Croyle, M.; Akeno, N.; Knauf, J.A.; Fabbro, D.; Chen, X.; Baumgartner, J.E.; Lane, H.A.; Fagin, J.A. RET/PTC-induced cell growth is mediated in part by epidermal growth factor receptor (EGFR) activation: Evidence for molecular and functional interactions between RET and EGFR. Cancer Res. 2008, 68, 4183–4191. [Google Scholar] [CrossRef] [PubMed]
- Masago, K.; Asato, R.; Fujita, S.; Hirano, S.; Tamura, Y.; Kanda, T.; Mio, T.; Katakami, N.; Mishima, M.; Ito, J. Epidermal growth factor receptor gene mutations in papillary thyroid carcinoma. Int. J. Cancer 2009, 124, 2744–2749. [Google Scholar] [CrossRef] [PubMed]
- Landriscina, M.; Pannone, G.; Piscazzi, A.; Toti, P.; Fabiano, A.; Tortorella, S.; Occhini, R.; Ambrosi, A.; Bufo, P.; Cignarelli, M. Epidermal growth factor receptor 1 expression is up-regulated in undifferentiated thyroid carcinomas in humans. Thyroid 2011, 21, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Yang, L.; Wang, N.; Li, L.; Xu, M.; Chen, G.G.; Liu, Z.M. High expression of GPER1, EGFR and CXCR1 is associated with lymph node metastasis in papillary thyroid carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3213–3223. [Google Scholar] [PubMed]
- Sethi, K.; Sarkar, S.; Das, S.; Rajput, S.; Mazumder, A.; Roy, B.; Patra, S.; Mohanty, B.; el-Naggar, A.K.; Mandal, M. Expressions of CK-19, NF-κB, E-cadherin, β-catenin and EGFR as diagnostic and prognostic markers by immunohistochemical analysis in thyroid carcinoma. J. Exp. Ther. Oncol. 2011, 9, 187–199. [Google Scholar] [PubMed]
- Knauf, J.A. Does the epidermal growth factor receptor play a role in the progression of thyroid cancer? Thyroid 2011, 21, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Lote, H.; Bhosle, J.; Thway, K.; Newbold, K.; O’Brien, M. Epidermal growth factor mutation as a diagnostic and therapeutic target in metastatic poorly differentiated thyroid carcinoma: A case report and review of the literature. Case Rep. Oncol. 2014, 7, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar]
- Illouz, F.; Laboureau-Soares, S.; Dubois, S.; Rohmer, V.; Rodien, P. Tyrosine kinase inhibitors and modifications of thyroid function tests: A review. Eur. J. Endocrinol. 2009, 160, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Santoro, M. Update: The status of clinical trials with kinase inhibitors in thyroid cancer. J. Clin. Endocrinol. Metab. 2014, 99, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Pacini, F. Where do we stand with targeted therapy of refractory thyroid cancer? Utility of RECIST criteria. Thyroid 2008, 18, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D. Preclinical and clinical development of the oral multikinase inhibitor sorafenib in cancer treatment. Drugs Today (Barc.) 2005, 41, 773–784. [Google Scholar] [CrossRef]
- Fallahi, P.; Ferrari, S.M.; Santini, F.; Corrado, A.; Materazzi, G.; Ulisse, S.; Miccoli, P.; Antonelli, A. Sorafenib and thyroid cancer. BioDrugs 2013, 27, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43–9006 inhibition of oncogenic RET mutants. J. Natl. Cancer Inst. 2006, 98, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D.; Clark, J.W.; Awada, A.; Moore, M.J.; Richly, H.; Hendlisz, A.; Hirte, H.W.; Eder, J.P.; Lenz, H.J.; Schwartz, B. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: A review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007, 12, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Abramson, V.; Troxel, A.B.; Nellore, A.; Puttaswamy, K.; Redlinger, M.; Ransone, K.; Mandel, S.J.; Flaherty, K.T.; Loevner, L.A.; O’Dwyer, P.J.; et al. Phase II trial of sorafenib in advanced thyroid cancer. J. Clin. Oncol. 2008, 26, 4714–4719. [Google Scholar] [CrossRef] [PubMed]
- Kloos, R.T.; Ringel, M.D.; Knopp, M.V.; Hall, N.C.; King, M.; Stevens, R.; Liang, J.; Wakely, P.E., Jr.; Vasko, V.V.; Saji, M.; et al. Phase II trial of sorafenib in metastatic thyroid cancer. J. Clin. Oncol. 2009, 27, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Troxel, A.B.; Redlinger, M.; Harlacker, K.; Redlinger, C.; Chalian, A.A.; Flaherty, K.T.; Loevner, L.A.; Mandel, S.J.; O’Dwyer, P.J. Effect of BRAFV600E on response to sorafenib in advanced thyroid cancer patients. In Proceedings of the ASCO Annual Meeting, Orlando, FL, USA, 29 May–2 June 2009.
