Intramitochondrial Ascorbic Acid Enhances the Formation of Mitochondrial Superoxide Induced by Peroxynitrite via a Ca2+-Independent Mechanism

Abstract
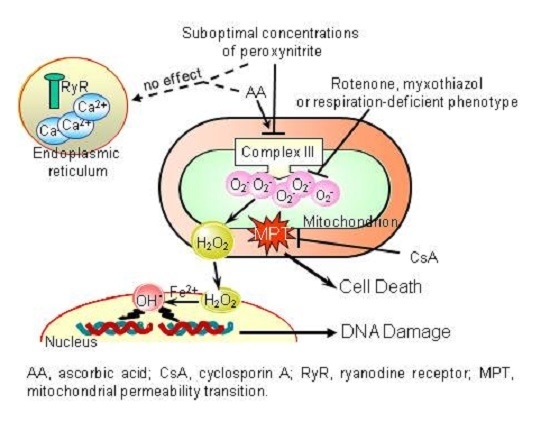
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
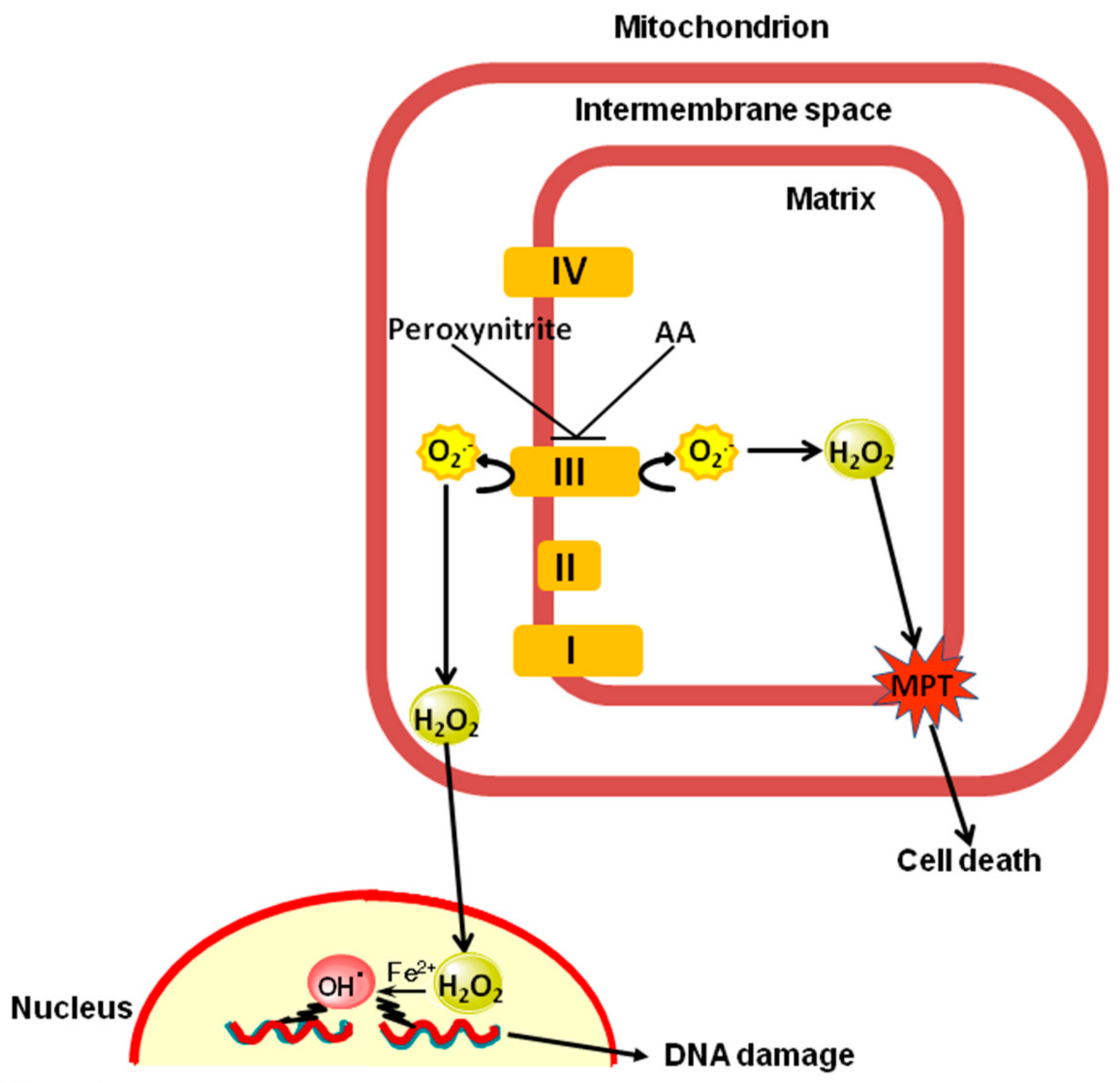
1. Introduction
2. Results
