
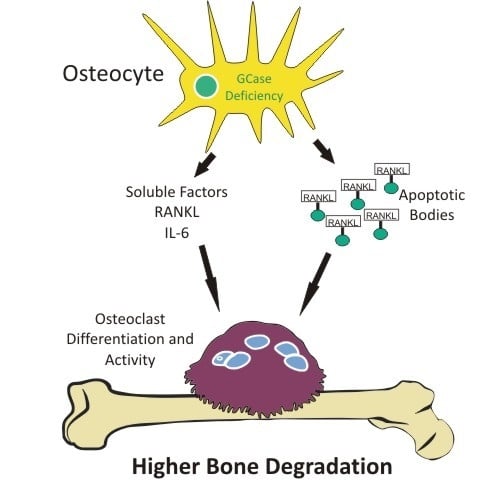
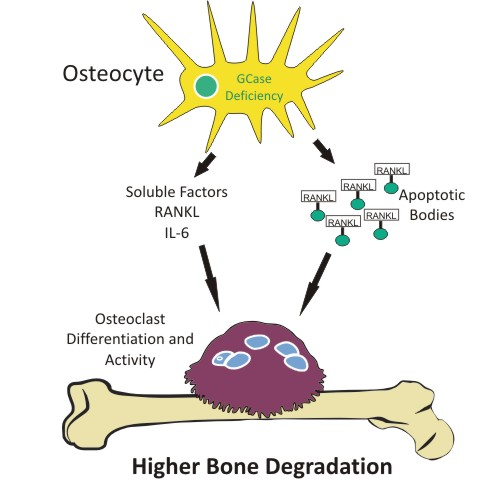
Osteocyte Alterations Induce Osteoclastogenesis in an In Vitro Model of Gaucher Disease

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
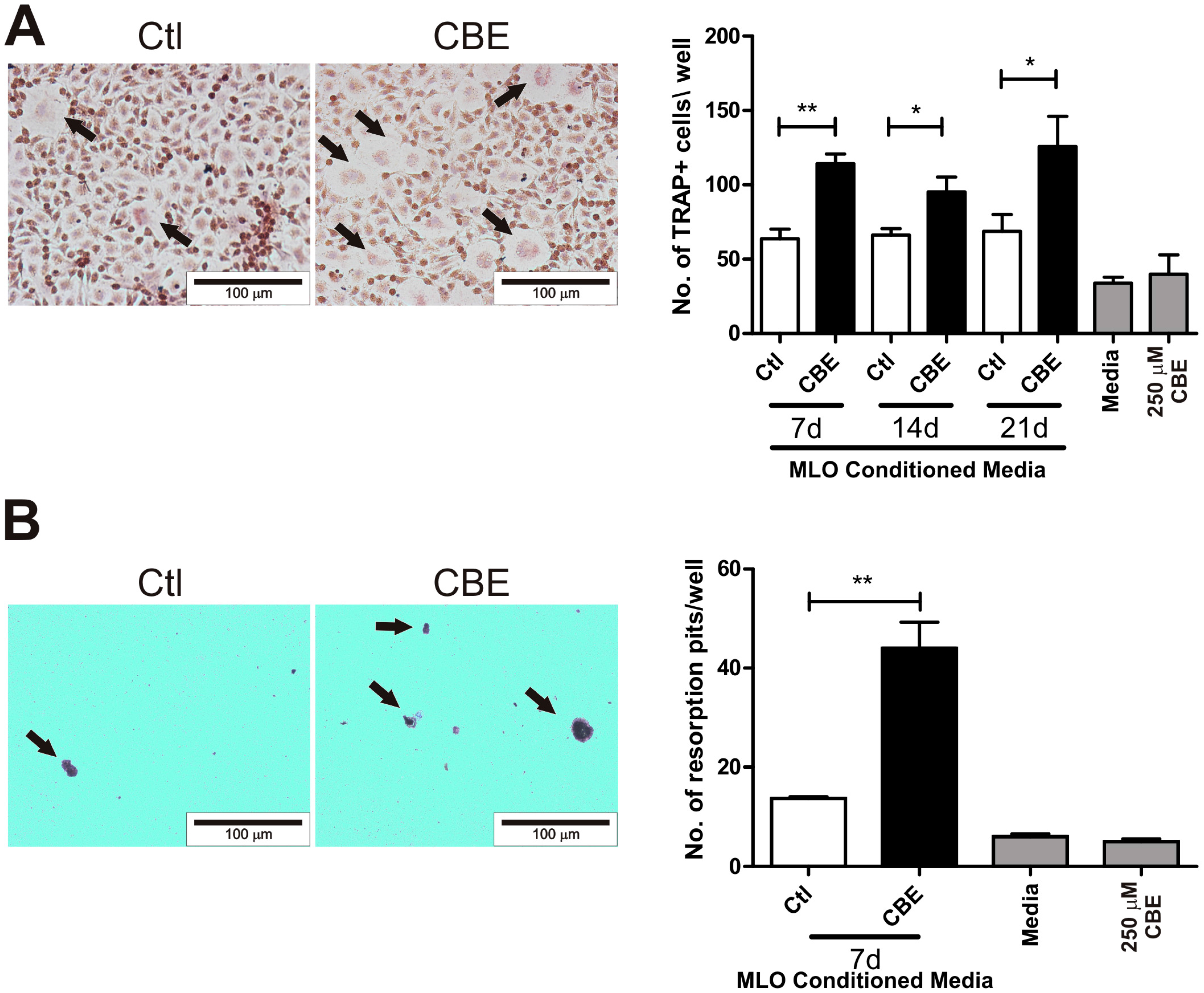
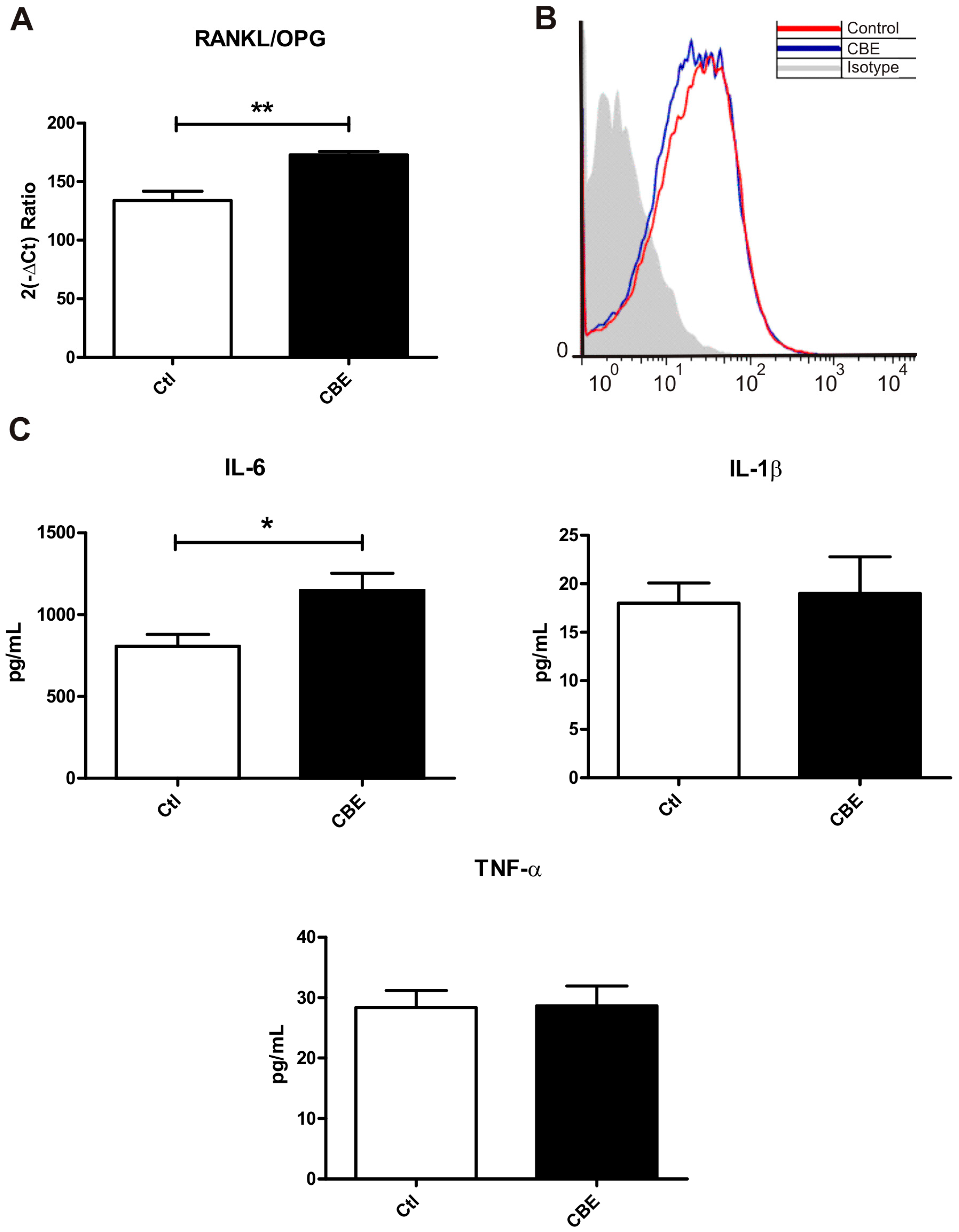
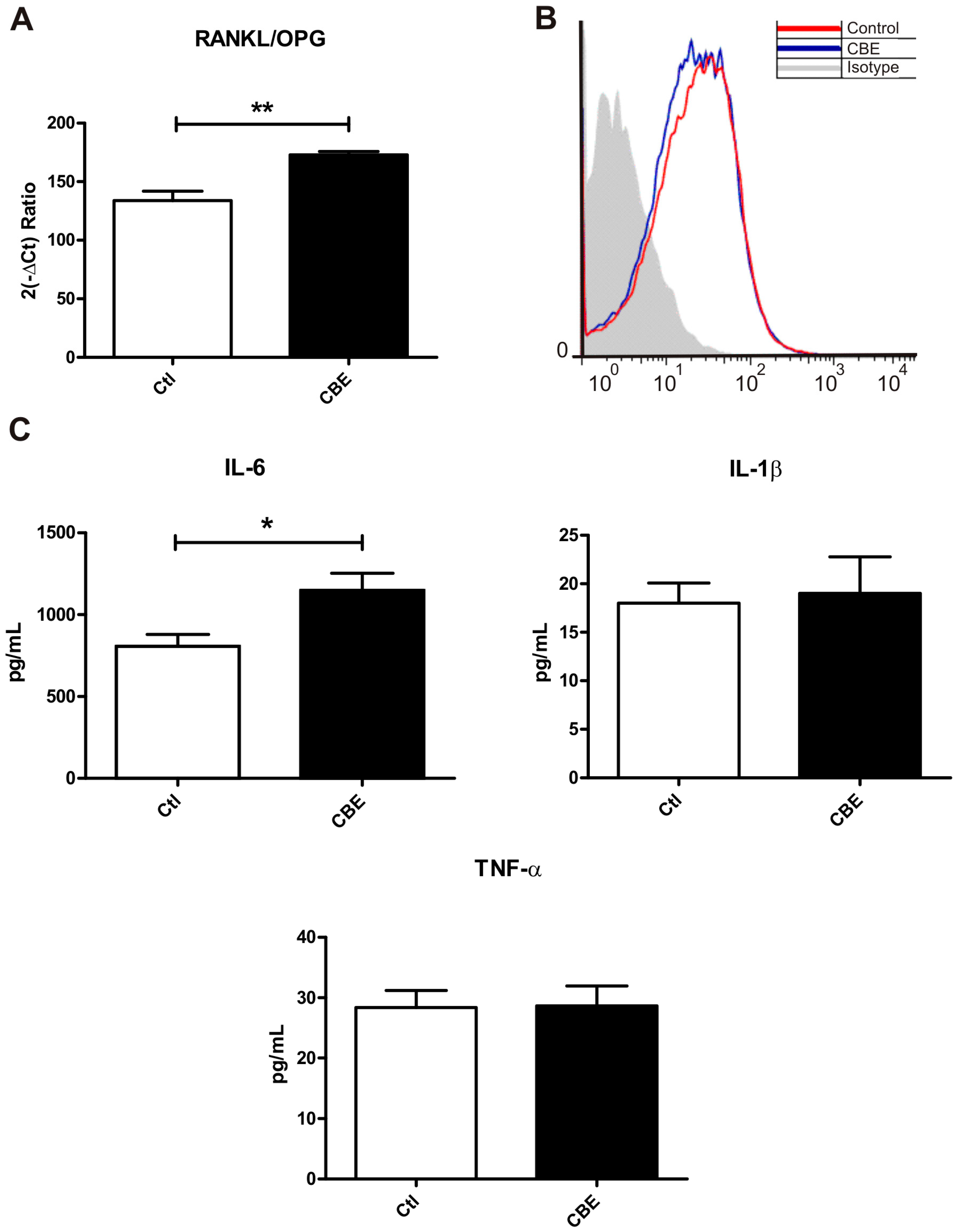
2.1. Conditioned Media from CBE-Treated Osteocytes Induces BMM-Derived Osteoclastogenesis
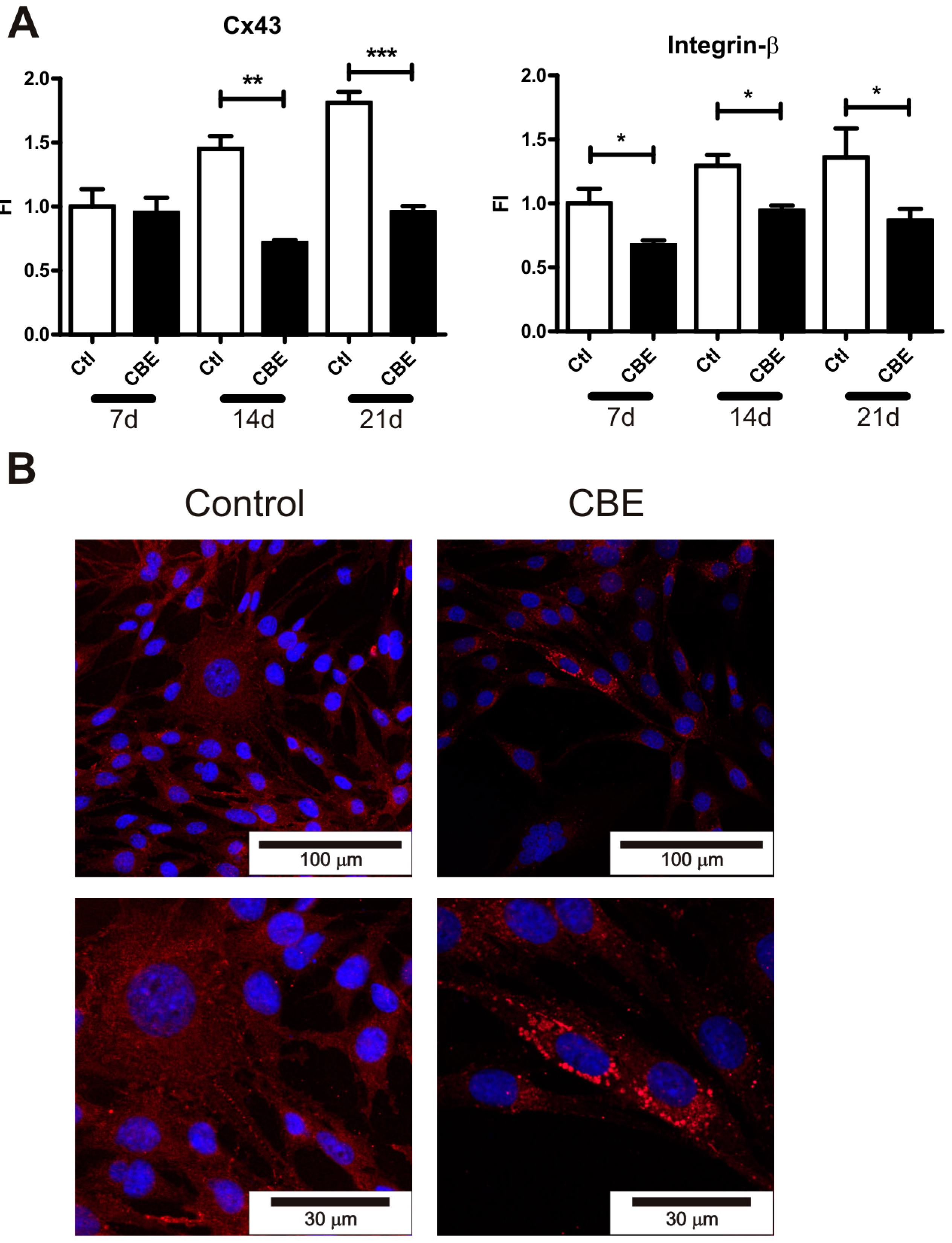
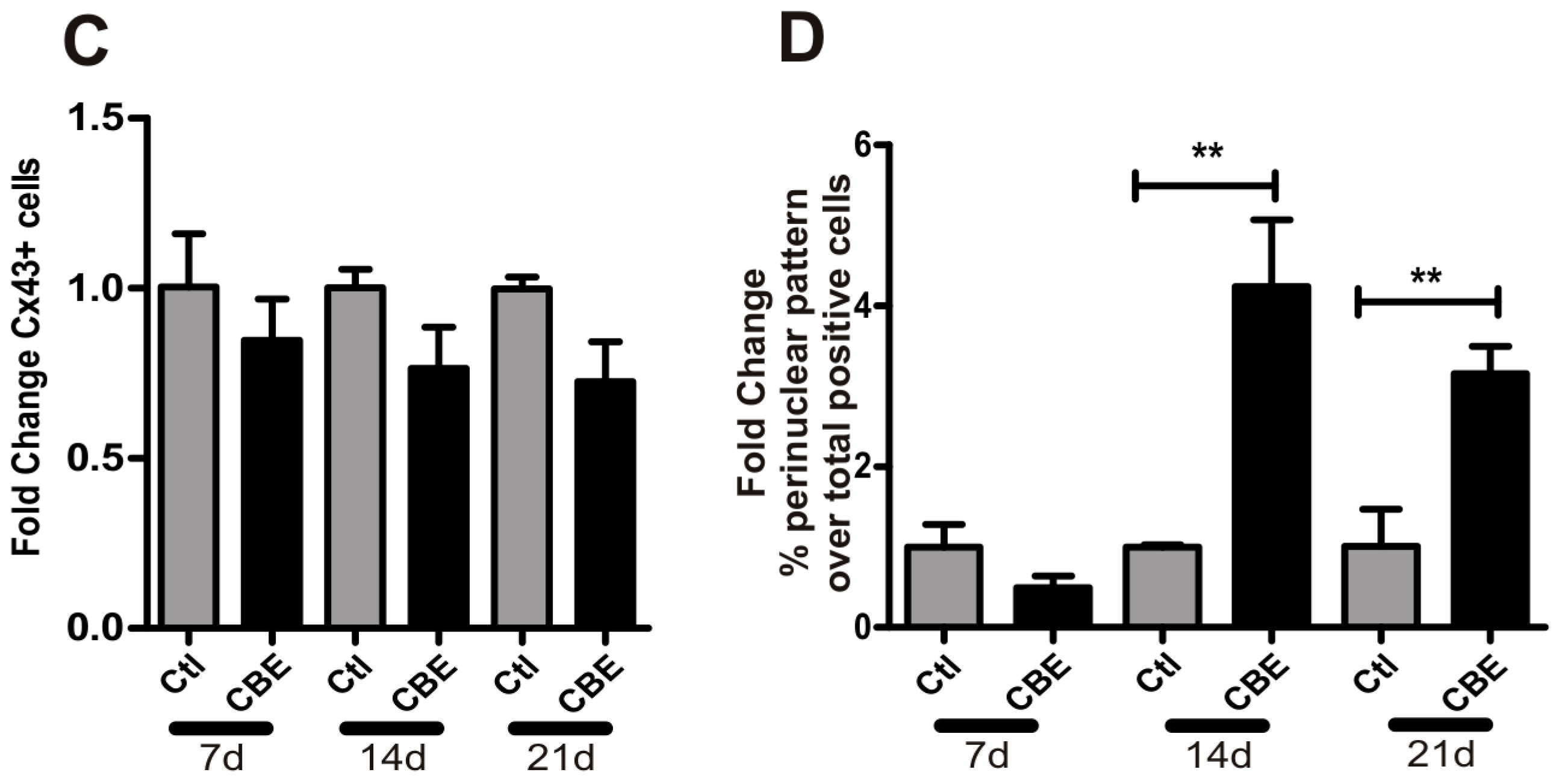
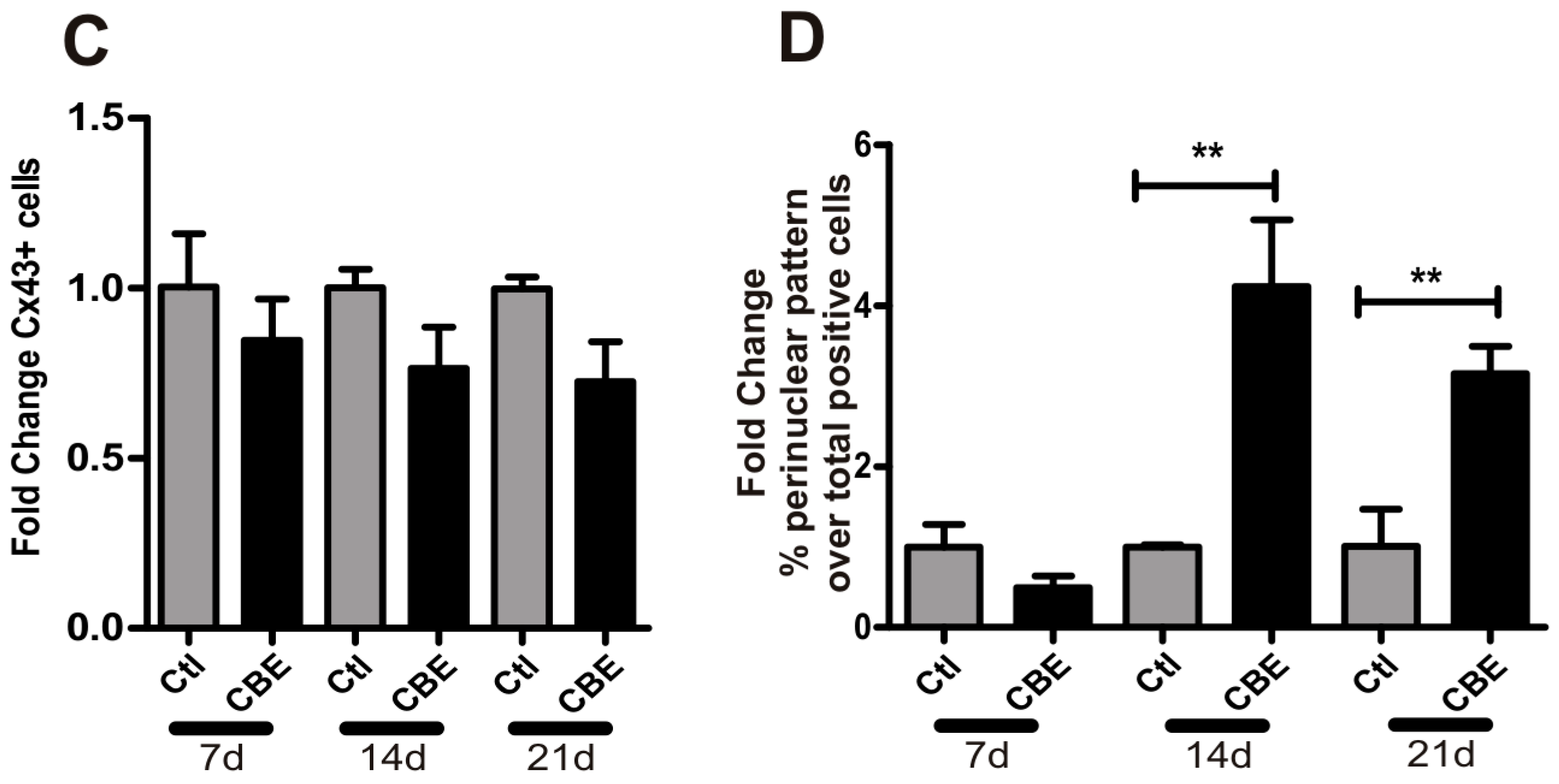
2.2. GCase Deficiency Reduces the Expression of Integrin-β and Cx43 and Alters Cx43 Distribution Pattern
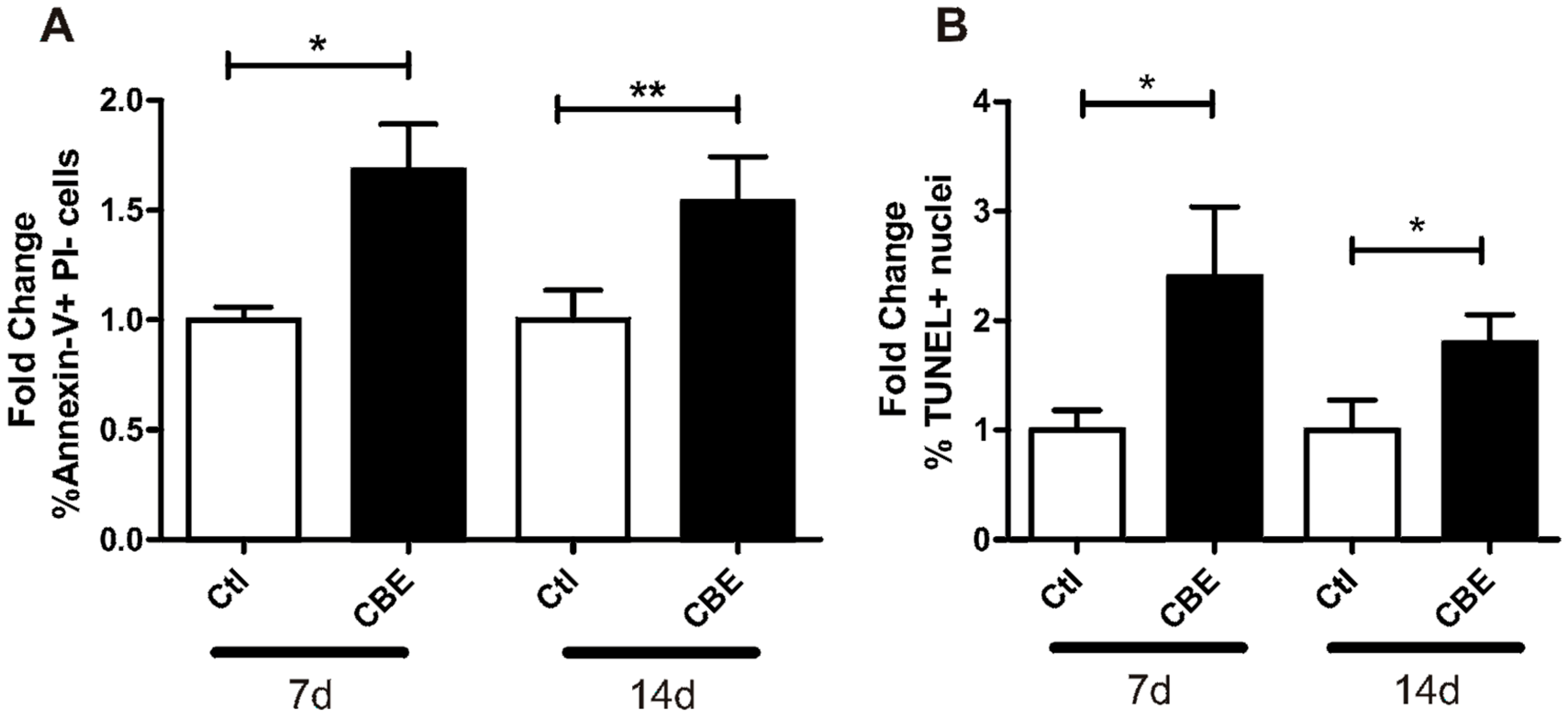
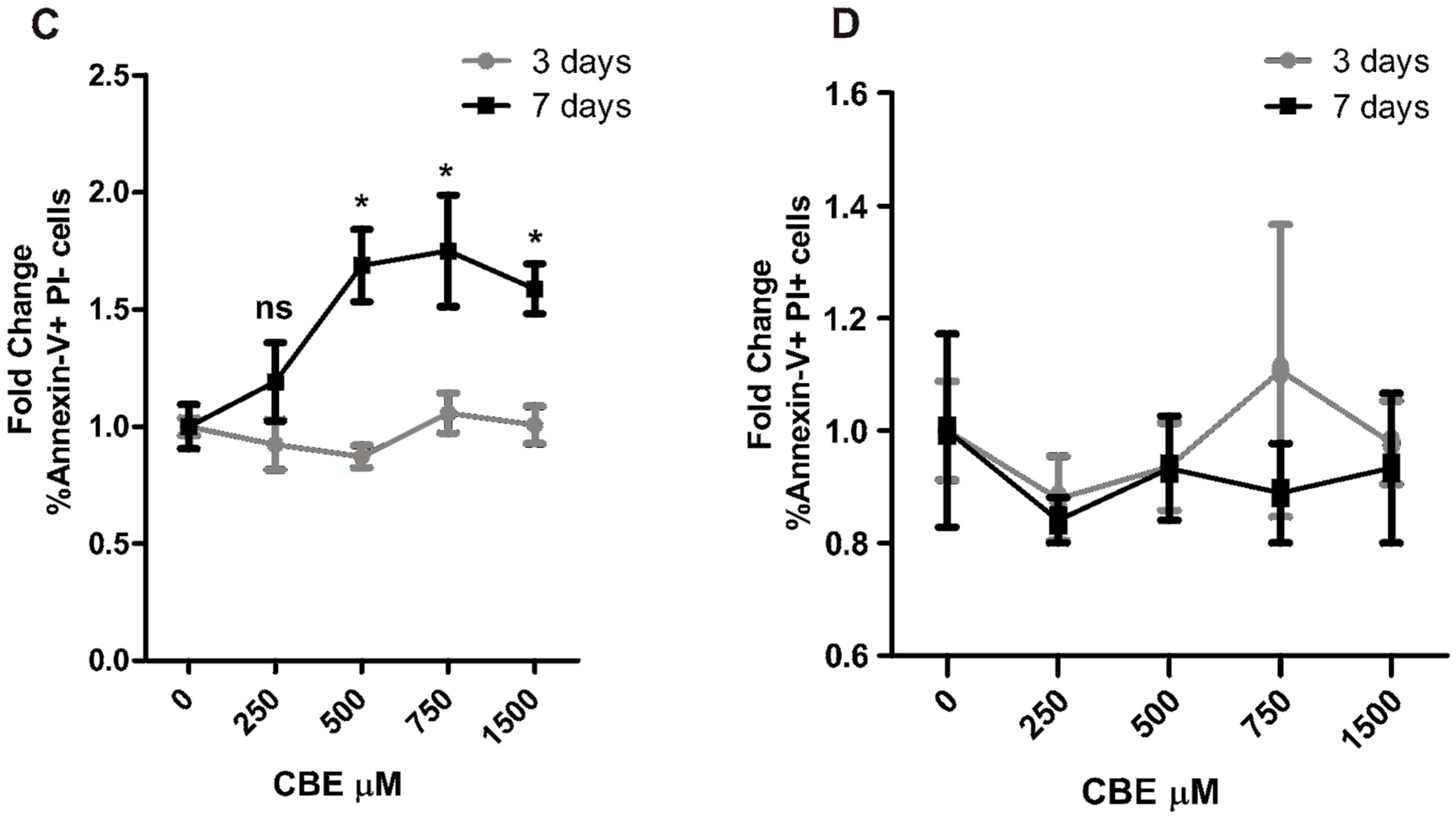
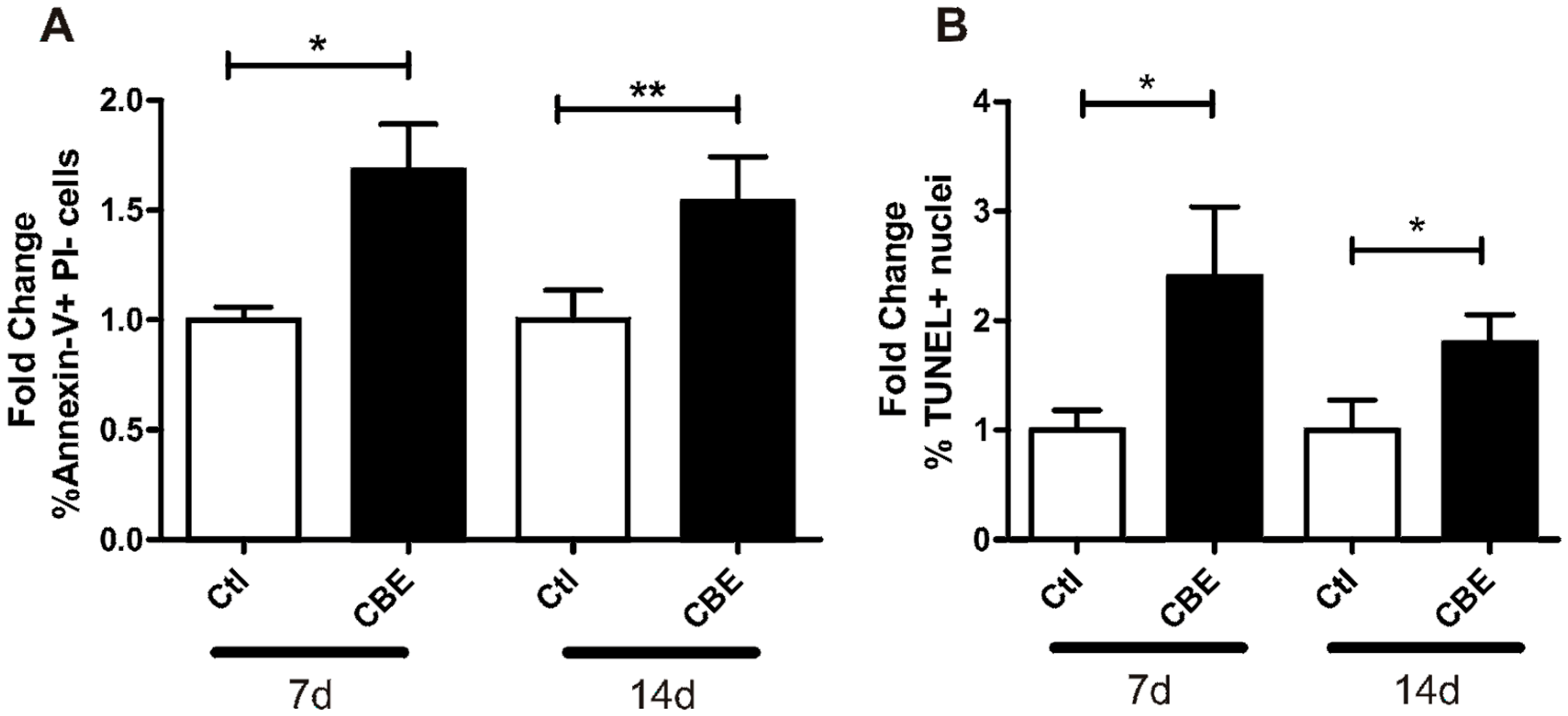
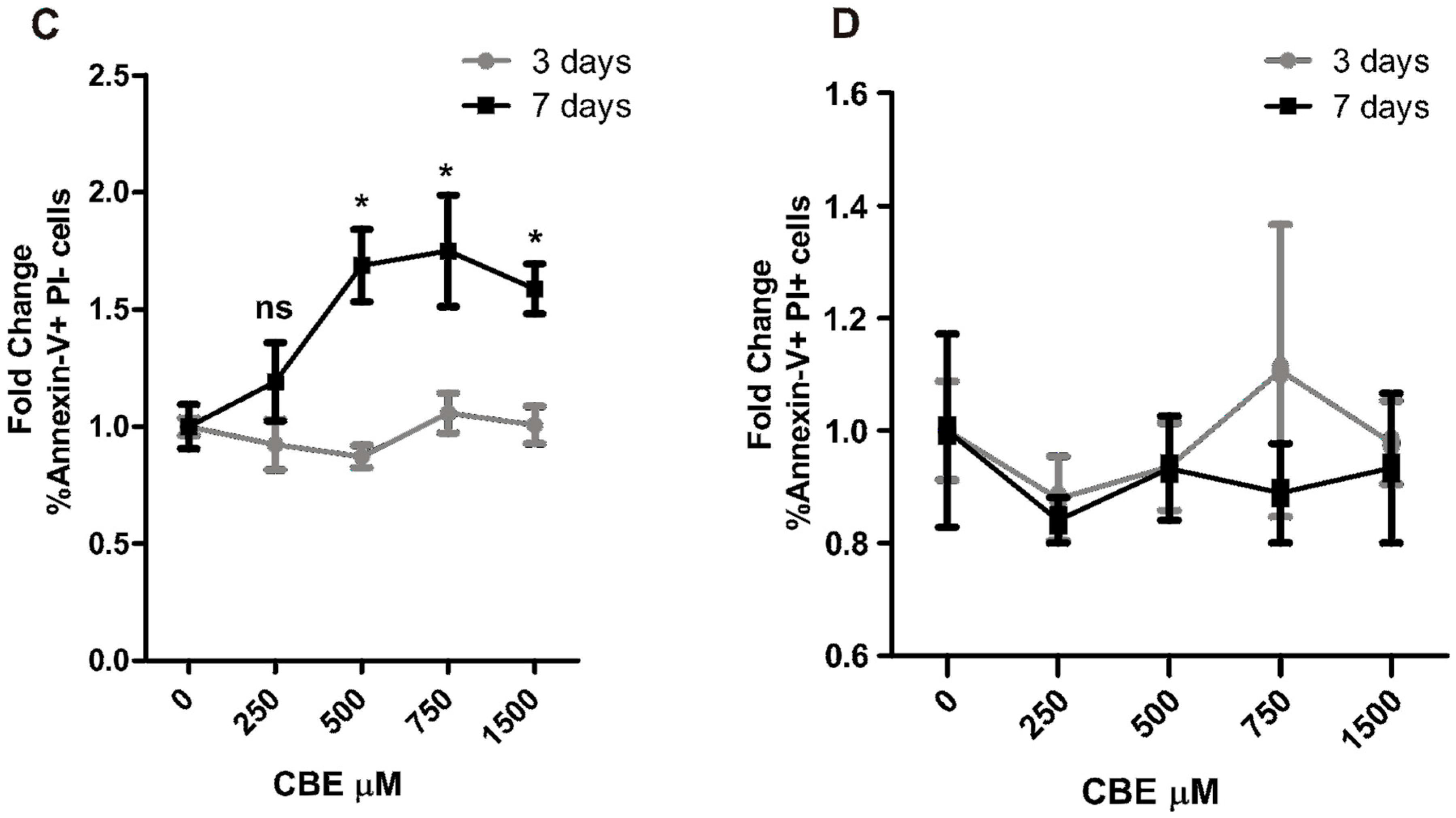
2.3. CBE Treatment Induces Osteocyte Apoptosis
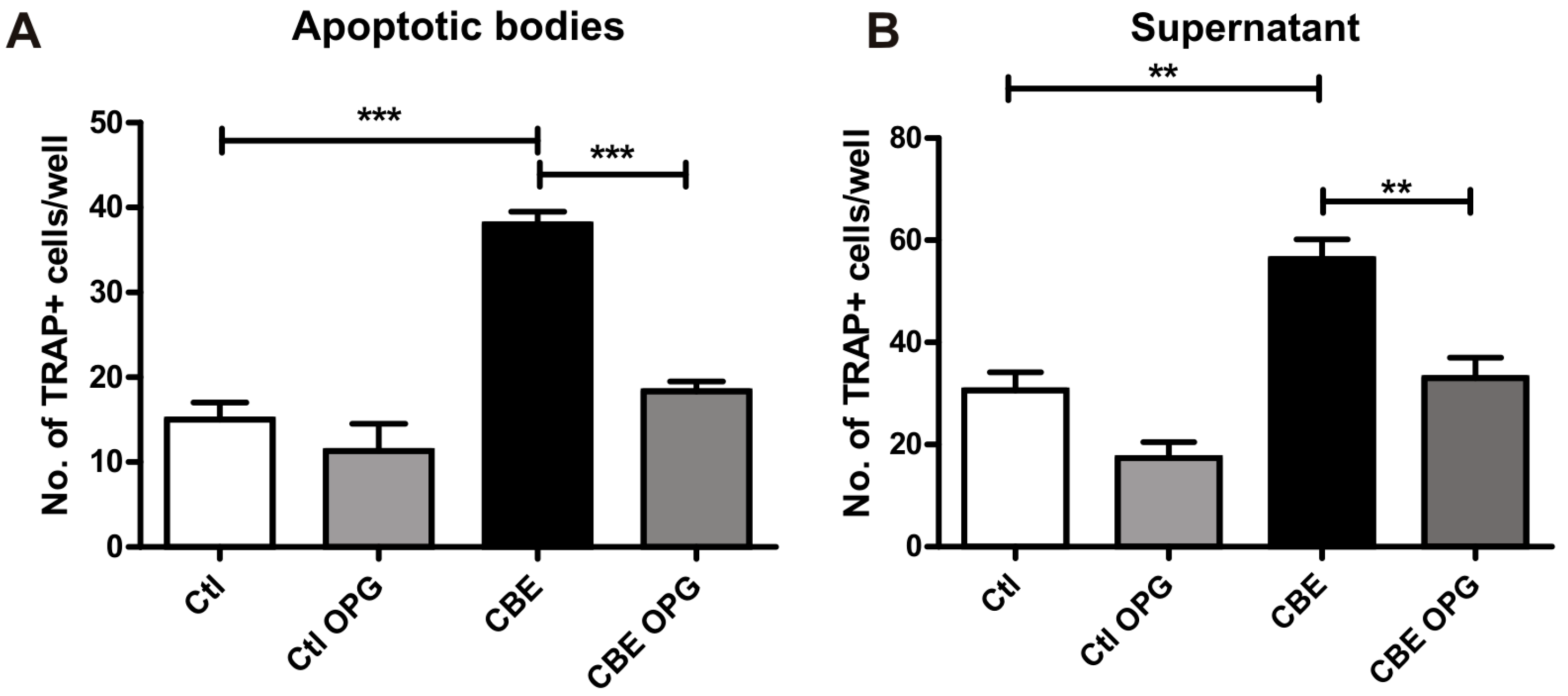
2.4. Osteoclastogenesis Induction by Conditioned Media from CBE-Treated Osteocytes Involves Apoptotic Bodies, RANKL and Soluble Factors
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture and Apoptotic Body Isolation
4.3. Osteoclast Formation Assay
4.4. Pit Formation Assay
4.5. Flow Cytometry
4.6. Annexin-V Staining
4.7. Real Time PCR
4.8. Immunofluorescence Microscopy
4.9. TUNEL
4.10. Cytokine Measurement