- Hoftijzer, H.; Heemstra, K.A.; Morreau, H.; Stokkel, M.P.; Corssmit, E.P.; Gelderblom, H.; Weijers, K.; Pereira, A.M.; Huijberts, M.; Kapiteijn, E.; et al. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur. J. Endocrinol. 2009, 161, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; Waguespack, S.G.; Bronstein, Y.; Williams, M.D.; Feng, L.; Hernandez, M.; Lopez, A.; Sherman, S.I.; Busaidy, N.L. Treatment with tyrosine kinase inhibitors for patients with differentiated thyroid cancer: The M.D. Anderson Experience. J. Clin. Endocrinol. Metab. 2010, 95, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Barbachano, Y.; Riddell, A.; Hickey, J.; Newbold, K.L.; Viros, A.; Harrington, K.J.; Marais, R.; Nutting, C.M. Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: A phase II study in a UK based population. Eur. J. Endocrinol. 2011, 165, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Keefe, S.M.; Troxel, A.B.; Rhee, S.; Puttaswamy, K.; O’Dwyer, P.J.; Loevner, L.A.; Mandel, S.J.; Brose, M.S. Phase II trial of sorafenib in patients with advanced thyroid cancer. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2011.
- Marotta, V.; Ramando, V.; Camera, L.; del Prete, M.; Fonti, R.; Esposito, R.; Calmieri, G.; Salvatore, M.; Vitale, M.; Colao, A.; et al. Sorafenib in advanced iodine-refractory differentiated thyroid cancer: Efficacy, safety and exploratory analysis of role of serum thyroglobulin and FDG-PET. Clin. Endocrinol. (Oxf.) 2012, 78, 760–767. [Google Scholar] [CrossRef]
- Schneider, T.C.; Abdulrahman, R.M.; Corssmit, E.P.; Morreau, H.; Smit, J.W.; Kapiteijn, E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: Final results of a phase II trial. Eur. J. Endocrinol. 2012, 167, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Pitoia, F. Response to sorafenib treatment in advanced metastatic thyroid cancer. Arq. Bras. Endocrinol. Metabol. 2014, 58, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Massicotte, M.H.; Brassard, M.; Claude-Desroches, M.; Borget, I.; Bonichon, F.; Giraudet, A.L.; Do Cao, C.; Chougnet, C.N.; Leboulleux, S.; Baudin, E.; et al. Tyrosine kinase inhibitor treatments in patients with metastatic thyroid carcinomas: A retrospective study of the TUTHYREF network. Eur. J. Endocrinol. 2014, 170, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I. Sunitinib. Expert. Opin. Pharmacother. 2007, 8, 2359–2369. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.R.; Leggas, M. Sunitinib malate for the treatment of metastatic renal cell carcinoma and gastrointestinal stromal tumors. Clin. Ther. 2007, 29, 1338–1353. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.I. Targeted therapy of thyroid cancer. Biochem. Pharmacol. 2010, 80, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Jo, Y.S.; Jung, H.S.; Chung, H.K.; Song, J.H.; Park, K.C.; Park, S.H.; Hwang, J.H.; Rha, S.Y.; Kweon, G.R.; et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J. Clin. Endocrinol. Metab. 2006, 91, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Fenton, M.S.; Marion, K.M.; Salem, A.K.; Hogen, R.; Naeim, F.; Hershman, J.M. Sunitinib inhibits MEK/ERK and SAPK/JNK pathways and increases sodium/iodide symporter expression in papillary thyroid cancer. Thyroid 2010, 20, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.J.; Mo, J.H.; Park, M.W.; Choi, I.J.; An, S.Y.; Jeon, E.H.; Ahn, S.H. Sunitinib inhibits papillary thyroid carcinoma with RET/PTC rearrangement but not BRAF mutation. Cancer Biol. Ther. 2011, 12, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.; Needles, B.M.; Cullen, K.J.; Wong, S.J.; Wade, J.L., III; Ivy, S.P.; Villaflor, V.M.; Seiwert, T.Y.; Nichols, K.; Vokes, E.E. Phase 2 study of sunitinib in refractory thyroid cancer. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 30 May–3 June 2008.
- Goulart, B.; Carr, L.; Martins, R.G.; Eaton, K.; Kell, E.; Wallace, S.; Capell, P.; Mankoff, D. Phase II study of sunitinib in iodine refractory, well-differentiated thyroid cancer (DTC) and metastatic medullary thyroid carcinoma (MTC). In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 30 May–3 June 2008.
- Ravaud, A.; de la Fouchardière, C.; Courbon, F.; Asselineau, J.; Klein, M.; Nicoli-Sire, P.; Bournaud, C.; Delord, J.; Weryha, G.; Catargi, B. Sunitinib in patients with refractory advanced thyroid cancer: The THYSU phase II trial. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 30 May–3 June 2008.
