Ab Initio Calculations of Co Shielding in Model Complexes

Department of Chemistry, The Open University, Milton Keynes, MK7 6AA, UK
Int. J. Mol. Sci. 2002, 3(8), 873-887; https://doi.org/10.3390/i3080873
Submission received: 26 November 2001
/
Accepted: 10 May 2002
/
Published: 31 August 2002
(This article belongs to the Special Issue Recent Advances in Nuclear Magnetic Shielding Theory)
Abstract
:Recent ab initio calculations of cobalt NMR shielding show that DFT-GIAO calculations using hybrid functionals are found to reproduce experimental values well. This method is used to calculate the variation of the cobalt NMR shielding tensor of sqaure pyramidal nitrosyl complexes with respect to the CoNO geometry and to differing basal ligands. The isotropic shielding is shown to have a large negative derivative with respect to CoX distance where X is a ligating atom.; the derivative with respect to NO distance is smaller but still significant. The zz component where z is along the CoN(NO) bond is more sensitive to the basal ligands but the other two principal components are sensitive to the CoNO geometry.
Introduction
The scope of cobalt NMR spectroscopy was illustrated in a recent review [1]. 59Co was one of the first nuclei to be studied using NMR spectroscopy. It is a nucleus with a high receptivity and the largest known chemical shift range of around 20 000 ppm. There are a large number of stable low-spin Co(III) complexes which are diamagnetic and hence well-suited to NMR studies. 59Co chemical shifts were also the subject of early theoretical considerations. Ramsey [2] derived an approximate formula for the paramagnetic term in the shielding of the cobalt atom in terms of ligand field theory.
where ΔE is the average excitation energy from the ground state to the excited states linked by the shielding operator, replaced in this case by the ligand field transition (1A1g→1T1g in the case of octahedral complexes) to a low-lying excited state. This led to a prediction that 59Co shieldings would correlate with the ligand field splitting energies, Δo, of cobalt complexes and such a correlation was found for measured spectra of simple complexes [3]. In later work [4,5,6], the r-3 term and the orbital angular momentum integral which determine the covalency or nephelauxetic effect were also shown to be important. In a series of Co(III) pentammine complexes, the angular momentum integral was shown to be of negligible influence in determining the change in chemical shift and a plot of isotropic chemical shift gave a better correlation with the ratio of the nephelauxetic factor, β35, to the excitation energy than with the reciprocal of the excitation energy alone [7]. The nephelauxetic factor, β, is a function of interelectron repulsion and is defined as the ratio of the Racah B parameter (obtained from optical spectra) in the complex and in the gaseous metal ion. Recently [8], Chan and Au-Yeung have shown that the angular momentum integral is related to the nuclear quadrupole coupling constant, QCC. For 59Co solution spectra, quadrupole relaxation is the dominant mechanism and QCC can be obtained from the half-width of the resonance. Correlations were obtained for complexes [Co(NH3)5X](3+n)+, [Co(CN)5X](2-n)- and trans-[Co(en)2X2](3+n)+ between the 59Co chemical shift and (Δν1/2)1/2/ΔEav where Δν1/2 is the solution linewidth and ΔEav is the energy of the 1A1g→1T1g transition. This suggests that the angular momentum integral is important in determining the chemical shift. The data for each type of complex lay on two lines, one for complexes with hard ligands, X, and one for those where X is a soft ligand. The differences between the hard and soft ligands is attributed to the increased covalency of the Co-X bond for soft ligands which causes a quenching of the angular momentum.
Full ab initio calculations of 59Co chemical shieldings have only recently been feasible however. The low energy excitations which give rise to the large chemical shifts observed and to the simplification of the denominator in Ramsey's formula pose problems for ab initio calculations and gave rise to large errors in Hartree-Fock calculations. For molecules containing light elements, the introduction of MP2 correlation corrections [e.g. 9,10] and multi-configurational methods [e.g. 11] have led to considerable success in calculating chemical shielding tensors in cases where there are low excitation energies. For a transition metal complex, however, very large computer resources would be needed for such methods. The advent of density functional methods has made possible the ab initio calculation of NMR shielding of transition metal complexes with reasonable accuracy with less computer resource.
The first density functional (DFT) calculations on cobalt complexes [12] were performed using the SOS-DFT-IGLO method of Malkin et al [13] in the deMon suite of programs [14,15,16]. The complexes studied covered both neutral and ionic species - [Co(CN)6]3- [Co(NO2)6]3-, [Co(NH3)6]3+, [Co(en)3]3+, [Co(NH3)4CO3]+and [Co(acac)3]. The best results were found for the Becke-Perdew [17] exchange correlation potential with the TZVP basis set of Malkin et al [13,14,15,16,18,19] on the cobalt and the IGLO-II basis set of Kutzelnigg [20] on he surrounding ligands. Although a considerable improvement on previous semi-empirical calculations [21], the calculated values were considerably more shielded than the experimental values e.g. -3159ppm for [Co(CN)6]3- compared to the currently accepted absolute shielding of -5400 ppm [6]. Possible reasons for the discrepancy were put forward including absence of 4f functions in the Co basis set and the description of the excited state in DFT theory.