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GD | Gaucher disease |
| GCase | Glucocerebrosidase |
| CBE | Conduritol-β-epoxide |
| Cx-43 | Connexin 43 |
| RANKL | Receptor activator of nuclear factor κ-B ligand |
| RANK | Receptor activator of nuclear factor κ-B |
| M-CSF | Macrophage colony stimulating factor |
| TNF-α | Tumor necrosis factor-α |
| IL-6 | Interleukin-6 |
| IL-1β | Interleukin-1β |
| BMM | Bone marrow derived macrophage |
| TRAP | Tartrate resistant acid phosphatase |
| PI | Propidium iodide |
| OPG | Osteoprotegerin |
| FACS | Fluorescence associated cell sorting |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
References
- Grabowski, G.A.; Kacena, K.; Cole, J.A.; Hollak, C.E.M.; Zhang, L.; Yee, J.; Mistry, P.K.; Zimran, A.; Charrow, J.; vom Dahl, S. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet. Med. 2009, 11, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Pavlova, E.; Tindall, J.; Stein, P.E.; Bearcroft, P.; Mehta, A.; Hughes, D.; Wraith, J.E.; Cox, T.M. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine 2011, 90, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M. Gaucher disease: Clinical profile and therapeutic developments. Biologics 2010, 4, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Downey, P.A.; Siegel, M.I. Bone biology and the clinical implications for osteoporosis. Phys. Ther. 2006, 86, 77–91. [Google Scholar] [PubMed]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef] [PubMed]