- Carr, L.L.; Mankoff, D.A.; Goulart, B.H.; Eaton, K.D.; Capell, P.T.; Kell, E.M.; Bauman, J.E.; Martins, R.G. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin. Cancer. Res. 2010, 16, 5260–5268. [Google Scholar] [CrossRef] [PubMed]
- Dìez, J.J.; Iglesias, P.; Alonso, T.; Grande, E. Activity and safety of sunitinib in patients with advanced radioactive iodine-refractory differentiated thyroid carcinoma in clinical practice. Endocrine 2014, 48, 582–588. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.W.; Plaza Menacho, I.; Schepers, H.; Drenth-Diephuis, L.J.; Osinga, J.; Plukker, J.T.; Links, T.P.; Eggen, B.J.; Hofstra, R.M. Cellular effects of imatinib on medullary thyroid cancer cells harboring multiple endocrine neoplasia Type 2A and 2B associated RET mutations. Surgery 2006, 139, 806–814. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.W.; Zonnenberg, B.A.; van Ufford-Mannesse, P.Q.; de Vries, M.M.; Links, T.P.; Lips, C.J.; Voest, E.E. A phase II trial of imatinib therapy for metastatic medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2007, 92, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Frank-Raue, K.; Fabel, M.; Delorme, S.; Haberkorn, U.; Raue, F. Efficacy of imatinib mesylate in advanced medullary thyroid carcinoma. Eur. J. Endocrinol. 2007, 157, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Halperin, D.M.; Phan, A.T.; Hoff, A.O.; Aaron, M.; Yao, J.C.; Hoff, P.M. A phase I study of imatinib, dacarbazine, and capecitabine in advanced endocrine cancers. BMC Cancer 2014, 14, 561. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar] [PubMed]
- Wells, S.A., Jr.; Gosnell, J.E.; Gagel, R.F.; Moley, J.; Pfister, D.; Sosa, J.A.; Skinner, M.; Krebs, A.; Vasselli, J.; Schlumberger, M. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Oncol. 2010, 28, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.G.; Paz-Ares, L.; Krebs, A.; Vasselli, J.; Haddad, R. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J. Clin. Endocrinol. Metab. 2010, 95, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Bastholt, L.; Krause, T.; de la Fouchardiere, C.; Tennvall, J.; Awada, A.; Gómez, J.M.; Bonichon, F.; Leenhardt, L.; Soufflet, C.; et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 2 trial. Lancet Oncol. 2012, 13, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Polverino, A.; Coxon, A.; Starnes, C.; Diaz, Z.; DeMelfi, T.; Wang, L.; Bready, J.; Estrada, J.; Cattley, R.; Kaufman, S.; et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and Kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res. 2006, 66, 8715–8721. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Kurzrock, R.; Mulay, M.; van Vugt, A.; Purdom, M.; Ng, C.; Silverman, J.; Koutsoukos, A.; Sun, Y.N.; Bass, M.B.; et al. Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2007, 25, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.I.; Wirth, L.J.; Droz, J.P.; Hofmann, M.; Bastholt, L.; Martins, R.G.; Licitra, L.; Eschenberg, M.J.; Sun, Y.N.; Juan, T.; et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N. Engl. J. Med. 2008, 359, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.J.; Elisei, R.; Bastholt, L.; Wirth, L.J.; Martins, R.G.; Locati, L.D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.P.; et al. Phase II study of safety and efficacy of motesanib in patients with progressive, symptomatic, advanced or metastatic medullary thyroid cancer. J. Clin. Oncol. 2009, 27, 3794–3801. [Google Scholar] [PubMed]
- Bass, M.B.; Sherman, S.I.; Schlumberger, M.J.; Davis, M.T.; Kivman, L.; Khoo, H.M.; Notari, K.H.; Peach, M.; Hei, Y.J.; Patterson, S.D. Biomarkers as predictors of response to treatment with motesanib in patients with progressive advanced thyroid cancer. J. Clin. Endocrinol. Metab. 2010, 95, 5018–5027. [Google Scholar] [CrossRef] [PubMed]
- Claret, L.; Lu, J.F.; Sun, Y.N.; Bruno, R. Development of a modeling framework to simulate efficacy endpoints for motesanib in patients with thyroid cancer. Cancer Chemother. Pharmacol. 2010, 66, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. Axitinib: VEGF inhibition in advanced thyroid cancer. Lancet Oncol. 2014, 15, E310. [Google Scholar] [CrossRef] [PubMed]