Chan and Au-Yeung [22] compared the DFT-IGLO method used in this study with the DFT-GIAO option in Gaussian94 [23] using the Becke-Perdew functional on 13 hexacoordinated complexes of Co(III) involving the ligands en, NH3, SCN-,N3-, H2O, NO2-, CN-, Cl-, NO, ONO-, CO32- and S2O32-. They concluded that DFT-GIAO produced results closer to experiment than DFT-IGLO. Using the DFT-GIAO method with the hybrid functional B3LYP gave results even closer to experiment. A subsequent paper [24] calculated chemical shift spans and skews for several complexes using the DFT-GIAO-B3LYP method and 6-311G basis sets with varying numbers of polarisation functions and obtained satisfactory agreement with experiment.
More recent studies of the complexes studied in ref. 12 [25] using DFT-GIAO-B3LYP in Gaussian94 with Wachters basis set (62111111/3311111/3111) [26,27] on cobalt and the 6-31G* basis set on the ligand atoms also gave reasonable results. These authors found that the addition of f functions to the cobalt basis set and the use of larger basis sets gave little improvement.
The DFT-GIAO studies not only reproduce the chemical shift well but also yielded a remarkably good value for the absolute shielding of [Co(CN)6]3-. Godbout and Oldfield [25] plotted the calculated absolute chemical shieldings against the experimental chemical shifts and obtained a value for the absolute shielding of -5162 ppm as the intercept compared to the experimentally determined value of -5400 ppm.
The success of hybrid DFT methods has been analysed [28] and attributed to more diffuse virtual orbitals, coupling due to Hartree-Fock exchange and the increase of the HOMO-LUMO gap relative to that obtained using pure DFT methods.
Because of their use as models for biological systems such as Vitamin B12 and the isoelectronic Fe(II) hemes, cobalt complexes with macrocyclic ligands such as porphins have been of considerable recent interest. A recent paper [29] reports a calculation of cobalt chemical shielding for the porphin complex [Co(TDCPP)(MeIm)2]+ in which the TDCPP (tetra(dichlorophenyl)porphyrin) group forms a plane around the cobalt and two methylimidazole groups occupy positions above and below this plane. The calculations used the DFT-GIAO-B3LYP method with a 6-311G basis set on the lighter atoms, the Maclean-Chandler basis set [30,31] on chlorine and the Wachters-Hay all-electron basis set [26,32] for cobalt. Although chemical shift tensors are more difficult to calculate than the isotropic shift, these authors obtained very close agreement with the experimental chemical shift tensor. Zhou et al [33] considered a set of complexes in which [Co(CN)6]3- was bound to protonated polyammonium macrocyclics. They reproduced the 59Co chemical shifts by altering the Co-C and C≡N distances and the symmetry of [Co(CN)6]3- to fit the geometry found in the macrocycles.
There have been some studies of how 59Co shielding varies with changes in geometry. Experimentally the variation of shielding with geometry shows up as temperature dependence and isotope shifts. Cobalt chemical shifts have extremely large temperature dependence and large isotope shifts indicating a strong dependence on geometry. In simple cases, such data can be analysed to obtain derivatives of shielding with respect to bond extensions and angle deformations. By analysing isotope shifts, Jameson et al [34] found for [Co(CN)6]3- that the first order derivative of the shielding with respect to the Co-C bond distance was –7 500ppm Å-1, that is the cobalt nucleus becomes more deshielded as the Co-C distance increases. The thermal shielding derivative of [Co(NO2)6]3- has been measured [34] as -2.85ppm K-1, over twice that observed for [Co(CN)6]3-, but the vibrational motion is more complicated than that of the cyano complex and there is the possibility of internal rotation of the nitrito group so that the thermal shielding derivative is not easily linked to the derivative of the shielding with Co-N distance. Godbout and Oldfield [25] have calculated the variation of cobalt shielding with Co-C bond distance in [Co(CN)6]3- and with Co-N distance for [Co(NO2)6]3-. They used a geometry-optimised structure for the free ion with a range of Co-C or Co-N distances. In both cases, the plots were linear with slopes of –4 856 ppm Å-1 for [Co(CN)6]3- and –6 180 ppm Å-1 for [Co(NO2)6]3-. They thus correctly predict that the slopes are negative, 1-2 orders of magnitude larger than the shielding derivatives of first row atoms and that the magnitude of the derivative for the nitrito complex is larger than that for the cyano-complex.
We report here a study of 5-coordinate cobalt nitrosyl complexes with particular focus on the response of 59Co shielding to variation in the geometry of the CoNO grouping.
Square-pyramidal nitrosyl porphin complexes have been studied as models for NO and O2 binding to heme-like systems. The CoNO group can adopt two distinguishable geometries, linear with a CoNO angle of 180° and bent where the CoNO angle is about 130°, resembling the FeOO arrangement in heme. Godbout and Oldfield [25] noted that they had determined the 59Co shielding in a nitrosyl cobalt tetraphenylporphin as -13 530 ppm. They remarked that this was considerably more deshielded than anticipated. However this value yields a chemical shift of 8086 ppm which is in fact very close to the measured value [35] of 7 909 ppm for cobalt nitrosyl tetraphenylporphinate in solution.
In addition to the data on porphinato complexes, there are also experimental measurements of solution chemical shifts for Co in nitrosyl complexes with polydentate ligands coordinating through N and O and nitrosyl dithiocarbamato complexes.
Because of the lower symmetry, square-pyramidal nitrosyl complexes would be expected to show greater asymmetry than the complexes previously studied and the different components of the shielding tensor can be shown to respond differently to geometry changes.
