Enhanced Thermostability of Glucose Oxidase through Computer-Aided Molecular Design

 , and
, and
Abstract
:1. Introduction
2. Results
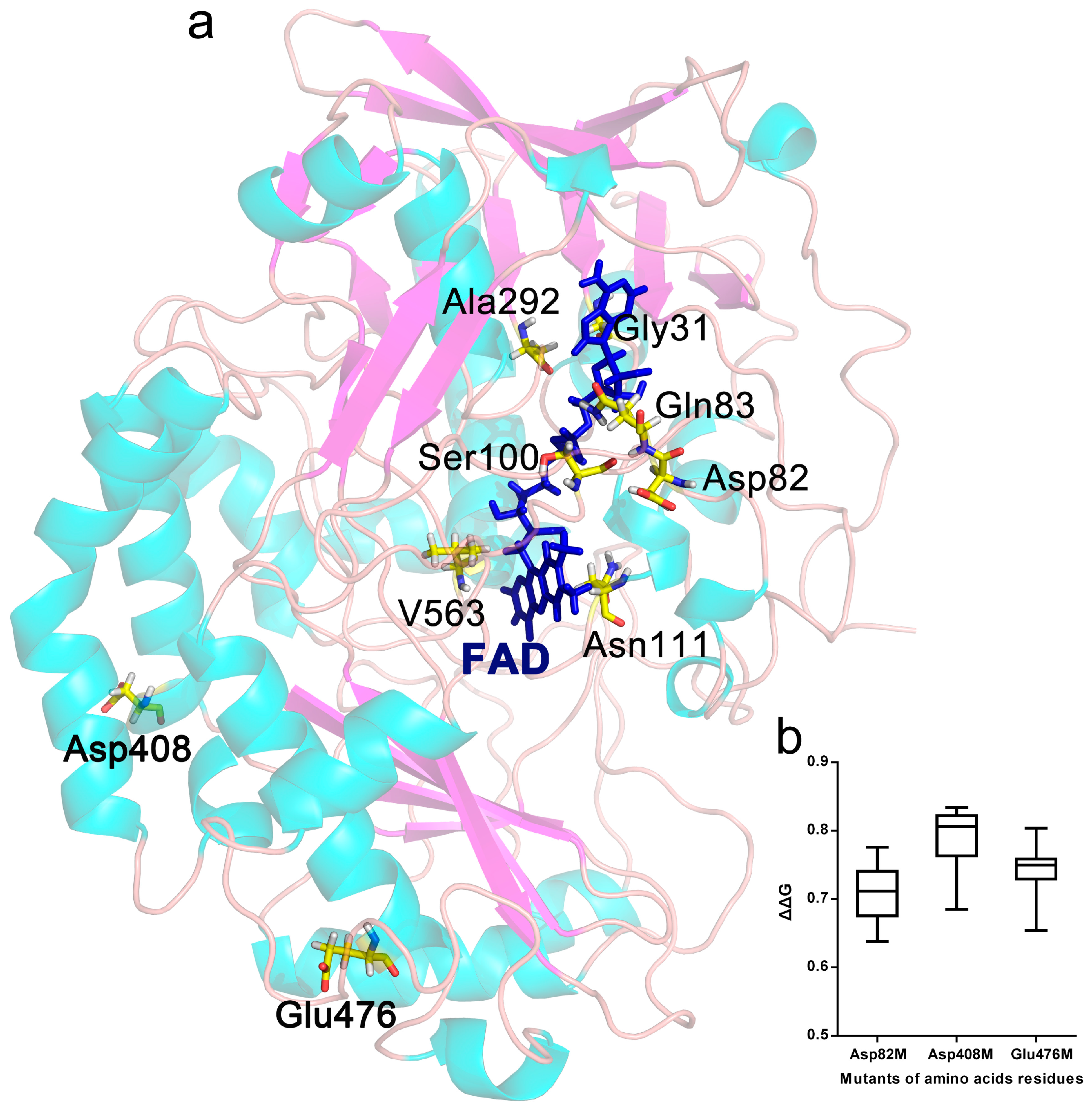
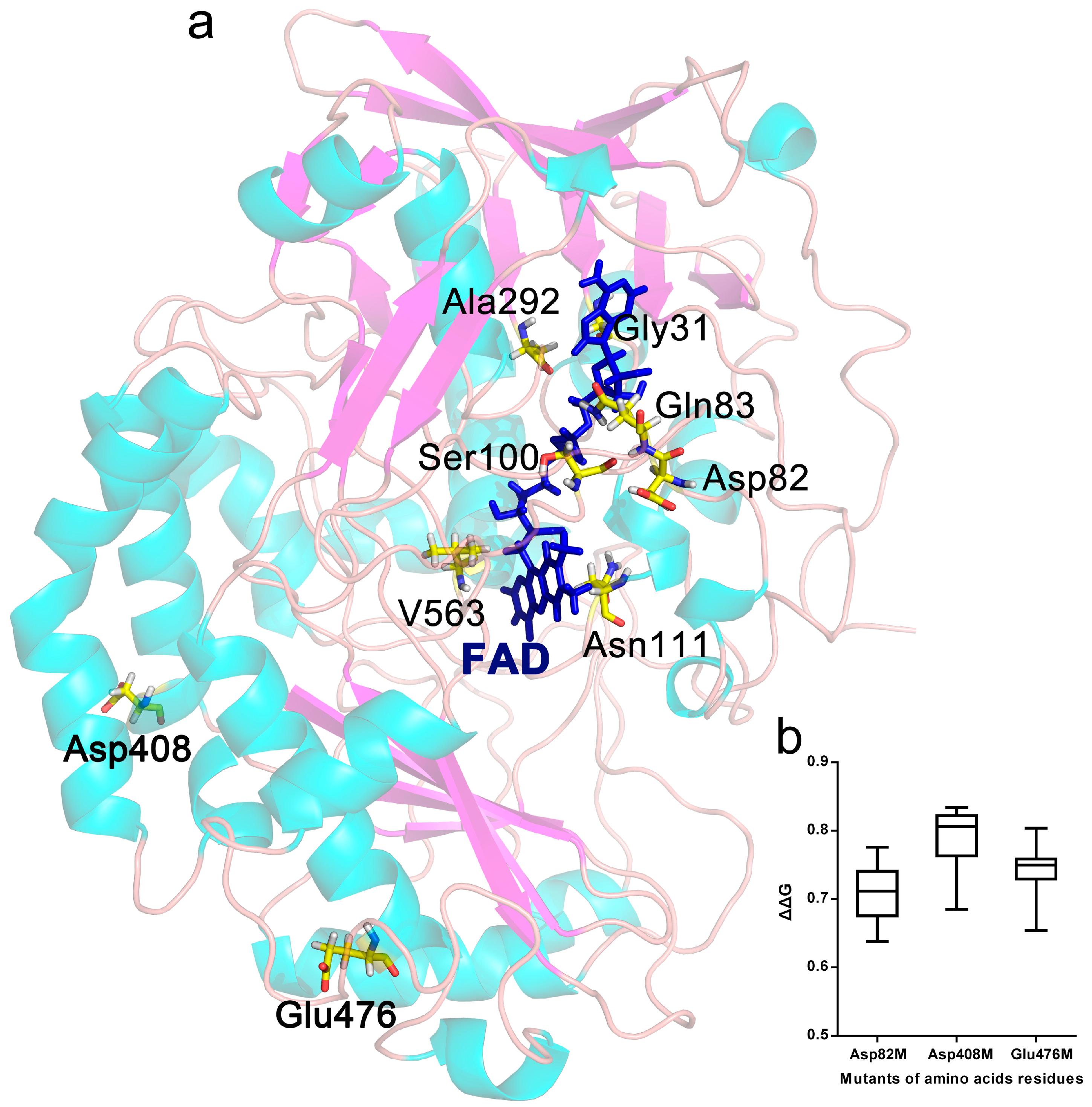
2.1. Selection of Mutant Sites
2.2. Mutant Library Construction
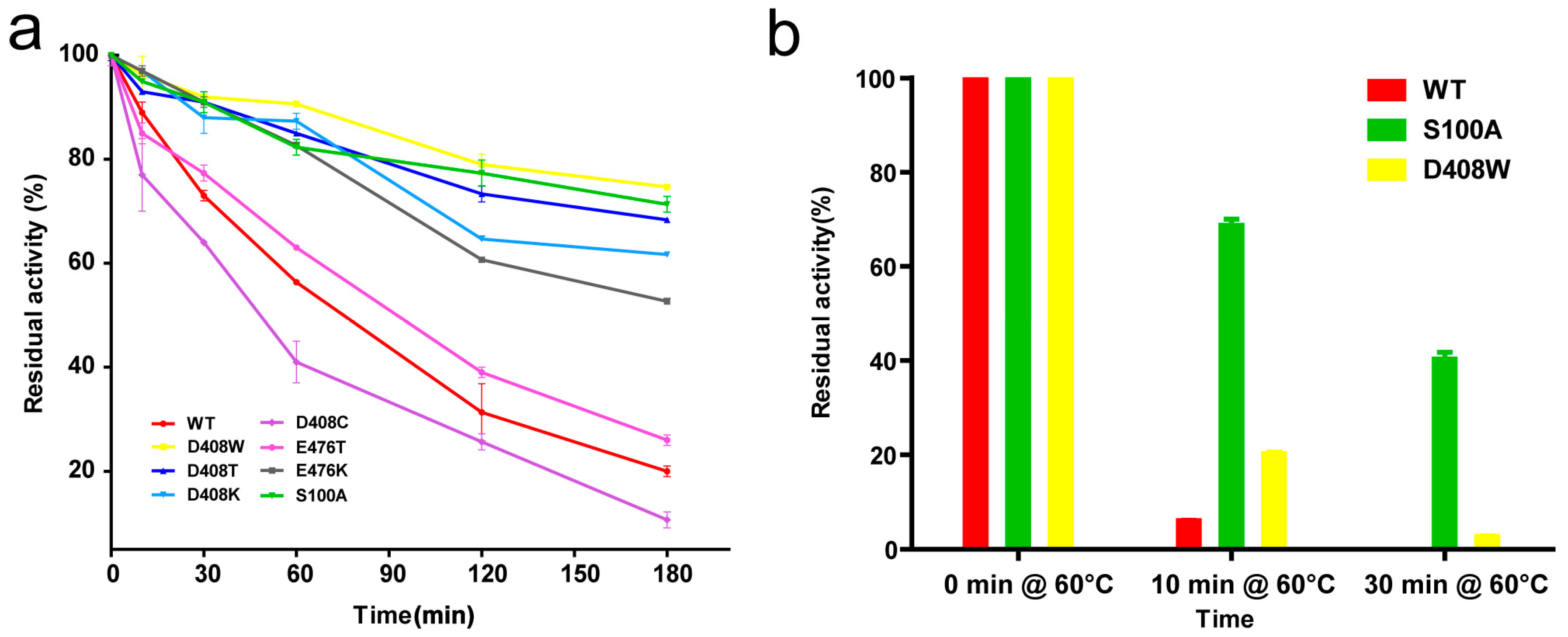
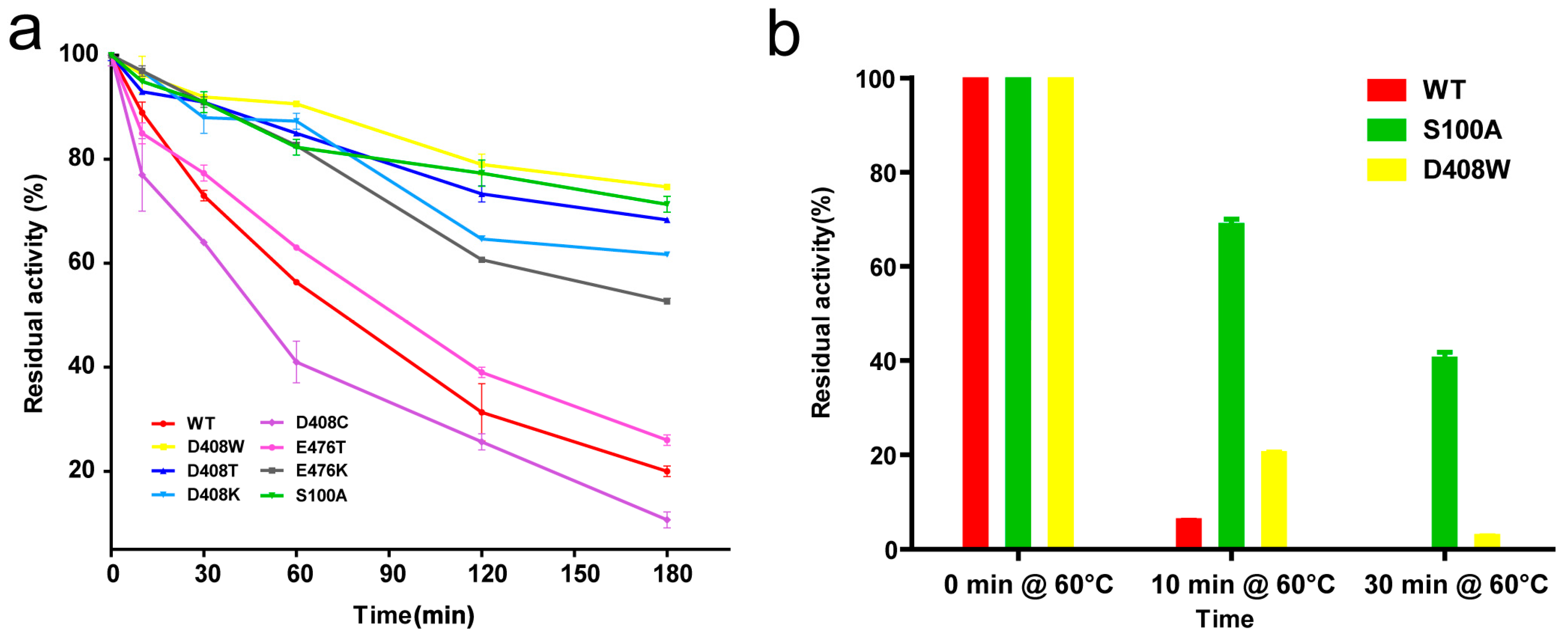
2.3. Thermostable Mutant Selection
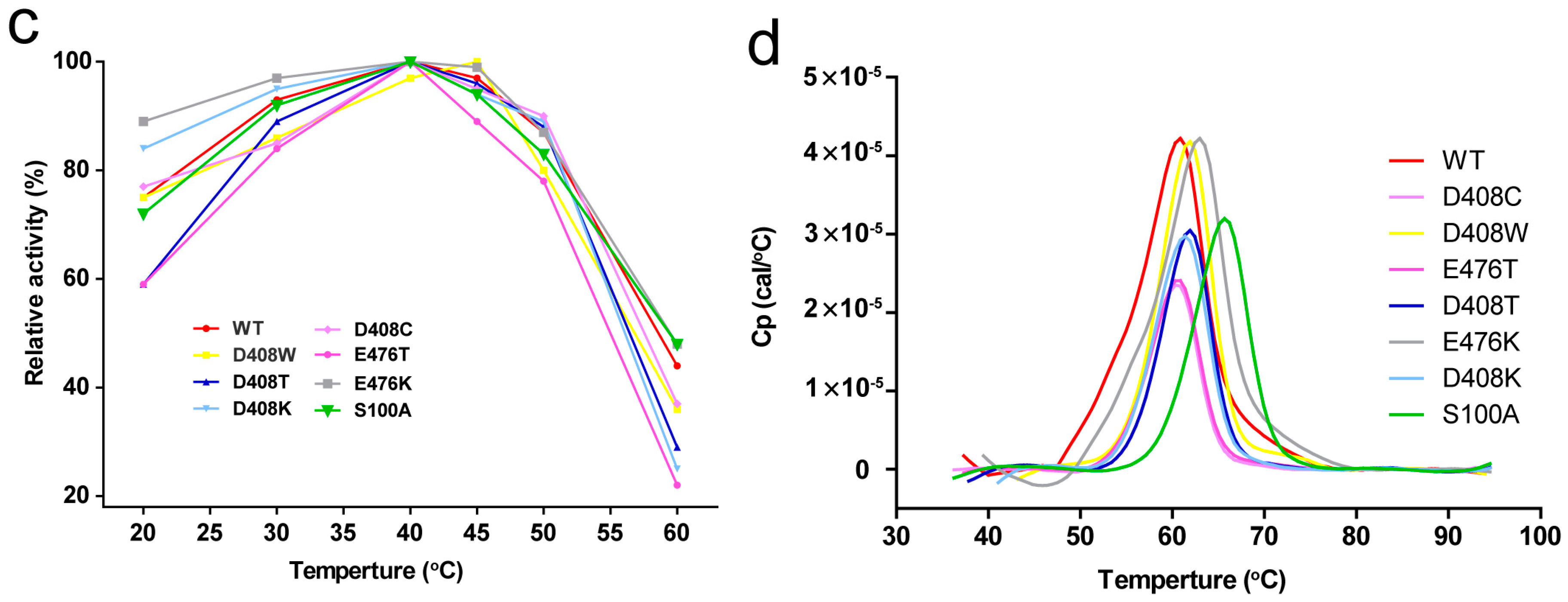
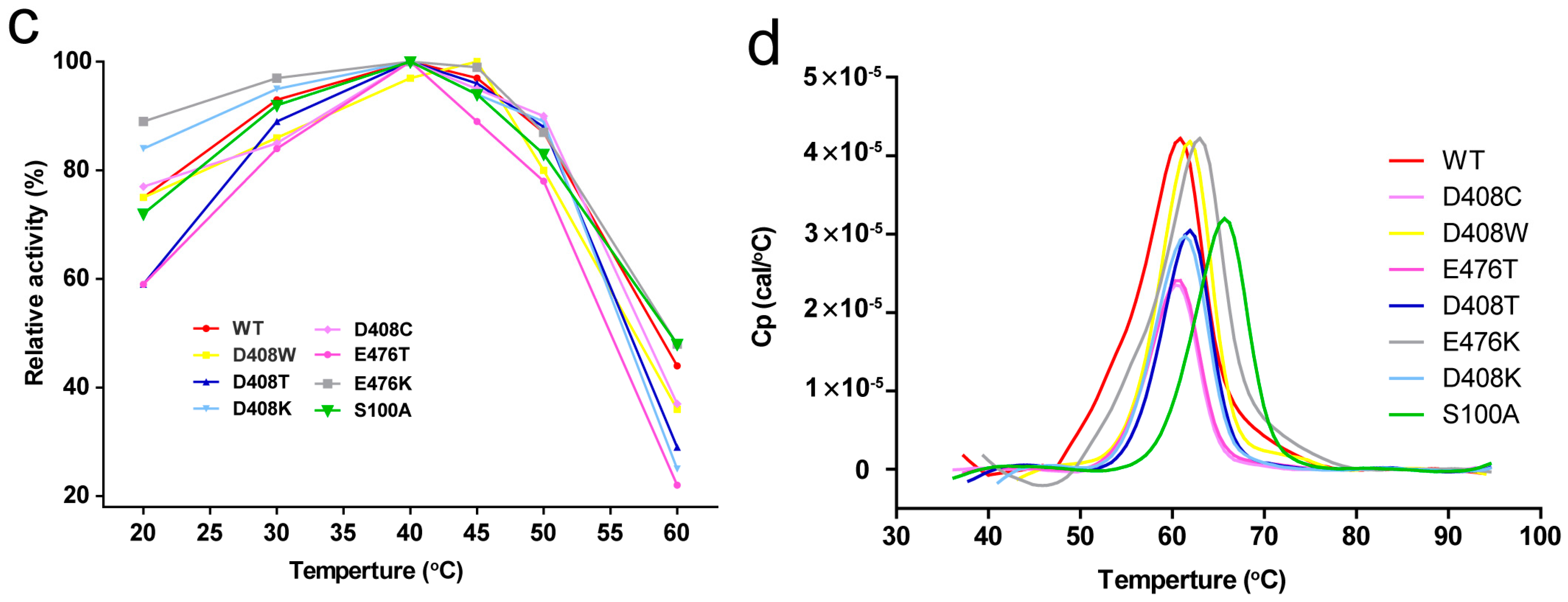
2.4. Thermostability and Optimum Temperature of the Mutants
2.5. Determination of Tm
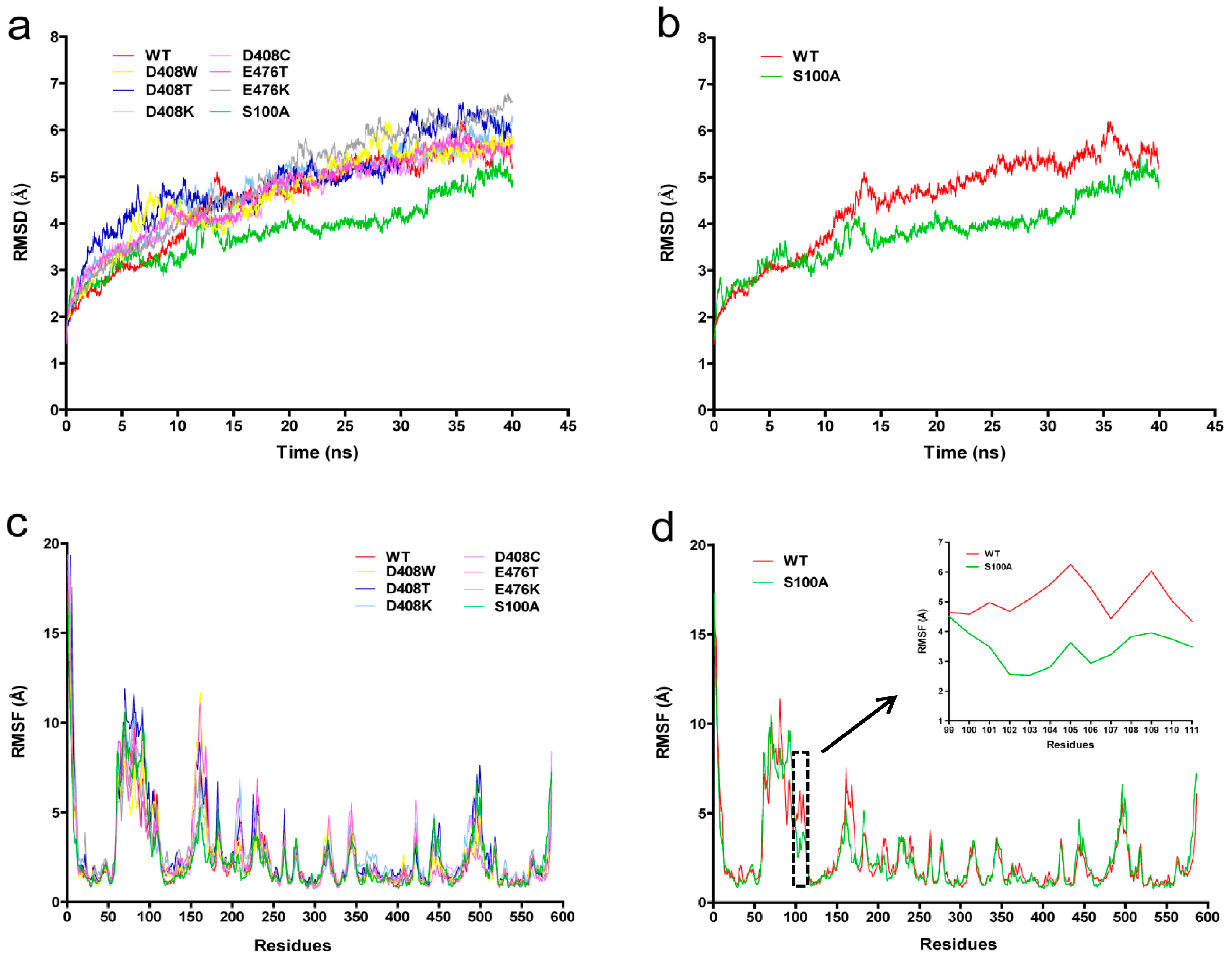
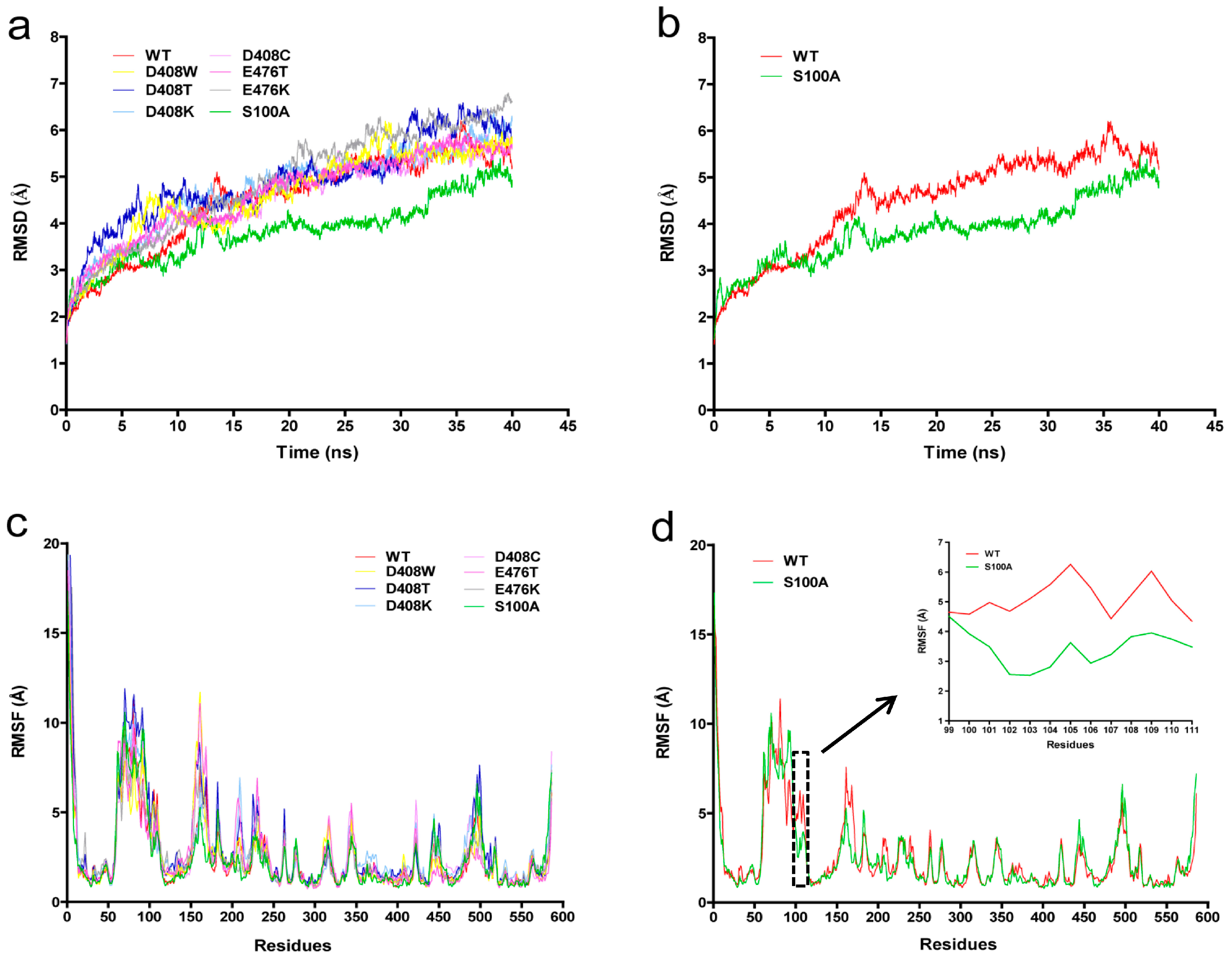
2.6. Protein Structure Analysis
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmids, and Media
4.2. Mutant Site Selection
4.3. Plasmid Construction
4.4. Positive Transformant Screening
4.5. Protein Expression and Purification
4.6. Enzyme Assay
4.7. Thermostability Assay
4.8. Mutant Protein Structure Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bankar, S.B.; Bule, M.V.; Singhal, R.S.; Ananthanarayan, L. Glucose oxidase—An overview. Biotechnol. Adv. 2009, 27, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Huo, F.; Wang, Y.; Fu, S. Application progresses of glucose oxidase (GOD) in feed. Anim. Husb. Feed Sci. 2015, 7, 298–300. [Google Scholar]
- Hatzinikolaou, D.G.; Macris, B.J. Factors regulating production of glucose oxidase by Aspergillus niger. Enzyme Microb. Tech. 1995, 17, 530–534. [Google Scholar] [CrossRef]
- Petruccioli, M.; Federici, F.; Bucke, C.; Keshavarz, T. Enhancement of glucose oxidase production by Penicillium variabile p16. Enzyme Microb. Technol. 1999, 24, 397–401. [Google Scholar] [CrossRef]
- Bonet, A.; Rosell, C.M.; Caballero, P.A.; Gómez, M.; Pérez-Munuera, I.; Lluch, M.A. Glucose oxidase effect on dough rheology and bread quality: A study from macroscopic to molecular level. Food Chem. 2006, 99, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Savas Anastassiadis, I.G.M. Gluconic acid production. Recent Pat. Biotechnol. 2007, 1, 167–180. [Google Scholar] [CrossRef]
- Kalisz, H.M.; Hendle, J.; Schmid, R.D. Structural and biochemical properties of glycosylated and deglycosylated glucose oxidase from Penicillium amagasakiense. Appl. Microbiol. Biotechnol. 