- Patson, B.; Cohen, R.B.; Olszanski, A.J. Pharmacokinetic evaluation of axitinib. Expert Opin. Drug Metab. Toxicol. 2012, 8, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar] [PubMed]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lowe, D.; Hallin, M.; Feeley, R.; Zou, H.; Rewolinski, D.; Wickman, G.; Chen, E.; Kim, Y.; Riney, S.; Reed, J.; et al. Characterization of potency and activity of the VEGF/PDGF receptor tyrosine kinase inhibitor AG013736. Proc. Am. Assoc. Cancer Res. 2002, 43, A5356. [Google Scholar]
- Rugo, H.S.; Herbst, R.S.; Liu, G.; Park, J.W.; Kies, M.S.; Steinfeldt, H.M.; Pithavala, Y.K.; Reich, S.D.; Freddo, J.L.; Wilding, G. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: Pharmacokinetic and clinical results. J. Clin. Oncol. 2005, 23, 5474–5483. [Google Scholar] [PubMed]
- Cohen, E.E.; Rosen, L.S.; Vokes, E.E.; Kies, M.S.; Forastiere, A.A.; Worden, F.P.; Kane, M.A.; Sherman, E.; Kim, S.; Bycott, P.; et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: Results from a phase II study. J. Clin. Oncol. 2008, 26, 4708–4713. [Google Scholar] [CrossRef] [PubMed]
- Rixe, O.; Bukowski, R.M.; Michaelson, M.D.; Wilding, G.; Hudes, G.R.; Bolte, O.; Motzer, R.J.; Bycott, P.; Liau, K.F.; Freddo, J.; et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: A phase II study. Lancet Oncol. 2007, 8, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.; Schiller, J.; Fruehauf, J.; Cohen, E.E.; Tarazi, J.C.; Rosbrook, B.; Ricart, A.D.; Olszanski, A.J.; Kim, S.; Spano, J. Association of diastolic blood pressure (dBP) ≥90 mmHg with overall survival (OS) in patients treated with axitinib (AG-013736). In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 30 May–3 June 2008.
- Alexandrescu, D.T.; McClure, R.; Farzanmehr, H.; Dasanu, C.A. Secondary erythrocytosis produced by the tyrosine kinase inhibitors sunitinib and sorafenib. J. Clin. Oncol. 2008, 26, 4047–4048. [Google Scholar] [CrossRef] [PubMed]
- Locati, L.D.; Licitra, L.; Agate, L.; Ou, S.H.; Boucher, A.; Jarzab, B.; Qin, S.; Kane, M.A.; Wirth, L.J.; Chen, C.; et al. Treatment of advanced thyroid cancer with axitinib: Phase 2 study with pharmacokinetic/pharmacodynamic and quality-of-life assessments. Cancer 2014, 120, 2694–2703. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J. Inhibitors targeting hepatocyte growth factor receptor and their potential therapeutic applications. Expert Opin. Ther. Pat. 2007, 17, 1035–1045. [Google Scholar] [CrossRef]
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F.; et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guessous, F.; Kofman, A.; Schiff, D.; Abounader, R. XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs 2010, 13, 112–121. [Google Scholar] [PubMed]
- Cabanillas, M.E.; Brose, M.S.; Ramies, D.A.; Lee, Y.; Miles, D.; Sherman, S.I. Antitumor activity of cabozantinib (XL184) in a cohort of patients (pts) with differentiated thyroid cancer (DTC). In Proceeding of the ASCO Annual Meeting, Chicago, IL, USA, 1–5 June 2012.
- Goldenberg, M.M. Pharmaceutical approval update. Pharm. Ther. 2013, 38, 86–95. [Google Scholar]
- Inoue, A.; Suzuki, T.; Fukuhara, T.; Maemondo, M.; Kimura, Y.; Morikawa, N.; Watanabe, H.; Saijo, Y.; Nukiwa, T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J. Clin. Oncol. 2006, 24, 3340–3346. [Google Scholar] [CrossRef] [PubMed]
- Schiff, B.A.; McMurphy, A.B.; Jasser, S.A.; Younes, M.N.; Doan, D.; Yigitbasi, O.G.; Kim, S.; Zhou, G.; Mandal, M.; Bekele, B.N.; et al. Epidermal growth factor receptor (EGFR) is overexpressed in anaplastic thyroid cancer, and the EGFR inhibitor gefitinib inhibits the growth of anaplastic thyroid cancer. Clin. Cancer Res. 2004, 10, 8594–8602. [Google Scholar] [CrossRef] [PubMed]
- Pennell, N.A.; Daniels, G.H.; Haddad, R.I.; Ross, D.S.; Evans, T.; Wirth, L.J.; Fidias, P.H.; Temel, J.S.; Gurubhagavatula, S.; Heist, R.S.; et al. A phase II study of gefitinib in patients with advanced thyroid cancer. Thyroid 2008, 18, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Wang-Rodriguez, J.; Chang, C.; Chen, J.S.; Pardo, F.S.; Aguilera, J.; Ongkeko, W.M. Gefitinib inhibition of drug resistance to doxorubicin by inactivating ABCG2 in thyroid cancer cell lines. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Canter, D.; Kutikov, A.; Golovine, K.; Makhov, P.; Simhan, J.; Uzzo, R.G.; Kolenko, V.M. Are all multi-targeted tyrosine kinase inhibitors created equal? An in vitro study of sunitinib and pazopanib in renal cell carcinoma cell lines. Can. J. Urol. 2011, 18, 5819–5825. [Google Scholar] [PubMed]