Computational method
The structures of the model complexes used were
![Ijms 03 00873 i001]() for the porphin complexes, with a N-Co-N angle of 76° and a Co-N-C angle of 137° ,
for the porphin complexes, with a N-Co-N angle of 76° and a Co-N-C angle of 137° ,
![Ijms 03 00873 i002]()


for the Schiff base complexes with an N-Co-O angle of 89°, a Co-N-C angle of 129.5°, a Co-O-C angle of 136° and a C-C-C angle of 162° and
![Ijms 03 00873 i003]() for the dithiocarbamato-complexes, with a Co-S-C angle of 68°. All three ligands were kept planar.
for the dithiocarbamato-complexes, with a Co-S-C angle of 68°. All three ligands were kept planar.

The arrangement of the basal ligands was kept symmetrical as shown and fixed for most calculations. Optimisation gave shorter Co-basal atom distances on the side nearest the nitrosyl oxygen when these distances were allowed to vary. Full optimisation of the CoNO group generally led to a tilt of the CoN axis away from the perpendicular to the plane, and together with the shorter Co-basal atom distance and the orientation of the nitrosyl oxygen over a basal ligand atom, this allowed stabilisation of the structure through O-basal ligand atom interaction. In experimental structures, the Co-basal atom distances are kept fixed in the symmetrical position by the rest of the molecule as in the porphinates and/or by the crystal packing, so it was considered a better model to retain the symmetry of the basal ligands. In most of the results reported here, only one parameter (bond distance or angle) was varied at a time keeping all others fixed. The nitrosyl oxygen was positioned between the basal atoms for some runs and over one basal atom for others. Both arrangements are found experimentally. However we also calculated changes in shielding for dithiocarbamato complexes allowing the CoNO geometry to relax as the Co-N distance and CoNO angle were changed.
Our calculations used the DFT-GIAO-B3LYP method in Gaussian98 [36] on the DEC8400/CompaqES40 cluster, Columbus, at the Rutherford Appleton Laboratory. The basis set used was Ahrlich's VTZ basis set [37] plus polarisation functions on all atoms. Convergence was found to be easier using the same basis set on all atoms. Triple zeta basis sets, as noted in earlier work described in the Introduction, give good results for Co shielding when used with hybrid functionals. The absolute shielding of [Co(CN)6]3- using the optimised geometry with a Co-C distance of 191pm and C≡N distance of 117pm was calculated to be –5 882ppm and using the experimental geometry for K3[Co(CN)6] of Co-C 190pm and C≡N 113pm –5 360ppm with this method and basis set. These values are very close to the experimentally determined one and give us confidence in our method.
Results
The calculated shielding tensors for our model complexes are shown in Table 1 along with the Mulliken charges on Co. Ω is the span and κ the skew. In terms of the principal components of the tensor, σ11, σ22 and σ33 where σ11 ≤ σ22 ≤ σ33, Ω = σ33 - σ11and κ = 3 × (σiso - σ22)/Ω.
We define molecular axes so that the CoN(NO) bond forms the z axis and the y axis is between the basal ligands. For the majority of the porphin and thiocarbamato complexes where the CoNO group is in the xz plane and the CoNO angle is in the range 115-140°, one principal component, usually σ33 but in some cases σ22, is σyy. The 1 axis is rotated by an angle of 5-15° from the x axis towards the z axis in the xz plane for the dithiocarbamato complexes. For the porphin complexes the 1 axis is rotated by a slightly larger angle (12-20°) from the z axis towards the x axis. The Schiff base complexes and those where the CoNO group is not in the xz plane are less symmetrical and none of the principal axes coincides with x, y or z. The principal axes of the Schiff base complexes are however close to those of the porphin complexes. For large CoNO angles, σxx and σyy become more nearly equal as the complex approaches C2v symmetry.