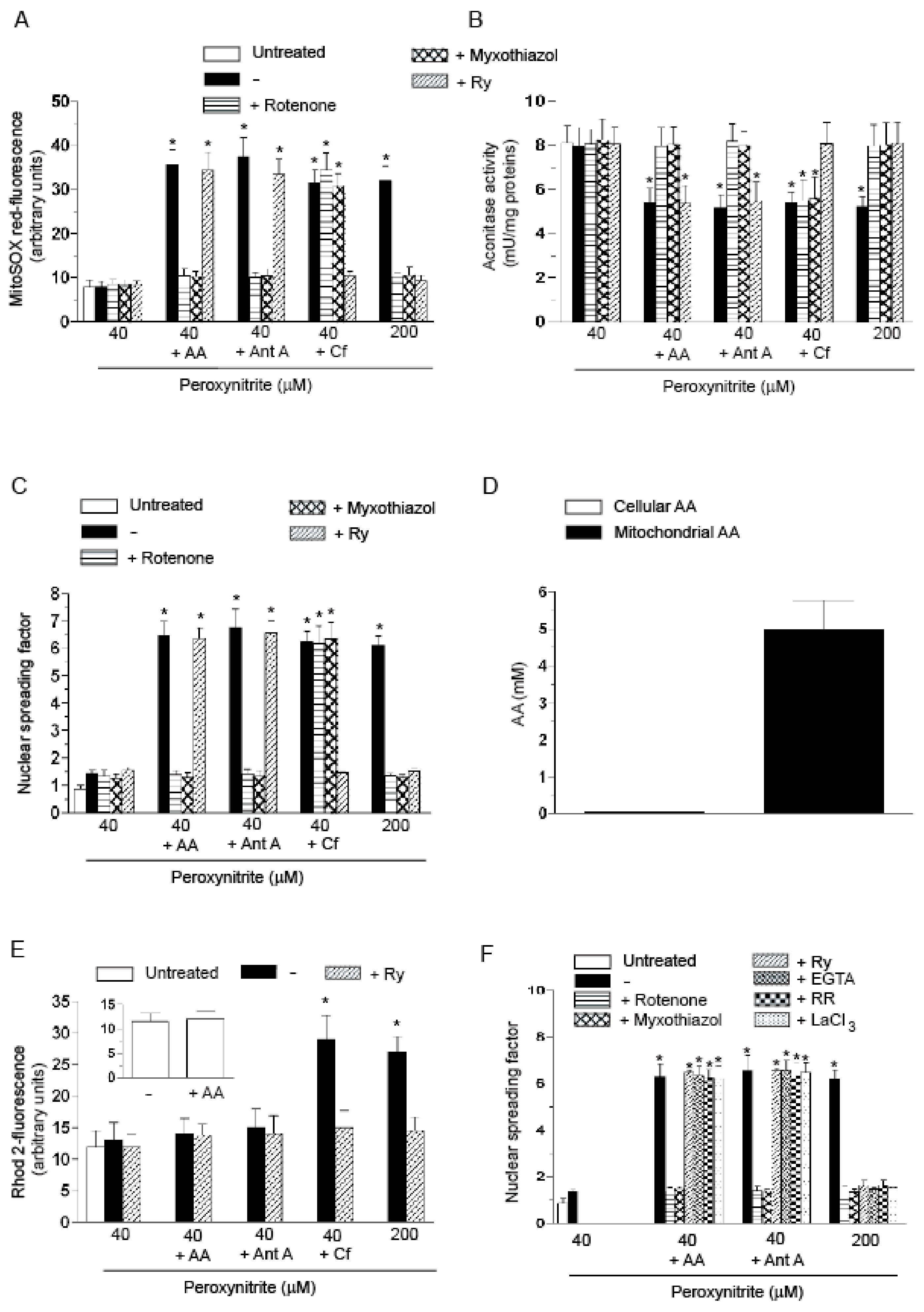
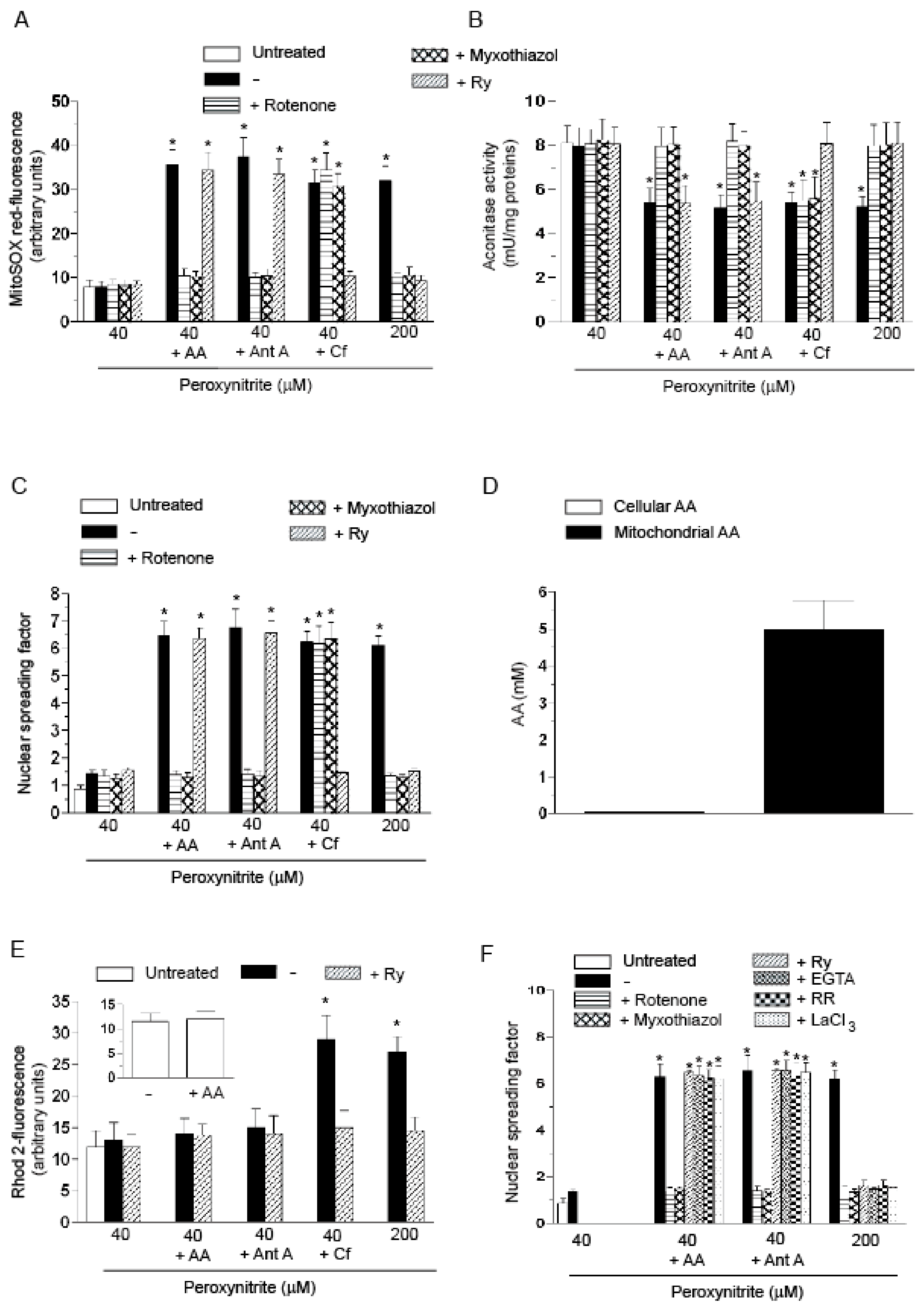
2.1. Relationships between Mitochondrial Superoxide Formation, with the Ensuing Downstream DNA Strand Scission, and Mitochondrial Ca2+ Accumulation in Cells Exposed to AA and Peroxynitrite
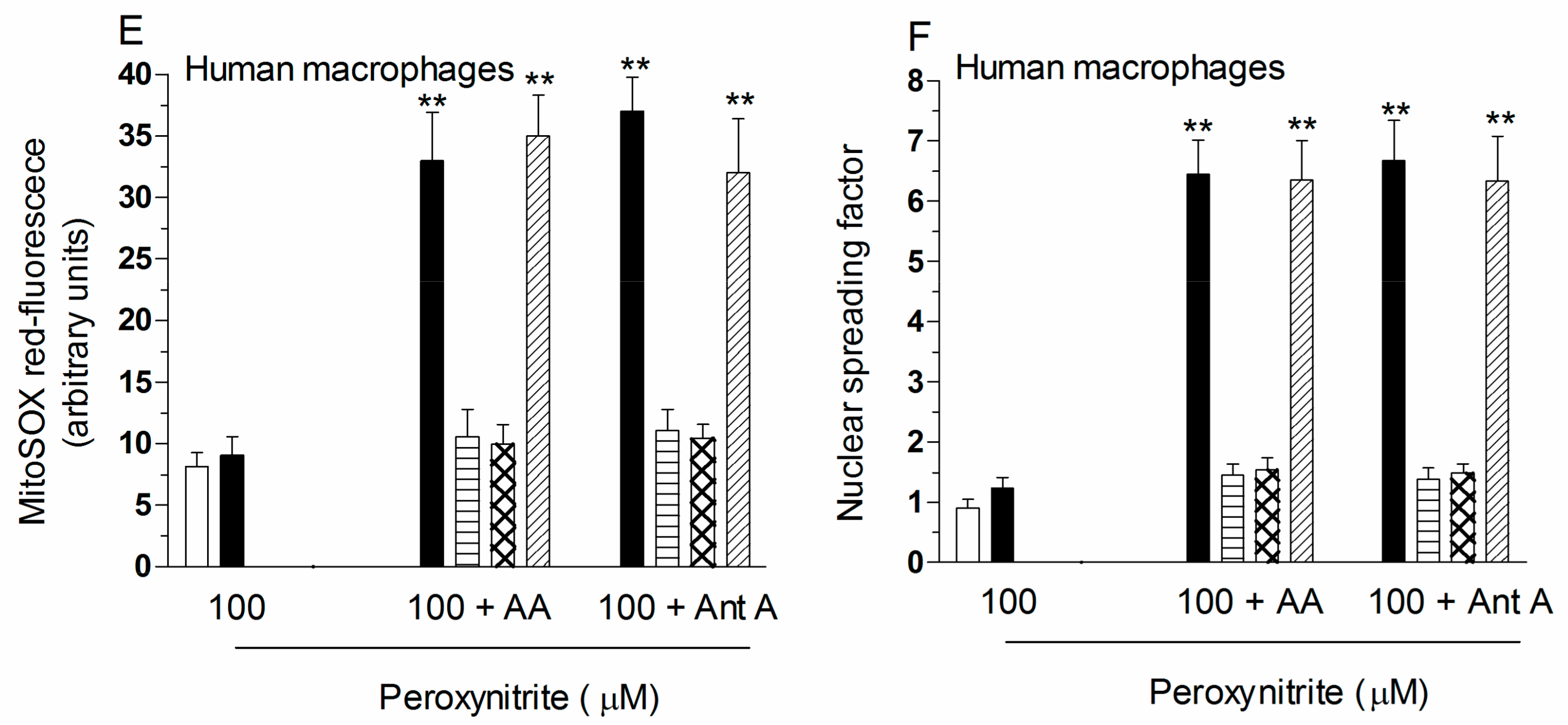
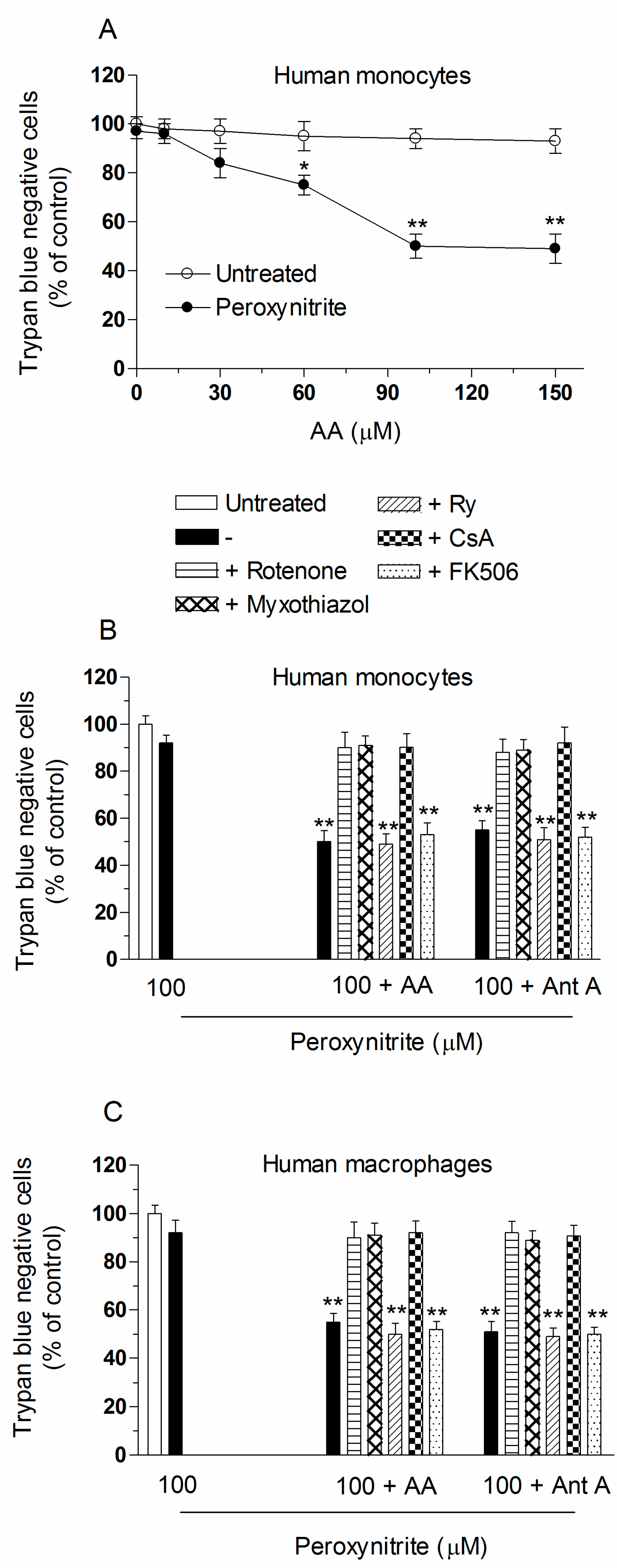