- Bible, K.C.; Suman, V.J.; Molina, J.R.; Smallridge, R.C.; Maples, W.J.; Menefee, M.E.; Rubin, J.; Sideras, K.; Morris, J.C., III; McIver, B.; et al. Endocrine malignancies disease oriented group; mayo clinic cancer center; mayo phase 2 consortium. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: Results of a phase 2 consortium study. Lancet Oncol. 2010, 11, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Isham, C.R.; Bossou, A.R.; Negron, V.; Fisher, K.E.; Kumar, R.; Marlow, L.; Lingle, W.L.; Smallridge, R.C.; Sherman, E.J.; Suman, V.J.; et al. Pazopanib enhances paclitaxel-induced mitotic catastrophe in anaplastic thyroid cancer. Sci. Transl. Med. 2013, 5, 166ra3. [Google Scholar] [CrossRef] [PubMed]
- Glen, H.; Mason, S.; Patel, H.; Macleod, K.; Brunton, V.G. E7080, a multi-targeted tyrosine kinase inhibitor suppresses tumor cell migration and invasion. BMC Cancer 2011, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin. Cancer Res. 2008, 14, 5459–5465. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.I.; Jarzab, B.; Cabanillas, M.E.; Licitra, L.F.; Pacini, F.; Martins, R.; Robinson, B.; Ball, D.; McCaffrey, J.; Shah, M.H.; et al. A phase II trial of the multitargeted kinase inhibitor E7080 in advanced radioiodine (RAI)-refractory differentiated thyroid cancer (DTC). In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 30 May–3 June 2008.
- Li, C.; Lee, K.C.; Schneider, E.B.; Zeiger, M.A. BRAF V600E mutation and its association with clinicopathological features of papillary thyroid cancer: A meta-analysis. J. Clin. Endocrinol. Metab. 2012, 97, 4559–4570. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, E.; Garcia, B.; Santisteban, P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-β repression of the sodium/iodide symporter gene. J. Biol. Chem. 2004, 279, 3439–3446. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Laquerre, S.; Arnone, M.; Moss, K.; Yang, J.; Fisher, K.; Kane-Carson, L.S.; Smitheman, K.; Ward, J.; Heidrich, B.; Rheault, T.; et al. A selective Raf kinase inhibitor induces cell death and tumor regression of human cancer cell lines encoding B-RafV600E mutation. International Conference: Molecular Targets and Cancer Therapeutics, 2009; Boston, MA. Mol. Cancer Ther. 2009, 8, AB88. [Google Scholar]
- Sharma, A.; Shah, S.R.; Illum, H.; Dowell, J. Vemurafenib: Targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs 2012, 72, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Ruggeri, B.; Armstrong, R.C.; Rowbottom, M.W.; Jones-Bolin, S.; Gunawardane, R.N.; Dobrzanski, P.; Gardner, M.F.; Zhao, H.; Cramer, M.D.; et al. CEP-32496: A novel orally active BRAF(V600E) inhibitor with selective cellular and in vivo antitumor activity. Mol. Cancer Ther. 2012, 11, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Cohen, R.B.; Franklin, W.; Morris, C.; Wilson, D.; Molina, J.R.; Hanson, L.J.; Gore, L.; Chow, L.; Leong, S.; et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J. Clin. Oncol. 2008, 26, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.N.; Lucas, A.S.; Tanvetyanon, T.; Krzyzanowska, M.K.; Chung, C.H.; Murphy, B.A.; Gilbert, J.; Mehra, R.; Moore, D.T.; Sheikh, A.; et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin. Cancer Res. 2012, 18, 2056–2065. [Google Scholar] [CrossRef] [PubMed]
- Henderson, Y.C.; Chen, Y.; Frederick, M.J.; Lai, S.Y.; Clayman, G.L. MEK inhibitor PD0325901 significantly reduces the growth of papillary thyroid carcinoma cells in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Sherman, E.; Haque, S.; Korte, S.; Lisa, D.; Shen, R.; Wu, N.; Pfister, D. A phase I study of daily everolimus plus low-dose weekly cisplatin for patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 69, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Kouvaraki, M.A.; Liakou, C.; Paraschi, A.; Dimas, K.; Patsouris, E.; Tseleni-Balafouta, S.; Rassidakis, G.Z.; Moraitis, D. Activation of mTOR signaling in medullary and aggressive papillary thyroid carcinomas. Surgery 2011, 150, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Yeager, N.; Brewer, C.; Cai, K.Q.; Xu, X.X.; di Cristofano, A. Mammalian target of rapamycin is the key effector of phosphatidylinositol-3-OH-initiated proliferative signals in the thyroid follicular epithelium. Cancer Res. 2008, 68, 444–449. [Google Scholar] [CrossRef] [PubMed]