An analysis of the d energy levels in square-pyramidal nitrosyl complexes of iridium was obtained by Mingos [38] using the Wolfsberg-Helmholtz method. Using the orientation just described, the HOMO was the a' orbital including contributions from dz2, dxz and the π* and σ orbitals on NO and the LUMO an a" orbital with contributions from dyz and the π* on NO. These would be expected to provide a large contribution to the paramagnetic shielding, particularly σxx, for Co from dz2 to dyz which will be dependent on the Co-NO bond. However there is another contribution, primarily to σzz, from dxy to dx2-y2 which does not involve NO orbitals and will be highly dependent on the Co-basal ligand interaction [39]. Thus if this latter contribution predominates we should expect little dependence of the shielding on the CoNO geometry. Our calculations on the dithiocarbamato complexes indicate that the HOMO is the dz2/NO bonding a' orbital and the LUMO the dyz/NO π∗ a" orbital. Both orbitals become lower in energy as the CoN bond distance increases or the CoNO angle decreases but the
decrease in the HOMO energy is greater so that the energy gap also decreases. The Mulliken population on Co increases slightly with CoN distance and NO distance and decreases with CoNO angle, but is mainly determined by the basal ligands, increasing as the Co-basal ligand distance increases reflecting a lowering of covalency.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| r(Co-basal atom) /pm | r(Co-N(NO))/ pm | R(NO) /pm | ∠CoNO /° | σiso/ppm | σ11/ppm | σ22/ppm | σ33/ppm | Ω/ppm | κ | Mulliken charge |
| Porphin complexes | ||||||||||
| 197.6 | 173.8 | 101 | 120 | -15542 | -21851 | -15902 | -8871 | 12900 | 0.08 | 0.243 |
| “ | “ | 112 | “ | -16057 | -21373 | -16990 | -9807 | 11566 | 0.24 | 0.259 |
| “ | “ | “ | 130 | -16528 | -21809 | -17688 | -10086 | 10723 | 0.32 | 0.253 |
| “ | “ | “ | 140 | -17460 | -22853 | -18628 | -10898 | 11955 | 0.29 | 0.244 |
| “ | “ | 114 | 120 | -16157 | -21348 | -17153 | -9969 | 11379 | 0.26 | 0.261 |
| “ | “ | 116 | “ | -16257 | -21345 | -17300 | -10126 | 11219 | 0.28 | 0.263 |
| “ | 183.3 | 112 | “ | -17576 | -22245 | -18695 | -11789 | 10456 | 0.02 | 0.297 |
| Schiff base complexes | ||||||||||
| 195.0/183.3 | 173.0 | 101 | 125 | -13142 | -18634 | -13220 | -7571 | 11063 | 0.22 | 0.327 |
| “ | 173.8 | 112 | 120 | -13760 | -18432 | -14374 | -8474 | 9958 | 0.18 | 0.348 |
| “ | “ | 116 | 120 | -13993 | -18478 | -14759 | -8441 | 10037 | 0.23 | 0.352 |
| “ | “ | 112 | 130 | -14128 | -18889 | -14762 | -8734 | 10155 | 0.19 | 0.342 |
| “ | “ | 116 | 129 | -14337 | -19002 | -15021 | -8988 | 10074 | 0.20 | 0.347 |
| “ | “ | 112 | 140 | -14864 | -19872 | -15224 | -9496 | 10376 | 0.10 | 0.335 |
| “ | 180.6 | 112 | 127 | -14888 | -19207 | -15695 | -9762 | 9445 | 0.26 | 0.372 |
| “ | 183.3 | 101 | 130 | -14398 | -19196 | -14936 | -9062 | 10134 | 0.16 | 0.367 |
| “ | 193 | 101 | 125 | -15230 | -19466 | -15928 | -10296 | 9170 | 0.23 | 0.403 |
| “ | “ | 110 | 140 | -17685 | -22727 | -17220 | -13109 | 21408 | -0.07 | 0.417 |