1997, 47, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.T.; Harper, J.C.; Dolan, P.L.; Manginell, M.M.; Arango, D.C.; Rawlings, J.A.; Apblett, C.A.; Brozik, S.M. Rational redesign of glucose oxidase for improved catalytic function and stability. PLoS ONE 2012, 7, e37924. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Benedix, A.; Becker, C.M.; de Groot, B.L.; Caflisch, A.; BöCkmann, R.A. Predicting free energy changes using structural ensembles. Nat. Methods 2009, 6, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razvi, A.; Scholtz, J.M. Lessons in stability from thermophilic proteins. Protein Sci. 2006, 15, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Marín-Navarro, J.; Roupain, N.; Talens-Perales, D.; Polaina, J. Identification and structural analysis of amino acid substitutions that increase the stability and activity of Aspergillus niger glucose oxidase. PLoS ONE 2015, 10, e0144289. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, P.; Huang, L.; Chu, X.; Wu, N.; Fan, Y. Improving the thermostability of methyl parathion hydrolase from Ochrobactrum sp. M231 using a computationally aided method. Appl. Microbiol. Biotechnol. 2013, 97, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; An, J.; Yang, G.; Wu, G.; Zhang, Y.; Cui, L.; Feng, Y. Enhanced enzyme kinetic stability by increasing rigidity within the active site. J. Biol. Chem. 2014, 289, 7994–8006. [Google Scholar] [CrossRef] [PubMed]
- Hatzinikolaou, D.G.; Hansen, O.C.; Macris, B.J.; Tingey, A.; Kekos, D.; Goodenough, P.; Stougaard, P. A new glucose oxidase from Aspergillus niger: Characterization and regulation studies of enzyme and gene. Appl. Microbiol. Biotechnol. 1996, 46, 371–381. [Google Scholar] [PubMed]
- Kiess, M.; Hecht, H.J.; Kalisz, H.M. Glucose oxidase from Penicillium amagasakiense. Eur. J. Biochem. 1998, 252, 90. [Google Scholar] [CrossRef] [PubMed]
- O'Malley, J.J.; Weaver, J.L. Subunit structure of glucose oxidase from Aspergillus niger. Biochemistry 1972, 11, 3527–3532. [Google Scholar] [CrossRef] [PubMed]
- Caves, M.S.; Derham, B.K.; Jezek, J.; Freedman, R.B. The mechanism of inactivation of glucose oxidase from Penicillium amagasakiense under ambient storage conditions. Enzyme Microb. Technol. 