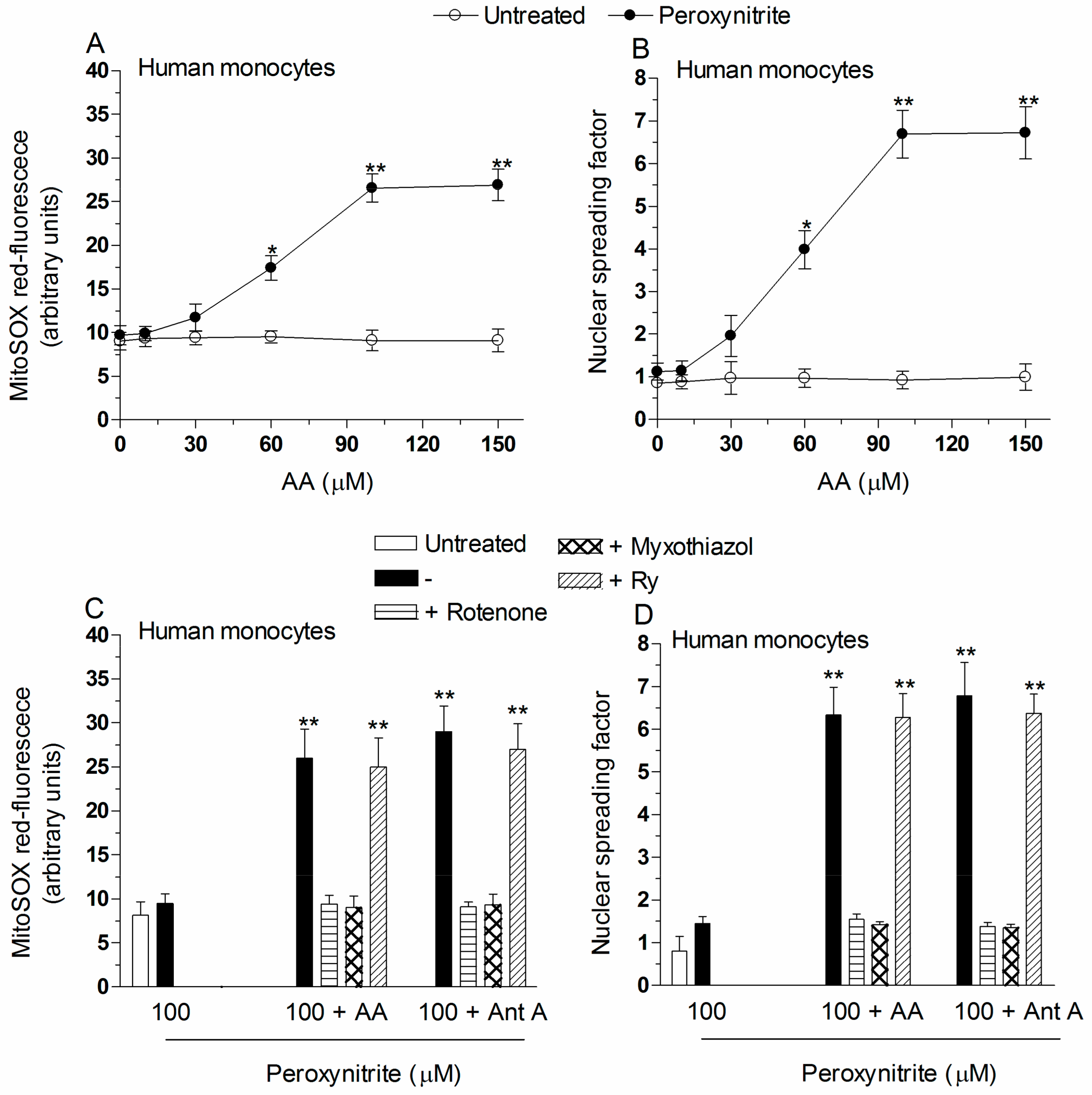
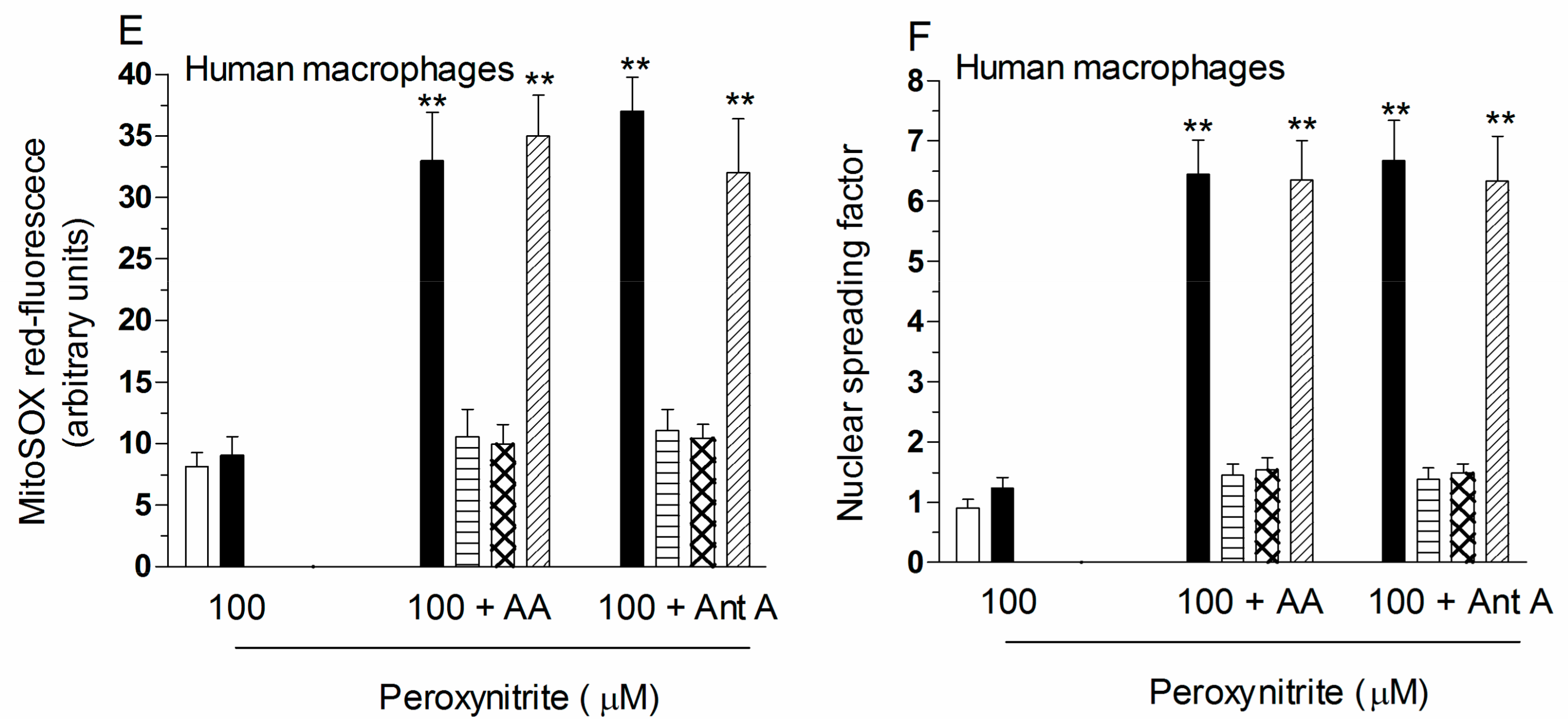
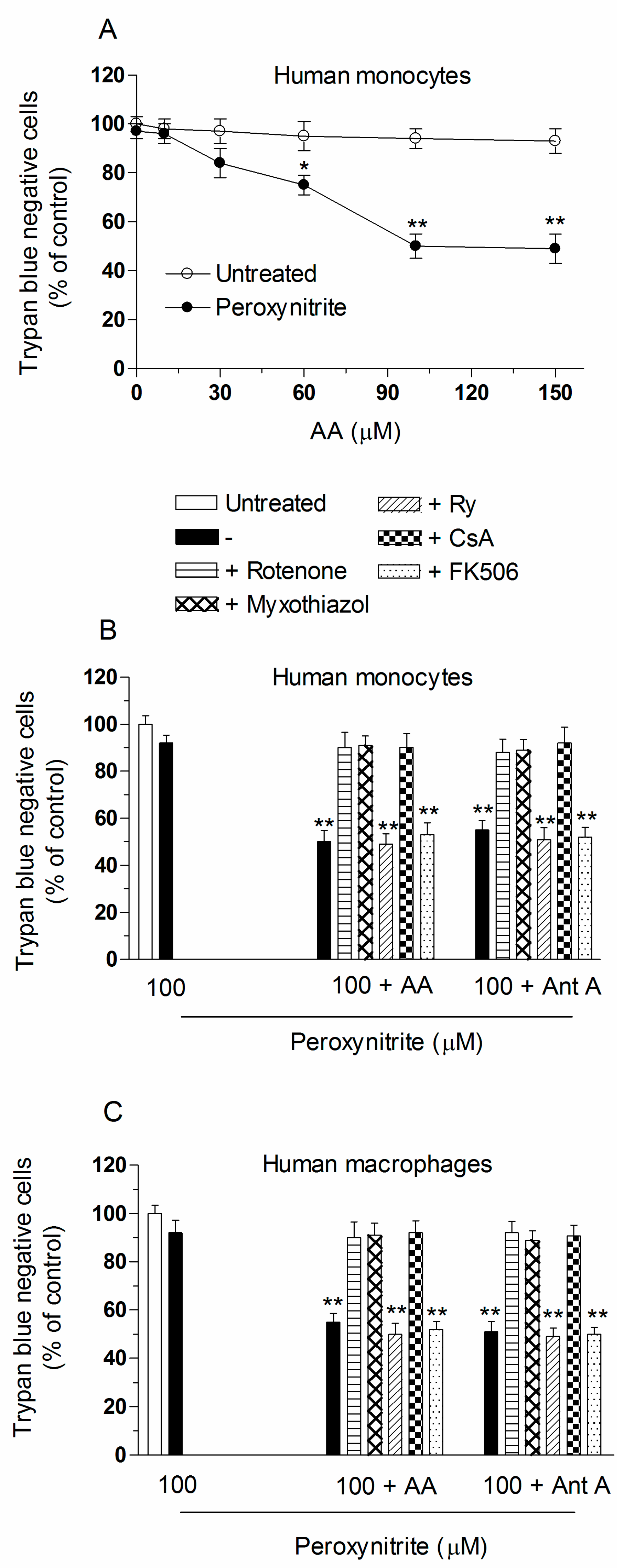
2.2. Mitochondrial Superoxide Formation and DNA Strand Scission in Human Monocytes and Macrophages Exposed to AA and Peroxynitrite
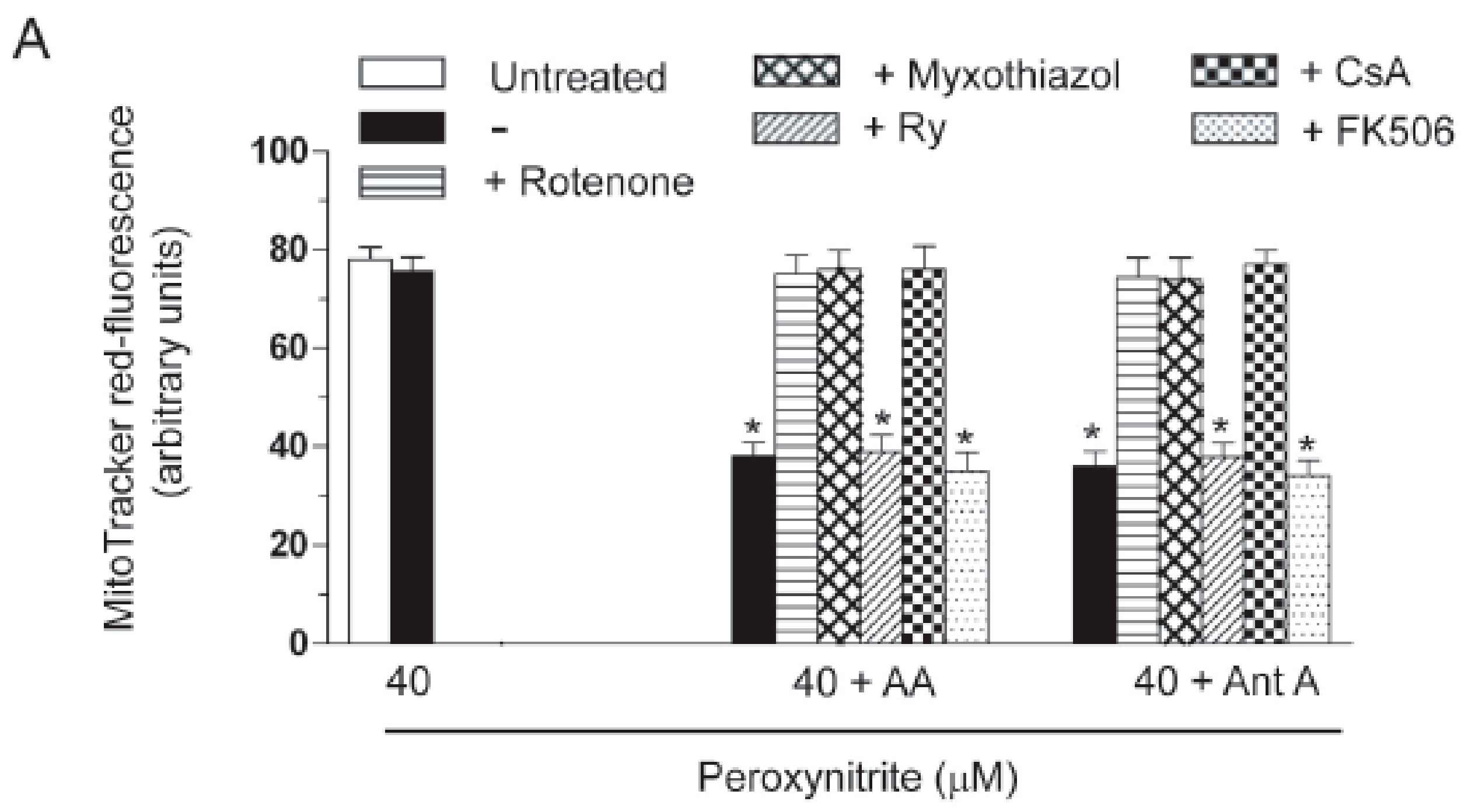
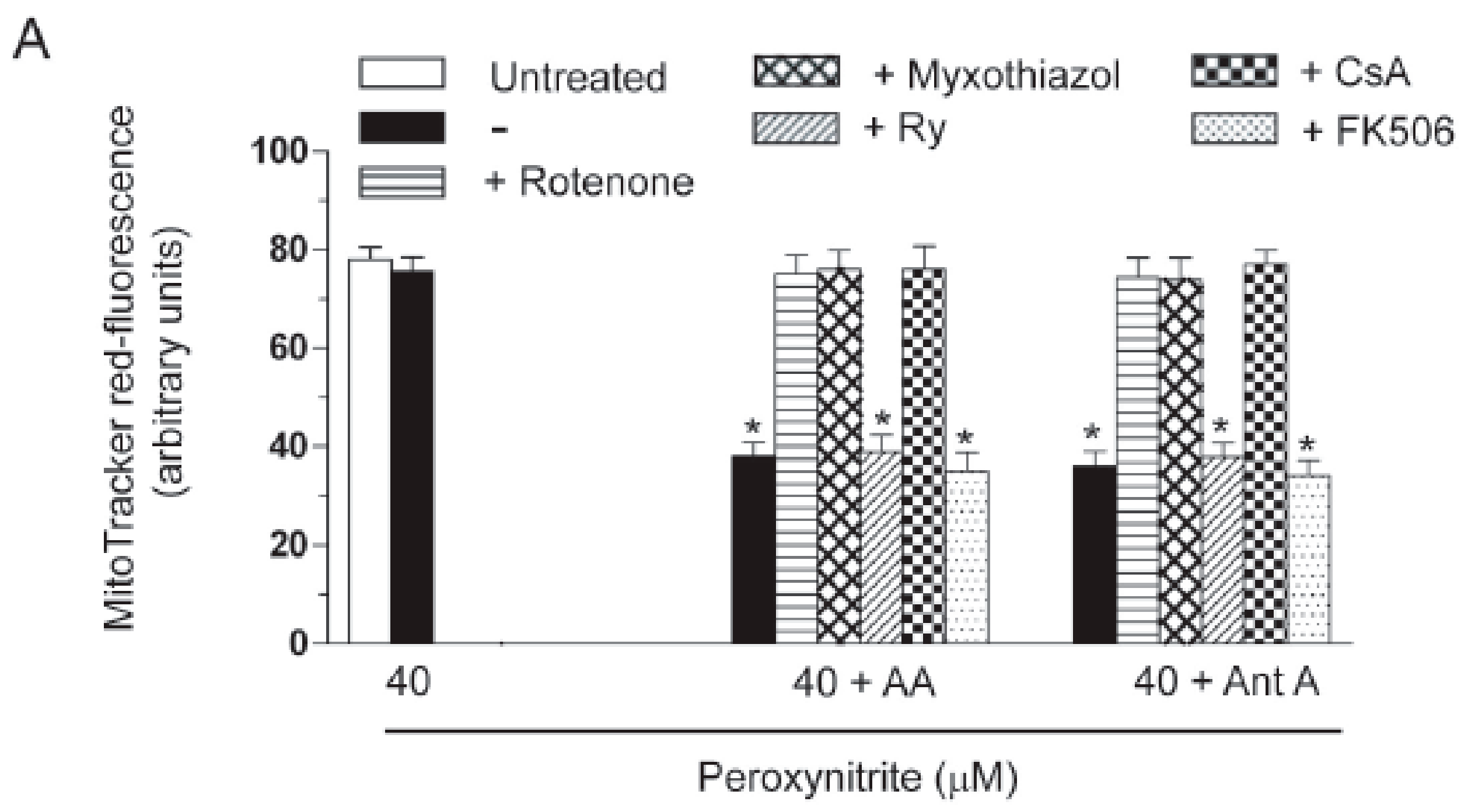