| “ | “ | 116 | 120 | -17274 | -23250 | -16977 | -11595 | 11655 | -0.08 | 0.432 |
| Dithiocarbamato complexes | ||||||||||
| 200 | 172.8 | 110 | 120 | -5713 | -9074 | -5388 | -2677 | 6397 | -0.15 | -0.241 |
| 210 | 172.8 | 101 | 115 | -6922 | -9798 | -6025 | -4944 | 4854 | -0.55 | -0.142 |
| “ | 177 | “ | 120 | -7443 | -11338 | -5963 | -5028 | 6310 | -0.70 | -0.129 |
| “ | 172.8 | 110* | 120 | -7761 | -11836 | -5871 | -5576 | 6266 | -0.90 | -0.135 |
| “ | “ | “ | “ | -7848 | -12553 | -5794 | -5198 | 7355 | -0.84 | -0.146 |
| “ | “ | “ | 130 | -8074 | -13072 | -5694 | -5456 | 7616 | -0.94 | -0.143 |
| “ | “ | “ | 140 | -8711 | -14238 | -6407 | -5488 | 8750 | -0.79 | -0.142 |
| “ | “ | “ | 150 | -10036 | -16659 | -8360 | -5087 | 11272 | -0.43 | -0.143 |
| “ | “ | “ | 160 | -12904 | -22024 | -12393 | -4296 | 17728 | -0.09 | -0.147 |
| “ | “ | “ | 170 | -19965 | -33982 | -22636 | -3279 | 30703 | 0.26 | -0.152 |
| 210 | 172.8 | 112 | 120 | -8023 | -12959 | -5777 | -5333 | 7626 | -0.88 | -0.145 |
| “ | 173.8 | “ | “ | -8085 | -12301 | -6136 | -5818 | 6483 | -0.90 | -0.127 |
| “ | 175 | 117* | 135 | -9422 | -15016 | -7733 | -5518 | 9498 | -0.53 | -0.124 |
| “ | 177 | 110 | 120 | -8380 | -13515 | -5845 | -5779 | 7736 | -0.98 | -0.124 |
| 210 | 183 | 101 | 120 | -8053 | -12367 | -6054 | -5737 | 6630 | -0.90 | -0.09 |
| “ | 110 | “ | -9191 | -14995 | -6684 | -5894 | 9101 | -0.83 | -0.093 | |
| “ | “ | 116 | “ | -9928 | -16694 | -7291 | -5798 | 10896 | -0.73 | -0.099 |
| “ | 193 | 110 | “ | -10685 | -17718 | -8411 | -5928 | 11790 | -0.58 | -0.046 |
| “ | “ | “ | 130 | -11096 | -18580 | -8880 | -5829 | 12751 | -0.52 | -0.040 |
| “ | “ | “ | 140 | -12264 | -21000 | -10216 | -5575 | 15425 | -0.40 | -0.036 |
| 226.5 | 172.8 | 110 | 120 | -11632 | -16300 | -11881 | -6715 | 9585 | 0.08 | 0.046 |
| 175 | 111* | 130 | -12536 | -16297 | -11985 | -9325 | 6972 | -0.24 | 0.057 | |
| “ | 175 | 116.6# | 129.5# | -12757 | -18754 | -11704 | -7814 | 10940 | -0.29 | 0.058 |
| “ | 183 | 116.25# | 127.3# | -14240 | -21366 | -12180 | -9174 | 12192 | -0.51 | 0.091 |
| “ | 193 | 115.86# | 124.8# | -16284 | -25185 | -12588 | -11077 | 14108 | -0.79 | 0.129 |
| “ | 187.85# | 116.23# | 120 | -14936 | -22490 | -12502 | -9817 | 12673 | -0.58 | 0.11 |
| “ | 183.57# | 116.12# | 130 | -14535 | -21980 | -12143 | -9481 | 12499 | -0.57 | 0.093 |
| “ | 180.8# | 115.89# | 140 | -14907 | -22965 | -11683 | -10072 | 12893 | -0.75 | 0.078 |
| “ | 186.04# | 112 | 127.1# | -14359 | -21167 | -12569 | -9342 | 0.100 | ||
| 240.0 | 173.8 | 101* | 120 | -16943 | -23093 | -14668 | -13067 | 10026 | -0.68 | 0.237 |
* O of nitrosyl over basal ligand atom# partially optimised geometry
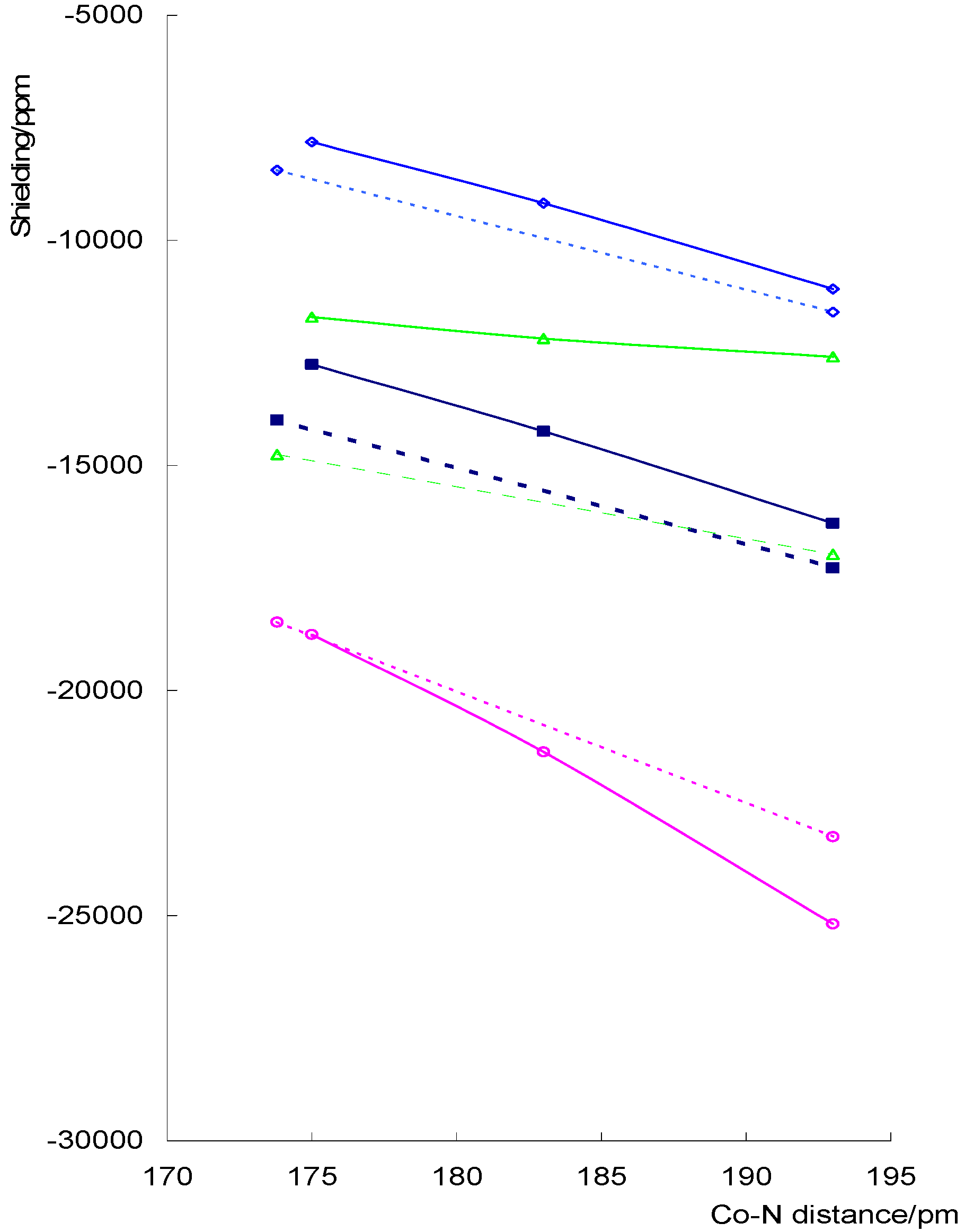
Figure 1 shows the variation of the isotropic shielding and the principal components with Co-N(NO) distance for both fixed NO distance and CoNO angle and when the CoNO geometry is allowed to relax. The NO distance for the series of runs in which the CoNO geometry is optimised is close to 1.16Å and so we have used a series of runs with an NO distance of 1.16Å for the fixed geometry comparison.