2011, 49, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Garajová, K.; Zimmermann, M.; Petrenčáková, M.; Dzurová, L.; Nemergut, M.; Škultéty, Ľ.; Žoldák, G.; Sedlák, E. The molten-globule residual structure is critical for reflavination of glucose oxidase. Biophys. Chem. 2017, 230, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, Z.; Zhang, Y.; Huang, H.; Li, M.; Zhou, L.; Tang, Y.; Yao, B.; Zhang, W. High-level expression of the Penicillium notatum glucose oxidase gene in Pichia pastoris using codon optimization. Biotechnol. Lett. 2012, 34, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Thaqi, M.; Zenelaj, I.; Jaka, F. Testing of pelleting efficiency, quality and effect of pelleted feed on broiler performance. In Proceedings of the 36th International Symposium ‘Actual tasks on agricultural engineering’, Opatija, Croatia, 10–13 February 2009; pp. 527–537. [Google Scholar]
- He, C.; Liu, J.; Xie, L.; Zhang, Q.; Li, C.; Gui, D.; Zhang, G.; Wu, C. Activity and thermal stability improvements of glucose oxidase upon adsorption on core-shell pmma-bsa nanoparticles. Langmuir 2009, 25, 13456–13460. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, M.; Gautam, A.; Nazor, J.; Momeu, C.; Prodanovic, R.; Schwaneberg, U. Directed evolution of glucose oxidase from Aspergillus niger for ferrocenemethanol-mediated electron transfer. Biotechnol. J. 2007, 2, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Ostafe, R.; Prodanovic, R.; Nazor, J.; Fischer, R. Ultra-high-throughput screening method for the directed evolution of glucose oxidase. Chem. Biol. 2014, 21, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Wohlfahrt, G.; Witt, S.; Hendle, J.; Schomburg, D.; Kalisz, H.M.; Hecht, H.J. 1.8 and 1.9 angstrom resolution structures of the Penicillium amagasakiense and Aspergillus niger glucose oxidases as a basis for modelling substrate complexes. Acta Crystallogr. D 1999, 55, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.A.; Wang, P.; Gao, S.; Chu, X.Y.; Wu, N.F.; Fan, Y.L. Enhanced thermostability of methyl parathion hydrolase from Ochrobactrum sp. M231 by rational engineering of a glycine to proline mutation. FEBS J. 2010, 277, 4901–4908. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.A.; de Marco, A.; Fortuna, S. Molecular dynamics simulations and docking enable to explore the biophysical factors controlling the yields of engineered nanobodies. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Picone, D. Molecular dynamics driven design of pH-stabilized mutants of MNEI, a sweet protein. PLoS ONE 2016, 11, e0158372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Wu, N.; Chu, X.; Fan, Y. Predicting changes in protein thermostability brought about by single- or multi-site mutations. BMC Bioinform. 2010, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- Valdivieso-Ugarte, M.; Ronchel, C.; Banuelos, O.; Velasco, J.; Adrio, J.L. Expression of an Aspergillus niger glucose oxidase in Saccharomyces cerevisiae and its use to optimize fructo-oligosaccharides synthesis. Biotechnol. Prog. 2006, 22, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Liu, B.; Zhang, Y.; Zhao, G.; Fan, X.; Ning, X.; Zhang, W. Production and secretion of Lactobacillus crispatus beta-galactosidase in Pichia pastoris. Protein Expr. Purif. 2013, 92, 88–93. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | t1/2 (min) at 55 °C | Tm (°C) |
|---|---|---|
| WT | 75.3 | 60.7 |
| D408W | 407.7 | 61.9 |
| D408T | 315.1 | 61.7 |
| D408K | 239.0 | 61.6 |
| D408C | 57.3 | 60.1 |
| E476T | 110.0 | 60.9 |
| E476K | 216.6 | 62.0 |
| S100A | 462.1 | 65.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, X.; Zhang, Y.; Yuan, T.; Li, Q.; Tian, J.; Guan, W.; Liu, B.; Zhang, W.; Xu, X.; Zhang, Y. Enhanced Thermostability of Glucose Oxidase through Computer-Aided Molecular Design. Int. J. Mol. Sci. 2018, 19, 425. https://doi.org/10.3390/ijms19020425
Ning X, Zhang Y, Yuan T, Li Q, Tian J, Guan W, Liu B, Zhang W, Xu X, Zhang Y. Enhanced Thermostability of Glucose Oxidase through Computer-Aided Molecular Design. International Journal of Molecular Sciences. 2018; 19(2):425. https://doi.org/10.3390/ijms19020425
Chicago/Turabian StyleNing, Xiaoyan, Yanli Zhang, Tiantian Yuan, Qingbin Li, Jian Tian, Weishi Guan, Bo Liu, Wei Zhang, Xinxin Xu, and Yuhong Zhang. 2018. "Enhanced Thermostability of Glucose Oxidase through Computer-Aided Molecular Design" International Journal of Molecular Sciences 19, no. 2: 425. https://doi.org/10.3390/ijms19020425