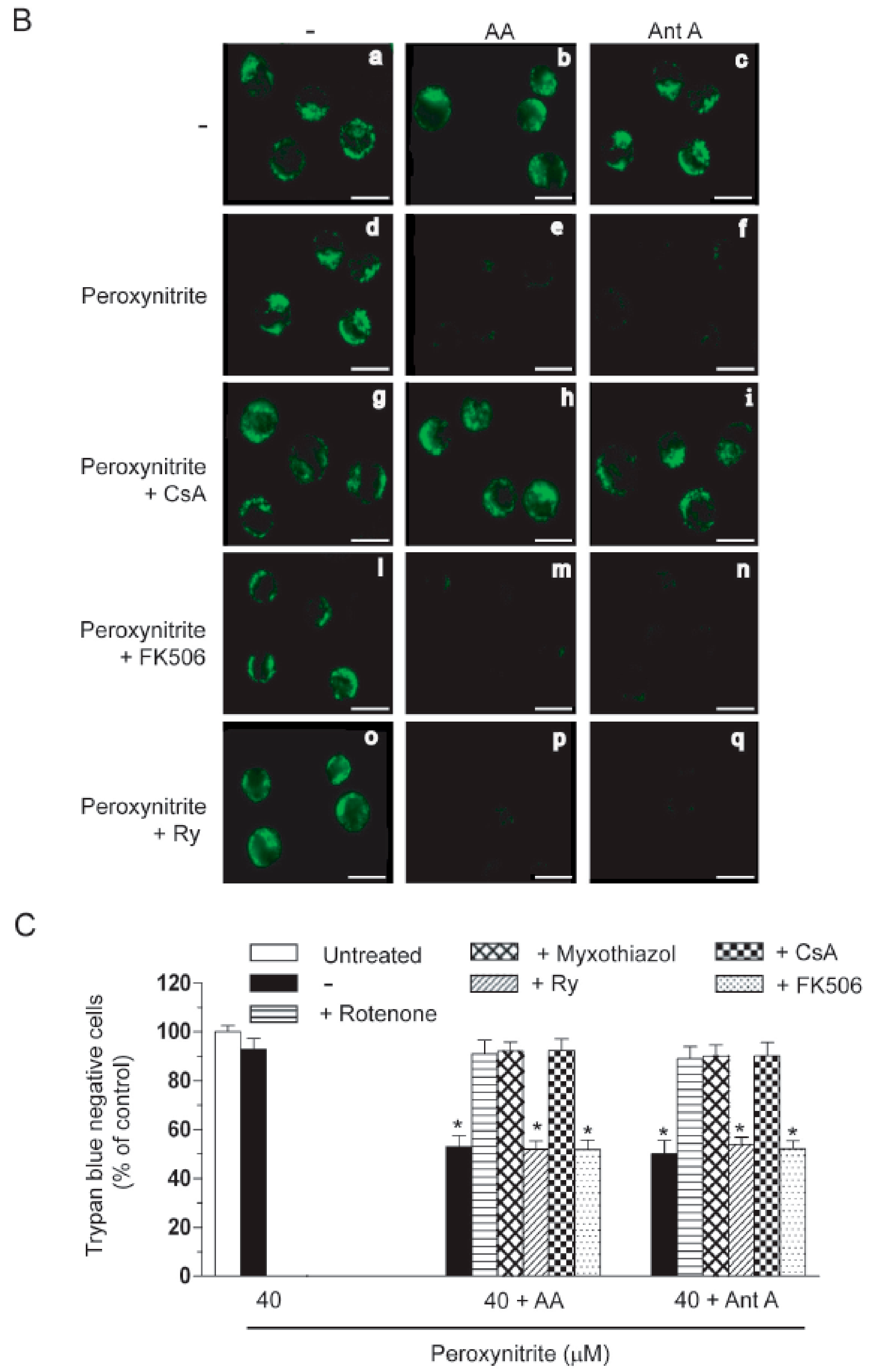
2.3. AA Promotes Peroxynitite-Dependent Mitochondrial Permeability Transition and Cytotoxicity via Ca2+-Independent Mechanisms
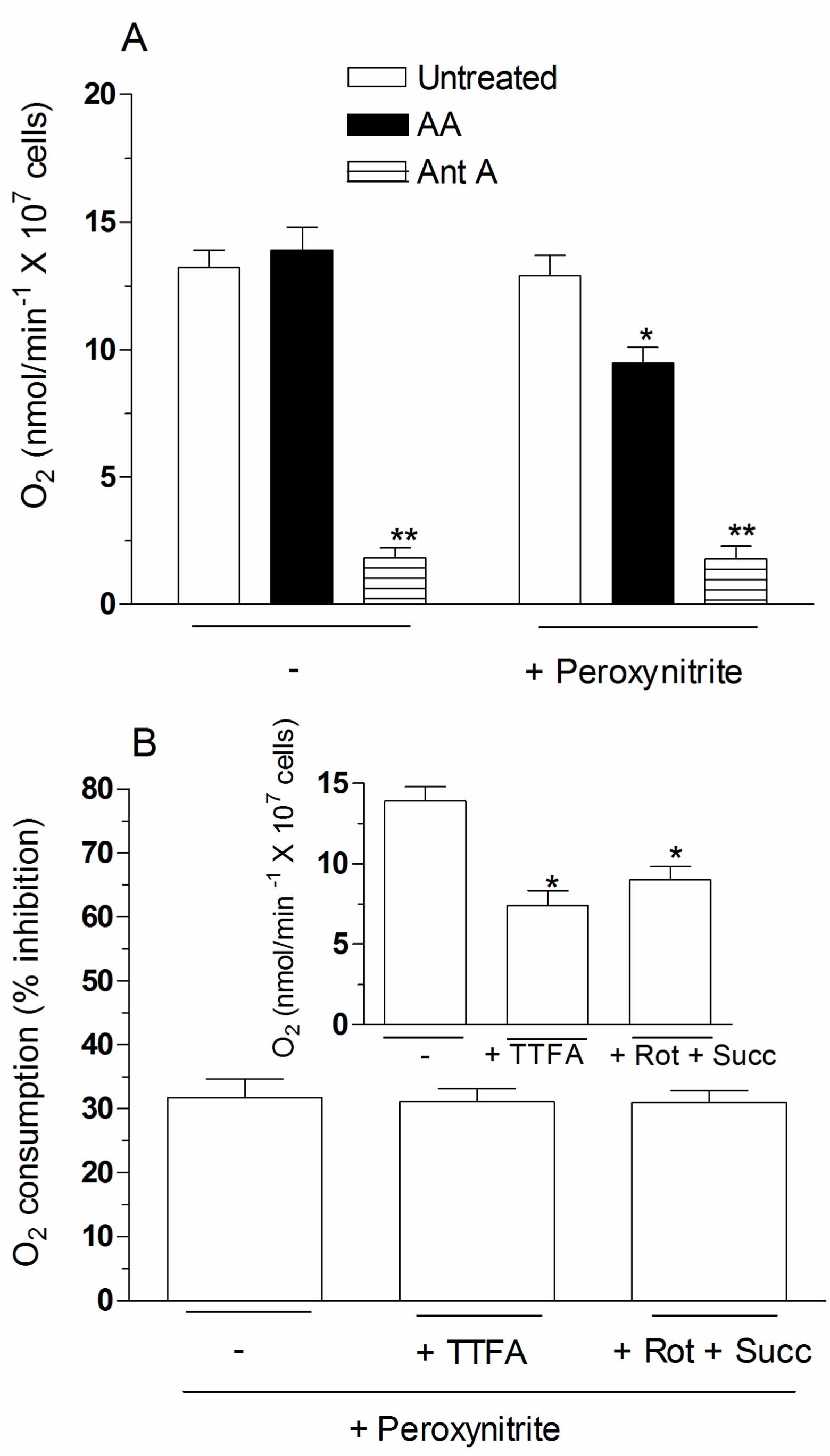
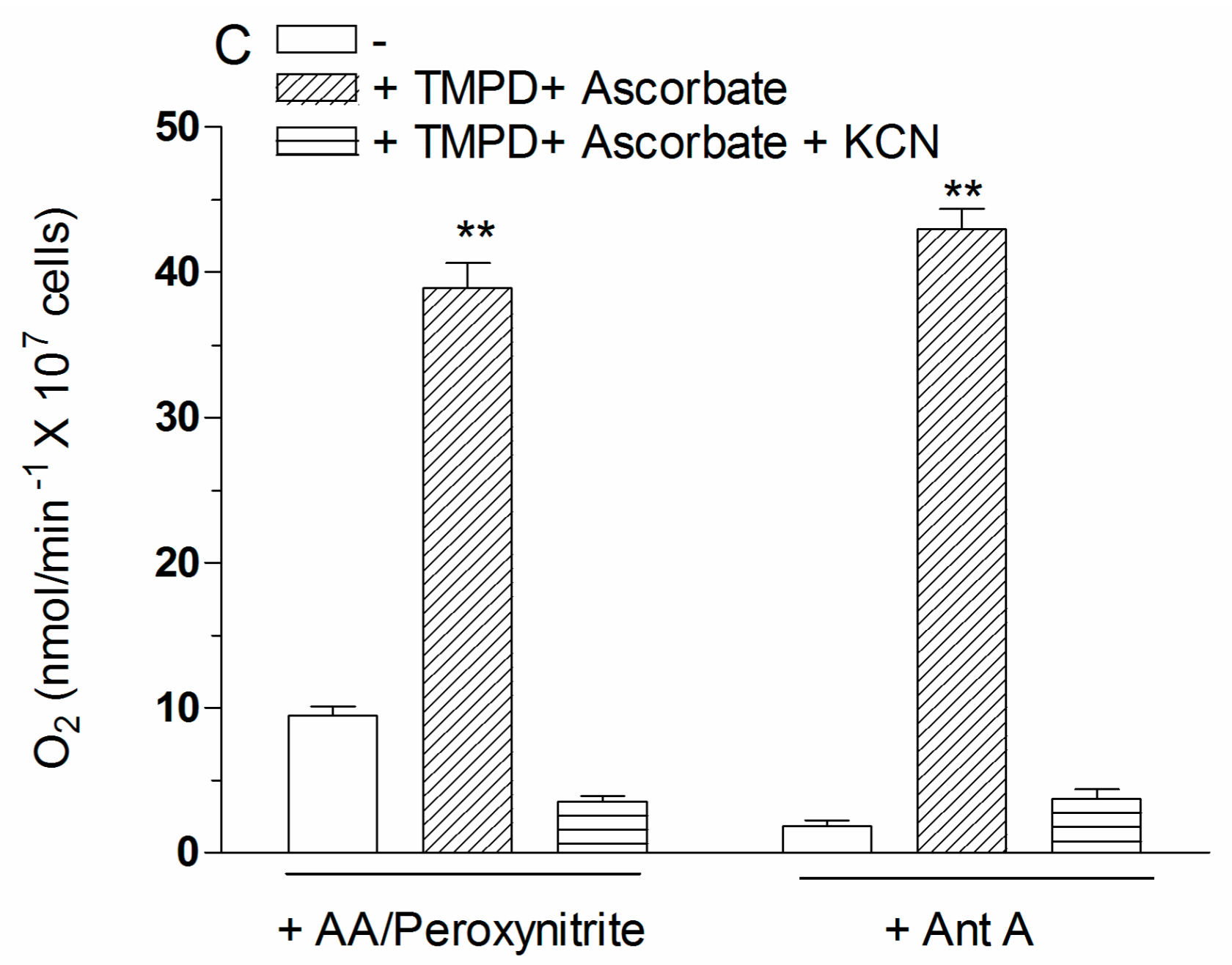
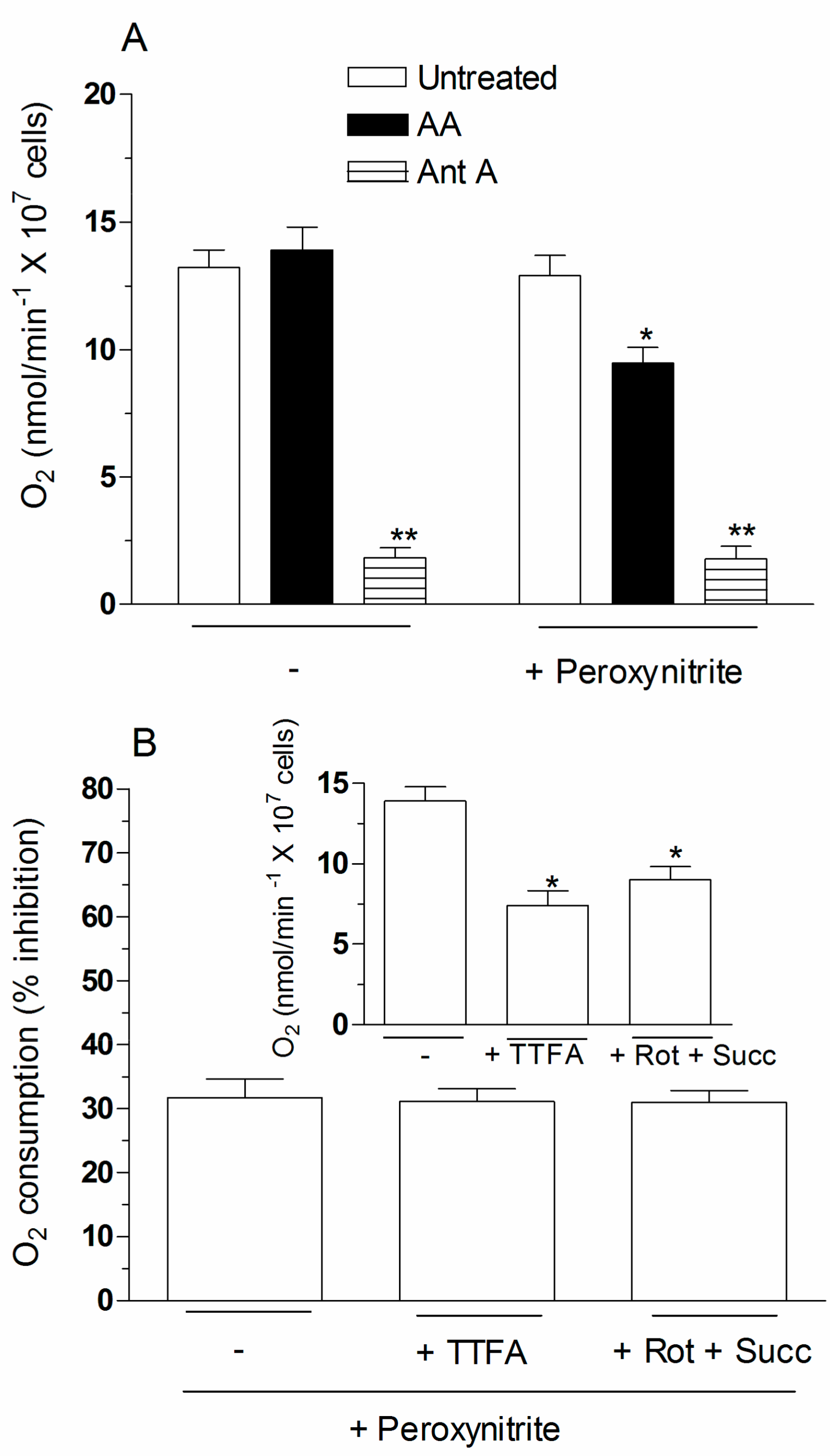
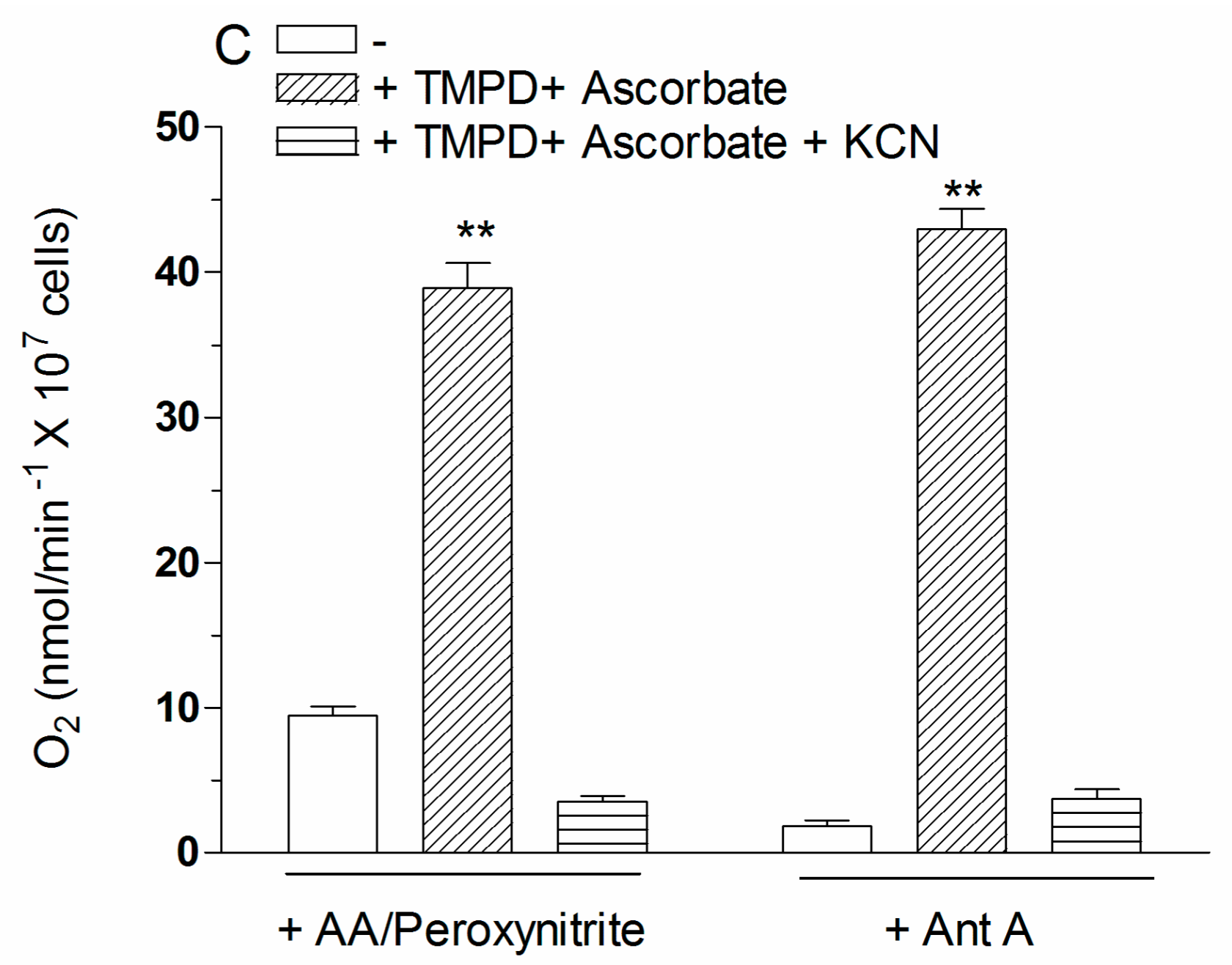