σ11 and σ33both decrease with increasing Co-N distance for the two sets of data shown. σ22 shows the least variation with Co-N distance as is expected for a component that is mainly σzz. Relaxing the CoNO geometry has little effect on the pattern of change. The notable difference in σ22 stems from the difference in the basal ligands.
The derivative of the isotropic shielding with respect to the Co-N distance is about –19 000 ppm Å-1 for the complexes in figure 1. Reference to the data in Table 1 shows a smaller derivative for complexes with shorter NO distances.
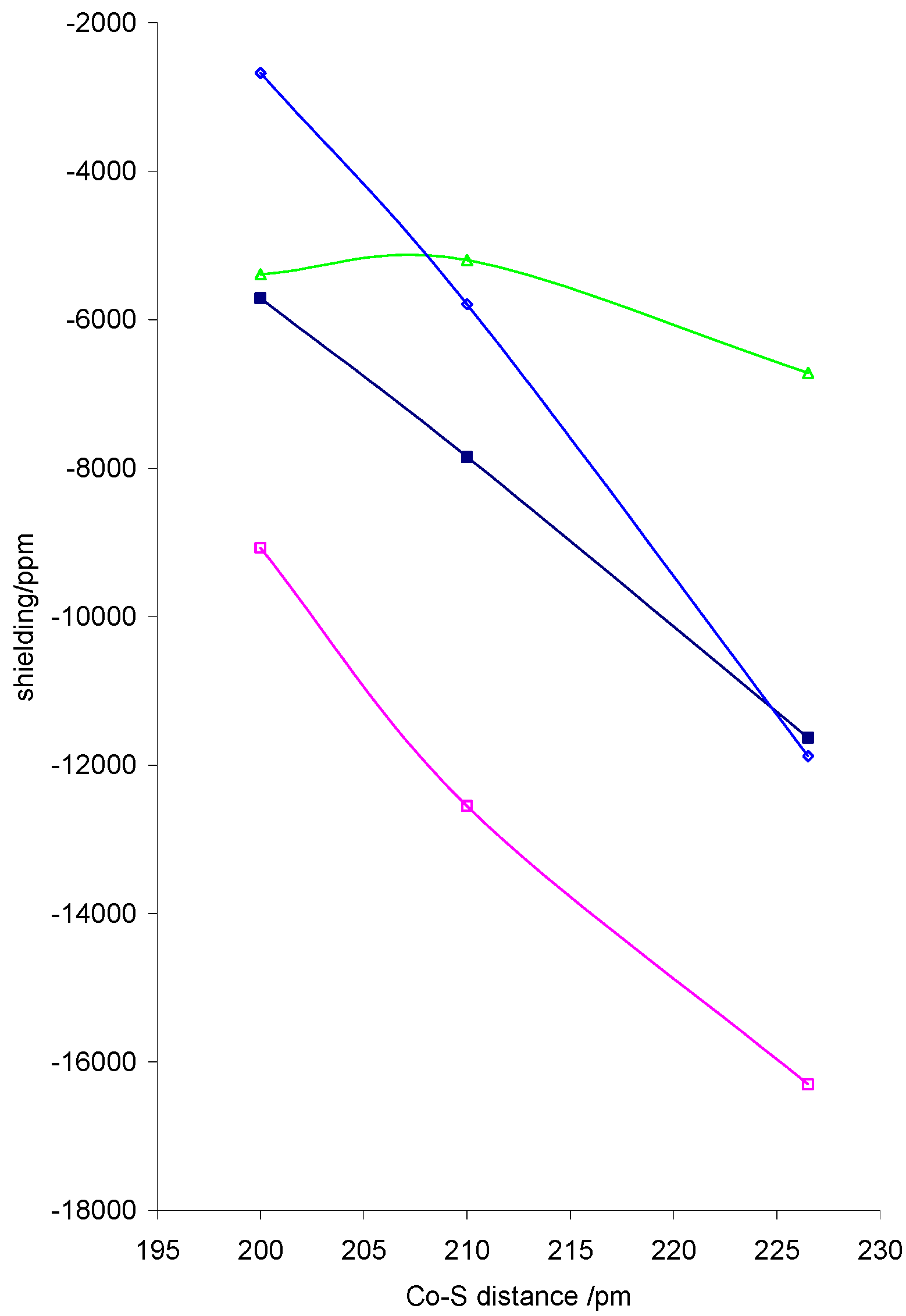
The isotropic shielding is also markedly dependent on the basal ligands. Adjusting the cobalt distance to the basal ligands in the dithiocarbamato complex from 2.0Å to 2.1Å to 2.65Å led to an
![Ijms 03 00873 g001]() increase in shielding equivalent to a derivative greater than –20 000 ppm Å-1 due to increases in σxx and σzz arising from decreased overlap of dz2 and dx2-y2 with basal ligand orbitals, as shown in figure 2. In this figure σyy and σzz refer to whichever of σ22 and σ33 are closest to these components, as the direction of the 2 and 3 axes change across the series. σzz is particularly sensitive to changes in Co-S distance as expected.
increase in shielding equivalent to a derivative greater than –20 000 ppm Å-1 due to increases in σxx and σzz arising from decreased overlap of dz2 and dx2-y2 with basal ligand orbitals, as shown in figure 2. In this figure σyy and σzz refer to whichever of σ22 and σ33 are closest to these components, as the direction of the 2 and 3 axes change across the series. σzz is particularly sensitive to changes in Co-S distance as expected.