- De Souza, E.C.; Padrón, A.S.; Braga, W.M.; de Andrade, B.M.; Vaisman, M.; Nasciutti, L.E.; Ferreira, A.C.; de Carvalho, D.P. MTOR down-regulates iodide uptake in thyrocytes. J. Endocrinol. 2010, 206, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Kogai, T.; Sajid-Crockett, S.; Newmarch, L.S.; Liu, Y.Y.; Brent, G.A. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J. Endocrinol. 2008, 199, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Faggiano, A.; Ramundo, V.; Dicitore, A.; Castiglioni, S.; Borghi, M.O.; Severino, R.; Ferolla, P.; Crinò, L.; Abbruzzese, A.; Sperlongano, P.; et al. Everolimus is an active agent in medullary thyroid cancer: A clinical and in vitro study. J. Cell Mol. Med. 2012, 16, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xing, J.; Trink, B.; Xing, M. BRAF mutation-selective inhibition of thyroid cancer cells by the novel MEK inhibitor RDEA119 and genetic-potentiated synergism with the mTOR inhibitor temsirolimus. Int. J. Cancer 2010, 127, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Luong, Q.T.; O’Kelly, J.; Braunstein, G.D.; Hershman, J.M.; Koeffler, H.P. Antitumor activity of suberoylanilide hydroxamic acid against thyroid cancer cell line in vitro and in vivo. Clin. Cancer Res. 2006, 12, 5570–5577. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Kloos, R.T.; Ringel, M.D.; Arbogast, D.; Collamore, M.; Zwiebel, J.A.; Grever, M.; Villalona-Calero, M.; Shah, M.H. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J. Clin. Endocrinol. Metab. 2009, 94, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Bakke, S.; Robey, R.W.; Kang, M.H.; Blagosklonny, M.V.; Bender, J.; Brooks, R.; Piekarz, R.L.; Tucker, E.; Figg, W.D.; et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin. Cancer Res. 2002, 8, 718–728. [Google Scholar] [PubMed]
- Harrison, S.J.; Bishton, M.; Bates, S.E.; Grant, S.; Piekarz, R.L.; Johnstone, R.W.; Dai, Y.; Lee, B.; Araujo, M.E.; Prince, H.M. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, Istodax®). Epigenomics 2012, 4, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Kitazono, M.; Robey, R.; Zhan, Z.; Sarlis, N.J.; Skarulis, M.C.; Aikou, T.; Bates, S.; Fojo, T. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na+/I− symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 2001, 86, 3430–3435. [Google Scholar] [PubMed]
- Sherman, E.J.; Su, Y.B.; Lyall, A.; Schöder, H.; Fury, M.G.; Ghossein, R.A.; Haque, S.; Lisa, D.; Shaha, A.R.; Tuttle, R.M.; et al. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid 2013, 23, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, P.; Ferrari, S.M.; Vita, R.; di Domenicantonio, A.; Corrado, A.; Benvenga, S.; Antonelli, A. Thyroid dysfunctions induced by tyrosine kinase inhibitors. Expert Opin. Drug Saf. 2014, 13, 723–733. [Google Scholar] [PubMed]
- Gild, M.L.; Bullock, M.; Robinson, B.G.; Clifton-Bligh, R. Multikinase inhibitors: A new option for the treatment of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, D.; Xing, M. The Akt inhibitor MK2206 synergizes, but perifosine antagonizes, the BRAF(V600E) inhibitor PLX4032 and the MEK1/2 inhibitor AZD6244 in the inhibition of thyroid cancer cells. J. Clin. Endocrinol. Metab. 2012, 97, E173–E182. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Fallahi, P.; Berti, P.; Materazzi, G.; Minuto, M.; Giannini, R.; Marchetti, I.; Barani, L.; Basolo, F.; et al. Thiazolidinediones and antiblastics in primary human anaplastic thyroid cancer cells. Clin. Endocrinol. (Oxf.) 2009, 70, 946–953. [Google Scholar] [CrossRef]
- Antonelli, A.; Bocci, G.; la Motta, C.; Ferrari, S.M.; Fallahi, P.; Ruffilli, I.; di Domenicantonio, A.; Fioravanti, A.; Sartini, S.; Minuto, M.; et al. CLM94, a novel cyclic amide with anti-VEGFR-2 and antiangiogenic properties, is active against primary anaplastic thyroid cancer in vitro and in vivo. J. Clin. Endocrinol. Metab. 2012, 97, E528–E536. [Google Scholar] [CrossRef] [PubMed]
- Newell, D.R. Flasks, fibres and flanks—Pre-clinical tumour models for predicting clinical antitumour activity. Br. J. Cancer 2001, 84, 1289–1290. [Google Scholar] [CrossRef] [PubMed]