- McNamara, L.M.; Majeska, R.J.; Weinbaum, S.; Friedrich, V.; Schaffler, M.B. Attachment of osteocyte cell processes to the bone matrix. Anat. Rec. (Hoboken) 2009, 292, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Loiselle, A.E.; Jiang, J.X.; Donahue, H.J. Gap junction and hemichannel functions in osteocytes. Bone 2013, 54, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Bivi, N.; Condon, K.W.; Allen, M.R.; Farlow, N.; Passeri, G.; Brun, L.R.; Rhee, Y.; Bellido, T.; Plotkin, L.I. Cell autonomous requirement of connexin 43 for osteocyte survival: Consequences for endocortical resorption and periosteal bone formation. J. Bone Miner Res. 2012, 27, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gu, S.; Riquelme, M.A.; Burra, S.; Callaway, D.; Cheng, H.; Guda, T.; Schmitz, J.; Fajardo, R.J.; Werner, S.L.; et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J. Bone Miner. Res. 2015, 30, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I. Apoptotic osteocytes and the control of targeted bone resorption. Curr. Osteoporos. Rep. 2014, 12, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogianni, G.; Mann, V.; Noble, B.S. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J. Bone Miner. Res. 2008, 23, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; Yuen, T.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [PubMed]
- Zancan, I.; Bellesso, S.; Costa, R.; Salvalaio, M.; Stroppiano, M.; Hammond, C.; Argenton, F.; Filocamo, M.; Moro, E. Glucocerebrosidase deficiency in zebrafish affects primary bone ossification through increased oxidative stress and reduced Wnt/β-catenin signaling. Hum. Mol. Genet. 2015, 24, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Mucci, J.M.; Suqueli García, F.; de Francesco, P.N.; Ceci, R.; di Genaro, S.; Fossati, C.A.; Delpino, M.V.; Rozenfeld, P.A. Uncoupling of osteoblast-osteoclast regulation in a chemical murine model of gaucher disease. Gene 2013, 532. [Google Scholar] [CrossRef] [PubMed]
- Mucci, J.M.; Scian, R.; de Francesco, P.N.; García, F.S.; Ceci, R.; Fossati, C.A.; Delpino, M.V.; Rozenfeld, P.A. Induction of osteoclastogenesis in an in vitro model of Gaucher disease is mediated by T cells via TNF-α. Gene 2012, 509. [Google Scholar] [CrossRef] [PubMed]
- Mucci, J.M.; Cuello, M.F.; Kisinovsky, I.; Larroude, M.; Delpino, M.V.; Rozenfeld, P.A. Proinflammatory and proosteoclastogenic potential of peripheral blood mononuclear cells from Gaucher patients: Implication for bone pathology. Blood Cells Mol. Dis. 2015, 55, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lecourt, S.; Vanneaux, V.; Cras, A.; Freida, D.; Heraoui, D.; Herbi, L.; Caillaud, C.; Chomienne, C.; Marolleau, J.