2.4. The Enhancing Effects of AA Are Mediated by an Increased Susceptibility of Complex III to Inhibition by Peroxynitrite
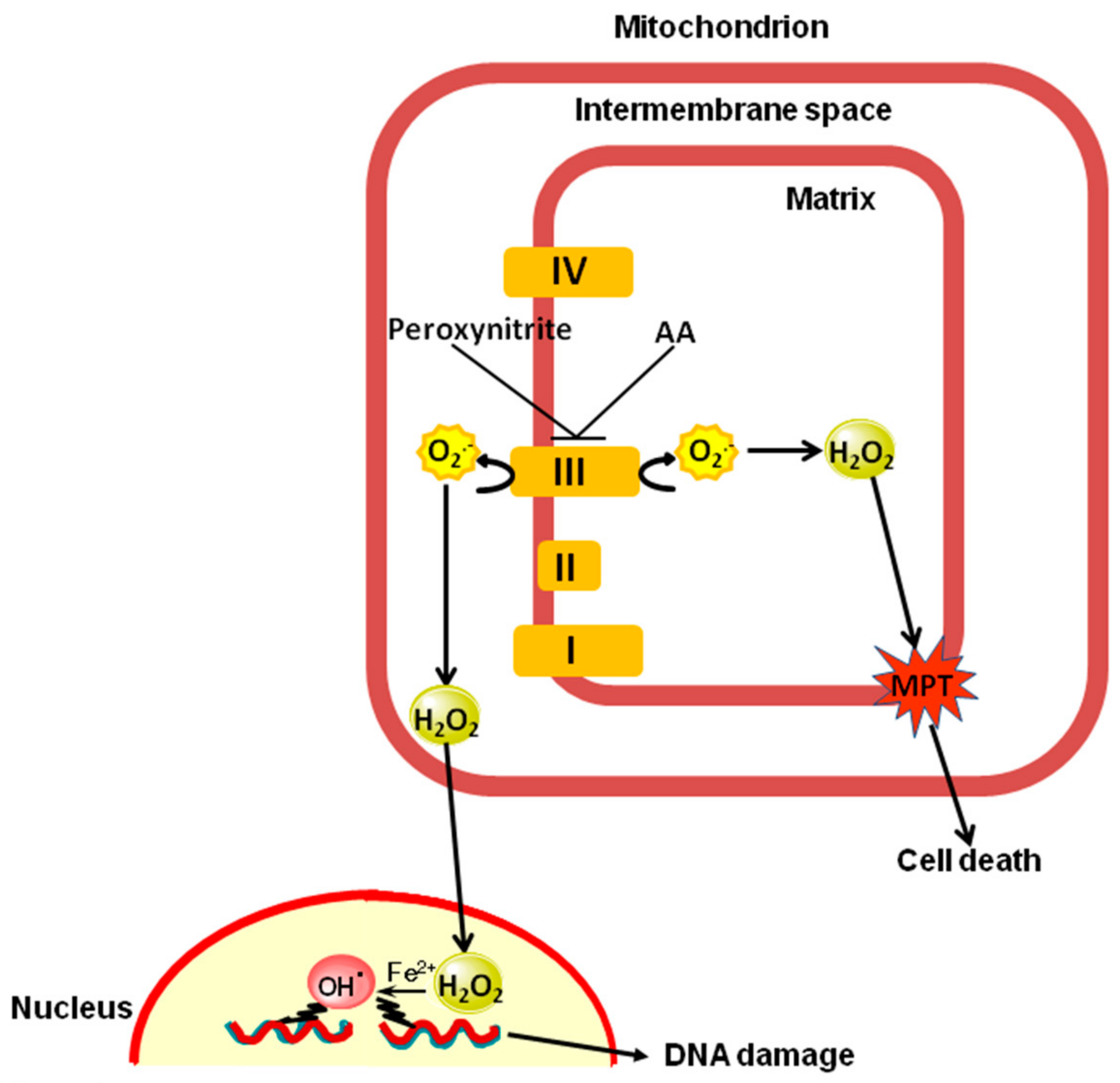
3. Discussion
- The enhancing effects of AA, detected in terms of mitochondrial superoxide formation and downstream DNA strand scission, were sensitive to rotenone, or myxothiazol, as in the case of CRDM and RDM, but not CDM.
- The enhancing effects of AA were insensitive to Ry, as for RDM, but not CRDM or CDM.
- There was no evidence of increased mitochondrial accumulation of Ca2+ after treatment with AA and/or peroxynitrite, as in the case of RDM. Increased mitochondrial accumulation of Ca2+ was instead associated with either CRDM or CDM.
- The DNA strand scission mediated by peroxynitrite in cells permeabilized after pre-exposure to AA, while suppressed by rotenone or myxothiazol, was insensitive to Ry, EGTA or inhibitors of mitochondrial Ca2+ uptake. All these treatments were instead effective in CRDM.