- Schroyens, W.; Tueni, E.; Dodion, P.; Bodecker, R.; Stoessel, F.; Klastersky, J. Validation of clinical predictive value of in vitro colorimetric chemosensitivity assay in head and neck cancer. Eur. J. Cancer 1990, 26, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A. Molecular profiling and ways towards personalized medicine in advanced differentiated thyroid cancer. Curr. Genomics 2014, 15, 161. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Fallahi, P.; Berti, P.; Materazzi, G.; Marchetti, I.; Ugolini, C.; Basolo, F.; Miccoli, P.; Ferrannini, E. Evaluation of the sensitivity to chemotherapeutics or thiazolidinediones of primary anaplastic thyroid cancer cells obtained by fine-needle aspiration. Eur. J. Endocrinol. 2008, 159, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Fallahi, P.; Berti, P.; Materazzi, G.; Barani, L.; Marchetti, I.; Ferrannini, E.; Miccoli, P. Primary cell cultures from anaplastic thyroid cancer obtained by fine-needle aspiration used for chemosensitivity tests. Clin. Endocrinol. (Oxf.) 2008, 69, 148–152. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; la Motta, C.; Bocci, G.; Corrado, A.; Materazzi, G.; Galleri, D.; Piaggi, S.; Danesi, R.; da Settimo, F.; et al. Antineoplastic activity of the multitarget tyrosine kinase inhibitors CLM3 and CLM94 in medullary thyroid cancer in vitro. Surgery 2014, 156, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Bocci, G.; la Motta, C.; Ferrari, S.M.; Fallahi, P.; Corrado, A.; Fioravanti, A.; Sartini, S.; Orlandi, P.; Piaggi, S.; et al. CLM29, a multi-target pyrazolopyrimidine derivative, has anti-neoplastic activity in medullary thyroid cancer in vitro and in vivo. Mol. Cell. Endocrinol. 2014, 393, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Bocci, G.; Fallahi, P.; la Motta, C.; Ferrari, S.M.; Mancusi, C.; Fioravanti, A.; di Desidero, T.; Sartini, S.; Corti, A.; et al. CLM3, a multitarget tyrosine kinase inhibitor with antiangiogenic properties, is active against primary anaplastic thyroid cancer in vitro and in vivo. J. Clin. Endocrinol. Metab. 2014, 99, E572–E581. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Bocci, G.; la Motta, C.; Ferrari, S.M.; Fallahi, P.; Fioravanti, A.; Sartini, S.; Minuto, M.; Piaggi, S.; Corti, A.; et al. Novel pyrazolopyrimidine derivatives as tyrosine kinase inhibitors with antitumoral activity in vitro and in vivo in papillary dedifferentiated thyroid cancer. J. Clin. Endocrinol. Metab. 2011, 96, E288–E296. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Pandini, G.; Frasca, F.; Conte, E.; Murabito, A.; Sacco, A.; Genua, M.; Vigneri, R.; Belfiore, A. Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology 2006, 147, 4463–4475. [Google Scholar] [CrossRef] [PubMed]
- Marlow, L.A.; Reynolds, L.A.; Cleland, A.S.; Cooper, S.J.; Gumz, M.L.; Kurakata, S.; Fujiwara, K.; Zhang, Y.; Sebo, T.; Grant, C.; et al. Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth. Cancer Res. 2009, 69, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Bravo, S.B.; García-Rendueles, M.E.; Seoane, R.; Dosil, V.; Cameselle-Teijeiro, J.; López-Lázaro, L.; Zalvide, J.; Barreiro, F.; Pombo, C.M.; Alvarez, C.V. Plitidepsin has a cytostatic effect in human undifferentiated (anaplastic) thyroid carcinoma. Clin. Cancer Res. 2005, 11, 7664–7673. [Google Scholar] [CrossRef] [PubMed]
- Karwowski, J.K.; Nowels, K.W.; McDougall, I.R.; Weigel, R.J. Needle track seeding of papillary thyroid carcinoma from fine needle aspiration biopsy. A case report. Acta Cytol. 2002, 46, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Suda, T.; Inoue, T.; Fujiwara, Y.; Ishiguro, K. Needle track dissemination of follicular thyroid carcinoma following fine-needle aspiration biopsy: Report of a case. Surg. Today 2007, 37, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, D.P.; Ferreira, A.C. The importance of sodium/iodide symporter (NIS) for thyroid cancer management. Arq. Bras. Endocrinol. Metab. 2007, 51, 672–682. [Google Scholar] [CrossRef]
- Dohan, O.; de la Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The sodium/iodide symporter (NIS): Characterization, regulation, and medical significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Dohan, O.; Carrasco, N. Thyroidal iodide transport and thyroid cancer. Cancer Treat. Res. 2004, 122, 221–236. [Google Scholar] [PubMed]