-P.; Belmatoug, N.; et al. Bone Marrow Microenvironment in an In Vitro Model of Gaucher Disease: Consequences of Glucocerebrosidase Deficiency. Stem Cells Dev. 2011, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.; Baker, R.J.; Mehta, A.B.; Hughes, D.A. Enhanced differentiation of osteoclasts from mononuclear precursors in patients with Gaucher disease. Blood Cells Mol. Dis. 2013, 51, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Novack, D.V.; Mbalaviele, G. Osteoclasts-Key Players in Skeletal Health and Disease. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor necrosis factor-α induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef] [PubMed]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Lee, Y. The role of interleukin-17 in bone metabolism and inflammatory skeletal diseases. BMB Rep. 2013, 46, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Al-Dujaili, S.A.; Lau, E.; Al-Dujaili, H.; Tsang, K.; Guenther, A.; You, L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J. Cell. Biochem. 2011, 112, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M. Gaucher disease: Understanding the molecular pathogenesis of sphingolipidoses. J. Inherit. Metab. Dis. 2001, 24 (Suppl. 2), 106–121. [Google Scholar] [PubMed]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.-L.; Yuen, T.; Yang, R. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.J.; Findlay, D.M. Osteocyte regulation of bone mineral: A little give and take. Osteoporos. Int. 2012, 23, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Nakashima, T.; Takayanagi, H. Osteocyte control of osteoclastogenesis. Bone 2013, 54, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.R.; Uzer, G.; Brobst, K.E.; Xie, Z.; Sen, B.; Yen, S.S.; Styner, M.; Rubin, J. Osteocyte specific responses to soluble and mechanical stimuli in a stem cell derived culture model. Sci. Rep. 2015, 5, 11049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, W.R.; Modla, S.; Grindel, B.J.; Czymmek, K.J.; Kirn-Safran, C.B.; Wang, L.; Duncan, R.L.; Farach-Carson, M.C. Perlecan/Hspg2 deficiency alters the pericellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. J. Bone Miner. Res. 2011, 26, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Uzer, G.; Pongkitwitoon, S.; Ian, C.; Thompson, W.R.; Rubin, J.; Chan, M.E.; Judex, S. Gap Junctional Communication in Osteocytes Is Amplified by Low Intensity Vibrations In Vitro. PLoS ONE 2014, 9, e90840. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Ma, Y.; Li, X.; Wu, X.; Liu, W.; Li, X.; Shen, J.; Wang, H. Lipopolysaccharide increases IL-6 secretion via activation of the ERK1/2 signaling pathway to up-regulate RANKL gene expression in MLO-Y4 cells. Cell Biol. Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.K.Y.; Harris, S.; Ahuja, S.S.; Bonewald, L.F. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res. 2002, 17, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Pesce Viglietti, A.I.; Arriola Benitez, P.C.; Gentilini, M.V.; Velásquez, L.N.; Fossati, C.A.; Giambartolomei, G.H.; Delpino, M.V. Brucella abortus invasion of osteocytes modulates connexin 43 and integrin expression and induces osteoclastogenesis via receptor activator of NF-κB ligand and tumor necrosis factor α secretion. Infect. Immun. 2015, 84, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Mathov, I.; Aguirre, J.I.; Parfitt, A.M.; Manolagas, S.C.; Bellido, T. Mechanical stimulation prevents osteocyte apoptosis: Requirement of integrins, Src kinases, and ERKs. Am. J. Physiol. Cell Physiol. 2005, 289, C633–C643. [Google Scholar] [CrossRef] [PubMed]
- Lezcano, V.; Bellido, T.; Plotkin, L.I.; Boland, R.; Morelli, S. Role of connexin 43 in the mechanism of action of alendronate: Dissociation of anti-apoptotic and proliferative signaling pathways. Arch. Biochem. Biophys. 2012, 518, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Kim, E.Y.; Jung, S.-C. Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse. J. Korean Med. Sci. 2006, 21, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Kitatani, K.; Wada, M.; Perry, D.; Usui, T.; Sun, Y.; Obeid, L.M.; Yaegashi, N.; Grabowski, G.A.; Hannun, Y.A. Activation of p38 mitogen-activated protein kinase in Gaucher’s disease. PLoS ONE 2015, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Li, P.; Zhang, Q.; Schwarz, E.M.; Keng, P.; Arbini, A.; Boyce, B.F.; Xing, L. Tumor necrosis factor-α increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J. Biol. Chem. 2006, 281, 11846–11855. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondar, C.; Ormazabal, M.; Crivaro, A.; Ferreyra-Compagnucci, M.; Delpino, M.V.; Rozenfeld, P.A.; Mucci, J.M. Osteocyte Alterations Induce Osteoclastogenesis in an In Vitro Model of Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 112. https://doi.org/10.3390/ijms18010112
Bondar C, Ormazabal M, Crivaro A, Ferreyra-Compagnucci M, Delpino MV, Rozenfeld PA, Mucci JM. Osteocyte Alterations Induce Osteoclastogenesis in an In Vitro Model of Gaucher Disease. International Journal of Molecular Sciences. 2017; 18(1):112. https://doi.org/10.3390/ijms18010112
Chicago/Turabian StyleBondar, Constanza, Maximiliano Ormazabal, Andrea Crivaro, Malena Ferreyra-Compagnucci, María Victoria Delpino, Paula Adriana Rozenfeld, and Juan Marcos Mucci. 2017. "Osteocyte Alterations Induce Osteoclastogenesis in an In Vitro Model of Gaucher Disease" International Journal of Molecular Sciences 18, no. 1: 112. https://doi.org/10.3390/ijms18010112