Figure 1.
Variation of shielding with change in Co-N distance for a set of dithiocarbamato complexes for which the CoNO geometry is relaxed (solid lines) and for Schiff base complexes with CoNO 120° and an NO distance of 1.16Å (dotted lines) isotropic shielding, σ11, σ22 and ◊ σ33.
Figure 1.
Variation of shielding with change in Co-N distance for a set of dithiocarbamato complexes for which the CoNO geometry is relaxed (solid lines) and for Schiff base complexes with CoNO 120° and an NO distance of 1.16Å (dotted lines) isotropic shielding, σ11, σ22 and ◊ σ33.

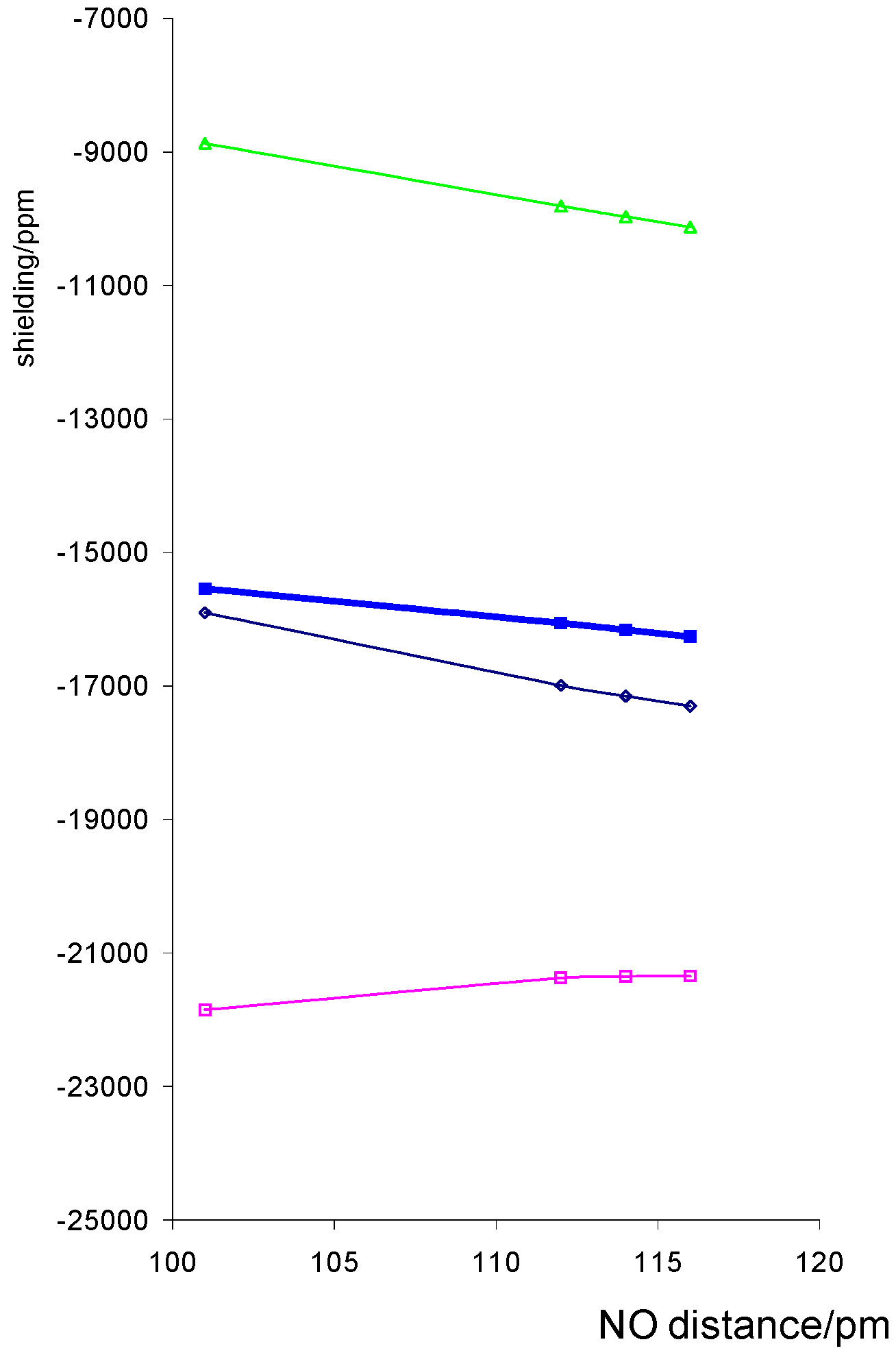
We would expect some effect from altering the NO distance because the nitrogen orbitals are used in bonding to O and Co and as the NO bond is weakened, the CoN bond is strengthened. The calculated shielding shows a decrease with increasing NO distance, figure 3, for all components except σ11.
For the porphin model shown the plots are much closer to linear with derivatives of –4 747 ppm Å-1 than those with respect to Co-ligand distance, although larger derivatives are found for some of the dithiocarbamato complexes.