- Li, W.; Ain, K.B. Human sodium-iodide symporter (hNIS) gene expression is inhibited by a trans-active transcriptional repressor, NIS-repressor, containing PARP-1 in thyroid cancer cells. Endocr. Relat. Cancer 2010, 17, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.E.; Read, M.L.; Turnell, A.S.; Watkins, R.J.; Watkinson, J.C.; Lewy, G.D.; Fong, J.C.; James, S.R.; Eggo, M.C.; Boelaert, K.; et al. A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J. Cell Sci. 2009, 122, 3393–3402. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, G.M.; Yatin, M.; Marcinek, R.; Ain, K.B. Restoration of iodide uptake in dedifferentiated thyroid carcinoma: Relationship to human Na+/I− symporter gene methylation status. J. Clin. Endocrinol. Metab. 1999, 84, 2449–2457. [Google Scholar] [PubMed]
- Castro, M.R.; Bergert, E.R.; Goellner, J.R.; Hay, I.D.; Morris, J.C. Immunohistochemical analysis of sodium iodide symporter expression in metastatic differentiated thyroid cancer: Correlation with radioiodine uptake. J. Clin. Endocrinol. Metab. 2001, 86, 5627–5632. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.J.; Fitzgerald, M.P.; Krager, K.; Domann, F.E. Increased iodine uptake in thyroid carcinoma after treatment with sodium butyrate and decitabine (5-Aza-dC). Otolaryngol. Head Neck Surg. 2007, 137, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Guo, R.; Xu, H.; Zhang, M.; Li, B. Retinoic acid and tributyrin induce in vitro radioiodine uptake and inhibition of cell proliferation in a poorly differentiated follicular thyroid carcinoma. Nucl. Med. Commun. 2011, 32, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Bojdani, E.; Xing, M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J. Clin. Endocrinol. Metab. 2010, 95, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Coelho, S.M.; Vaisman, M.; Carvalho, D.P. Tumour re-differentiation effect of retinoic acid: A novel therapeutic approach for advanced thyroid cancer. Curr. Pharm. Des. 2005, 11, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Kogai, T.; Brent, G.A. The sodium iodide symporter (NIS): Regulation and approaches to targeting for cancer therapeutics. Pharmacol. Ther. 2012, 135, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Tepmongkol, S.; Keelawat, S.; Honsawek, S.; Ruangvejvorachai, P. Rosiglitazone effect on radioiodine uptake in thyroid carcinoma patients with high thyroglobulin but negative total body scan: A correlation with the expression of peroxisome proliferator-activated receptor-gamma. Thyroid 2008, 18, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Fallahi, P.; Ulisse, S.; Ferrari, S.M.; Minuto, M.; Saraceno, G.; Santini, F.; Mazzi, V.; D’Armiento, M.; Miccoli, P. New targeted therapies for anaplastic thyroid cancer. Anticancer Agents Med. Chem. 2012, 12, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Lindsay, S.; Clark, O.H.; Woeber, K.A.; Hawkins, R.; Greenspan, F.S. Results of rosiglitazone therapy in patients with thyroglobulin-positive and radioiodine-negative advanced differentiated thyroid cancer. Thyroid 2009, 19, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Spitzweg, C. Gene therapy in thyroid cancer. Horm. Metab. Res. 2009, 41, 500–509. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fallahi, P.; Mazzi, V.; Vita, R.; Ferrari, S.M.; Materazzi, G.; Galleri, D.; Benvenga, S.; Miccoli, P.; Antonelli, A. New Therapies for Dedifferentiated Papillary Thyroid Cancer. Int. J. Mol. Sci. 2015, 16, 6153-6182. https://doi.org/10.3390/ijms16036153
Fallahi P, Mazzi V, Vita R, Ferrari SM, Materazzi G, Galleri D, Benvenga S, Miccoli P, Antonelli A. New Therapies for Dedifferentiated Papillary Thyroid Cancer. International Journal of Molecular Sciences. 2015; 16(3):6153-6182. https://doi.org/10.3390/ijms16036153
Chicago/Turabian StyleFallahi, Poupak, Valeria Mazzi, Roberto Vita, Silvia Martina Ferrari, Gabriele Materazzi, David Galleri, Salvatore Benvenga, Paolo Miccoli, and Alessandro Antonelli. 2015. "New Therapies for Dedifferentiated Papillary Thyroid Cancer" International Journal of Molecular Sciences 16, no. 3: 6153-6182. https://doi.org/10.3390/ijms16036153