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Treatments
4.3. Measurement of AA Content
4.4. MitoSOX Red Oxidation
4.5. Aconitase Activity
4.6. Measurement of DNA Single-Strand Breakage by the Alkaline Halo Assay
4.7. Cytotoxicity Assay
4.8. Measurement of Mitochondrial Ca2+
4.9. MitoTracker Red CMXRos, Calcein Staining and Imaging
4.10. Measurement of Oxygen Consumption
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Szabó, E.; Gergely, P.; Szabó, C. Peroxynitrite-induced cytotoxicity: Mechanism and opportunities for intervention. Toxicol. Lett. 2003, 140–141, 113–124. [Google Scholar] [CrossRef]
- Szabó, C.; Ohshima, H. DNA damage induced by peroxynitrite: Subsequent biological effects. Nitric Oxide 1997, 1, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Tommasini, I.; Fiorani, M.; Cantoni, O. Essential role of the mitochondrial respiratory chain in peroxynitrite-induced strand scission of genomic DNA. IUBMB Life 2000, 50, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Sciorati, C.; Clementi, E.; Cantoni, O. Peroxynitrite mobilizes calcium ions from ryanodine-sensitive stores, a process associated with the mitochondrial accumulation of the cation and the enforced formation of species mediating cleavage of genomic DNA. Free Radic. Biol. Med. 2006, 41, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Fiorani, M.; Azzolini, C.; Cantoni, O. A novel mechanism, uniquely dependent on mitochondrial calcium accumulation, whereby peroxynitrite promotes formation of superoxide/hydrogen peroxide and the ensuing strand scission of genomic DNA. Antioxid. Redox Signal. 2010, 13, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Cerioni, L.; Fiorani, M.; Cantoni, O. Differentiation-associated loss of ryanodine receptors: A strategy adopted by monocytes/macrophages to prevent the DNA single-strand breakage induced by peroxynitrite. J. Immunol. 2009, 183, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Cerioni, L.; Cantoni, O. Inhibition of complex III promotes loss of Ca2+ dependence for mitochondrial superoxide formation and permeability transition evoked by peroxynitrite. J. Cell Sci. 2007, 120, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Vitamin C: Antioxidant or pro-oxidant in vivo? Free Radic. Res. 1996, 25, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Cerioni, L.; Fiorani, M.; Azzolini, C.; Cantoni, O. Mitochondrial ascorbic acid is responsible for enhanced susceptibility of U937 cells to the toxic effects of peroxynitrite. Biofactors 2014, 40, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Azzolini, C.; Cerioni, L.; Guidarelli, A.; Cantoni, O. Superoxide dictates the mode of U937 cell ascorbic acid uptake and prevents the enhancing effects of the vitamin to otherwise nontoxic levels of reactive oxygen/nitrogen species. J. Nutr. Biochem. 2013, 24, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Azzolini, C.; Cerioni, L.; Scotti, M.; Guidarelli, A.; Ciacci, C.; Cantoni, O. The mitochondrial transporter of ascorbic acid functions with high affinity in the presence of low millimolar concentrations of sodium and in the absence of calcium and magnesium. Biochim. Biophys. Acta 2015, 1848, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Hasko, G.; Hawkins, B.J.; Madesh, M.; Pacher, P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc. 2007, 2, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Scandroglio, F.; Tortora, V.; Radi, R.; Castro, L. Metabolic control analysis of mitochondrial aconitase: Influence over respiration and mitochondrial superoxide and hydrogen peroxide production. Free Radic. Res. 2014, 48, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J.; Levine, R.L.; Sohal, R.S. Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl. Acad. Sci. USA 1997, 94, 11168–11172. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Methods Enzymol. 2002, 349, 9–23. [Google Scholar] [PubMed]
- Azzolini, C.; Fiorani, M.; Cerioni, L.; Guidarelli, A.; Cantoni, O. Sodium-dependent transport of ascorbic acid in U937 cell mitochondria. IUBMB Life 2013, 65, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. Intracellular calcium homeostasis. Annu. Rev. Biochem. 1987, 56, 395–433. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Reed, D.J. Effect of extracellular Ca++ omission on isolated hepatocytes. II. Loss of mitochondrial membrane potential and protection by inhibitors of uniport Ca++ transduction. J. Pharmacol. Exp. Ther. 1988, 245, 501–507. [Google Scholar] [PubMed]
- Halestrap, A.P.; Connern, C.P.; Griffiths, E.J.; Kerr, P.M. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell Biochem. 1997, 174, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Henke, W.; Jung, K. Comparison of the effects of the immunosuppressive agents FK 506 and cyclosporin A on rat kidney mitochondria. Biochem. Pharmacol. 1993, 46, 829–832. [Google Scholar] [CrossRef]
- Tommasini, I.; Sestili, P.; Cantoni, O. Delayed formation of hydrogen peroxide mediates the lethal response evoked by peroxynitrite in U937 cells. Mol. Pharmacol. 2002, 61, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Guidarelli, A.; Fiorani, M.; Azzolini, C.; Cerioni, L.; Scotti, M.; Cantoni, O. U937 cell apoptosis induced by arsenite is prevented by low concentrations of mitochondrial ascorbic acid with hardly any effect mediated by the cytosolic fraction of the vitamin. Biofactors 2015, 41, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine. In Free Radicals in Biology and Medicine, 3rd ed.; Halliwell, B., Gutteridge, J.M.C., Eds.; Oxford University Press: Oxford, UK, 1999; pp. 1–25. [Google Scholar]
- Halliwell, B. Oxidative stress in cell culture: An under-appreciated problem? FEBS Lett. 2003, 540, 3–6. [Google Scholar] [CrossRef]
- Halliwell, B. Cell culture, oxidative stress, and antioxidants: Avoiding pitfalls. Biomed. J. 2014, 37, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Leshin, J.A.; Levine, M. Parenteral ascorbate as a cancer therapeutic: A reassessment based on pharmacokinetics. Antioxid. Redox Signal. 2013, 19, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Fritz, H.; Flower, G.; Weeks, L.; Cooley, K.; Callachan, M.; McGowan, J.; Skidmore, B.; Kirchner, L.; Seely, D. Intravenous vitamin C and cancer: A systematic review. Integr. Cancer Ther. 2014, 13, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Cadrobbi, J.; Marques, C.; Taper, H.; Habraken, Y.; Piette, J.; Calderon, P.B. Ascorbate potentiates the cytotoxicity of menadione leading to an oxidative stress that kills cancer cells by a non-apoptotic caspase-3 independent form of cell death. Apoptosis 2004, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Jones, G.D. Vitamin C modulation of H2O2-induced damage and iron homeostasis in human cells. Free Radic. Biol. Med. 2007, 43, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, C.M.; Badisa, R.B.; Soliman, K.F. Effect of ascorbic acid and hydrogen peroxide on mouse neuroblastoma cells. Mol. Med. Rep. 2012, 5, 1449–1452. [Google Scholar] [PubMed]
- Nienhuis, A.W.; Griffith, P.; Strawczynski, H.; Henry, W.; Borer, J.; Leon, M.; Anderson, W.F. Evaluation of cardiac function in patients with thalassemia major. Ann. N. Y. Acad. Sci. 1980, 344, 384–396. [Google Scholar] [CrossRef] [PubMed]
- McLaran, C.J.; Bett, J.H.; Nye, J.A.; Halliday, J.W. Congestive cardiomyopathy and haemochromatosis-rapid progression possibly accelerated by excessive ingestion of ascorbic acid. Aust. N. Z. J. Med. 1982, 12, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Gerster, H. High-dose vitamin C: A risk for persons with high iron stores? Int. J. Vitam. Nutr. Res. 1999, 69, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.; Jacobs, C.; Kaminski, T.; Halliwell, B.; Leeuwenburgh, C. Supplementation with vitamin C and N-acetyl-cysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic. Biol. Med. 2001, 31, 745–753. [Google Scholar] [CrossRef]
- Bryer, S.C.; Goldfarb, A.H. Effect of high dose vitamin C supplementation on muscle soreness, damage, function, and oxidative stress to eccentric exercise. Int. J. Sport Nutr. Exerc. Metab. 2006, 16, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Davies, M.J.; Webster, N.R. Ascorbyl radical formation in patients with sepsis: Effect of ascorbate loading. Free Radic. Biol. Med. 1996, 20, 139–143. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, G.; Craig, S.W.; Decker, G.L.; Lehninger, A.L. The cytoskeleton of digitonin-treated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1980, 77, 3430–3434. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Duflot, S.; Avigliano, L. Dehydroascorbic acid uptake in a human keratinocyte cell line (HaCaT) is glutathione-independent. Biochem. J. 2000, 345, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Guidarelli, A.; Blasa, M.; Azzolini, C.; Candiracci, M.; Piatti, E.; Cantoni, O. Mitochondria accumulate large amounts of quercetin: Prevention of mitochondrial damage and release upon oxidation of the extramitochondrial fraction of the flavonoid. J. Nutr. Biochem. 2010, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cruz, I.; Carcamo, J.M.; Golde, D.W. Vitamin C inhibits FAS-induced apoptosis in monocytes and U937 cells. Blood 2003, 102, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cobb, C.E.; Hill, K.E.; Burk, R.F.; May, J.M. Mitochondrial uptake and recycling of ascorbic acid. Arch. Biochem. Biophys. 2001, 387, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, O.; Guidarelli, A. Indirect mechanisms of DNA strand scission by peroxynitrite. Methods Enzymol. 2008, 440, 111–120. [Google Scholar] [PubMed]
- Trollinger, D.R.; Cascio, W.E.; Lemasters, J.J. Mitochondrial calcium transients in adult rabbit cardiac myocytes: Inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca2+-indicating fluorophores. Biophys. J. 2000, 79, 39–50. [Google Scholar] [CrossRef]
- Robinson, J.; Cooper, J.M. Method of determining oxygen concentrations in biological media, suitable for calibration of the oxygen electrode. Anal. Biochem. 1970, 33, 390–399. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guidarelli, A.; Cerioni, L.; Fiorani, M.; Cantoni, O. Intramitochondrial Ascorbic Acid Enhances the Formation of Mitochondrial Superoxide Induced by Peroxynitrite via a Ca2+-Independent Mechanism. Int. J. Mol. Sci. 2017, 18, 1686. https://doi.org/10.3390/ijms18081686
Guidarelli A, Cerioni L, Fiorani M, Cantoni O. Intramitochondrial Ascorbic Acid Enhances the Formation of Mitochondrial Superoxide Induced by Peroxynitrite via a Ca2+-Independent Mechanism. International Journal of Molecular Sciences. 2017; 18(8):1686. https://doi.org/10.3390/ijms18081686
Chicago/Turabian StyleGuidarelli, Andrea, Liana Cerioni, Mara Fiorani, and Orazio Cantoni. 2017. "Intramitochondrial Ascorbic Acid Enhances the Formation of Mitochondrial Superoxide Induced by Peroxynitrite via a Ca2+-Independent Mechanism" International Journal of Molecular Sciences 18, no. 8: 1686. https://doi.org/10.3390/ijms18081686