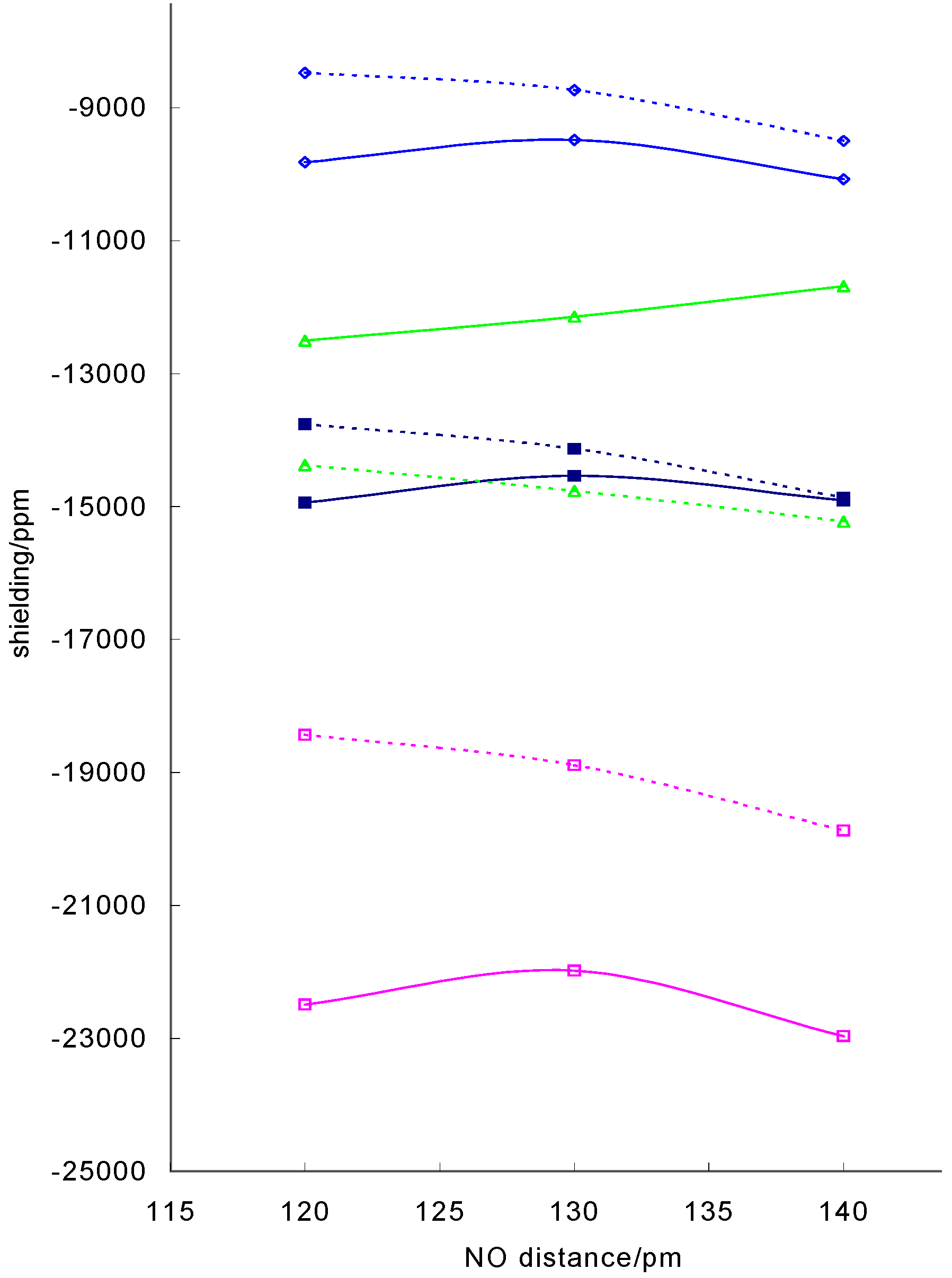
CoNO groups have the potential to wag (varying the angle) and swing (varying the position of the nitrosyl oxygen with respect to the base ligands). Varying the angle from 120° to 140°, the range of angles found in most bent Co(III) nitrosyl complexes, produced a decrease in shielding of about 1 000 – 2 000 ppm about 10% of the absolute shielding. This is exemplified in figure 4 where the isotropic
![Ijms 03 00873 g002]()
![Ijms 03 00873 g003]()
![Ijms 03 00873 g004]() shielding is plotted against CoNO angle for a model Schiff base model complex. Similar changes with change in angle were obtained for a porphin model complex and for dithiocarbamato and Schiff base complexes with different CoN and NO distances.
shielding is plotted against CoNO angle for a model Schiff base model complex. Similar changes with change in angle were obtained for a porphin model complex and for dithiocarbamato and Schiff base complexes with different CoN and NO distances.
Figure 2.
Calculated variation of Co shielding with Co-S distance for dithiocarbamato complexes with r(co-N) =1.728Å, r(NO) = 1.10Å and a CoNO angle of 120°. isotropic shielding, σ11, σyy and ◊ σzz.
Figure 2.
Calculated variation of Co shielding with Co-S distance for dithiocarbamato complexes with r(co-N) =1.728Å, r(NO) = 1.10Å and a CoNO angle of 120°. isotropic shielding, σ11, σyy and ◊ σzz.
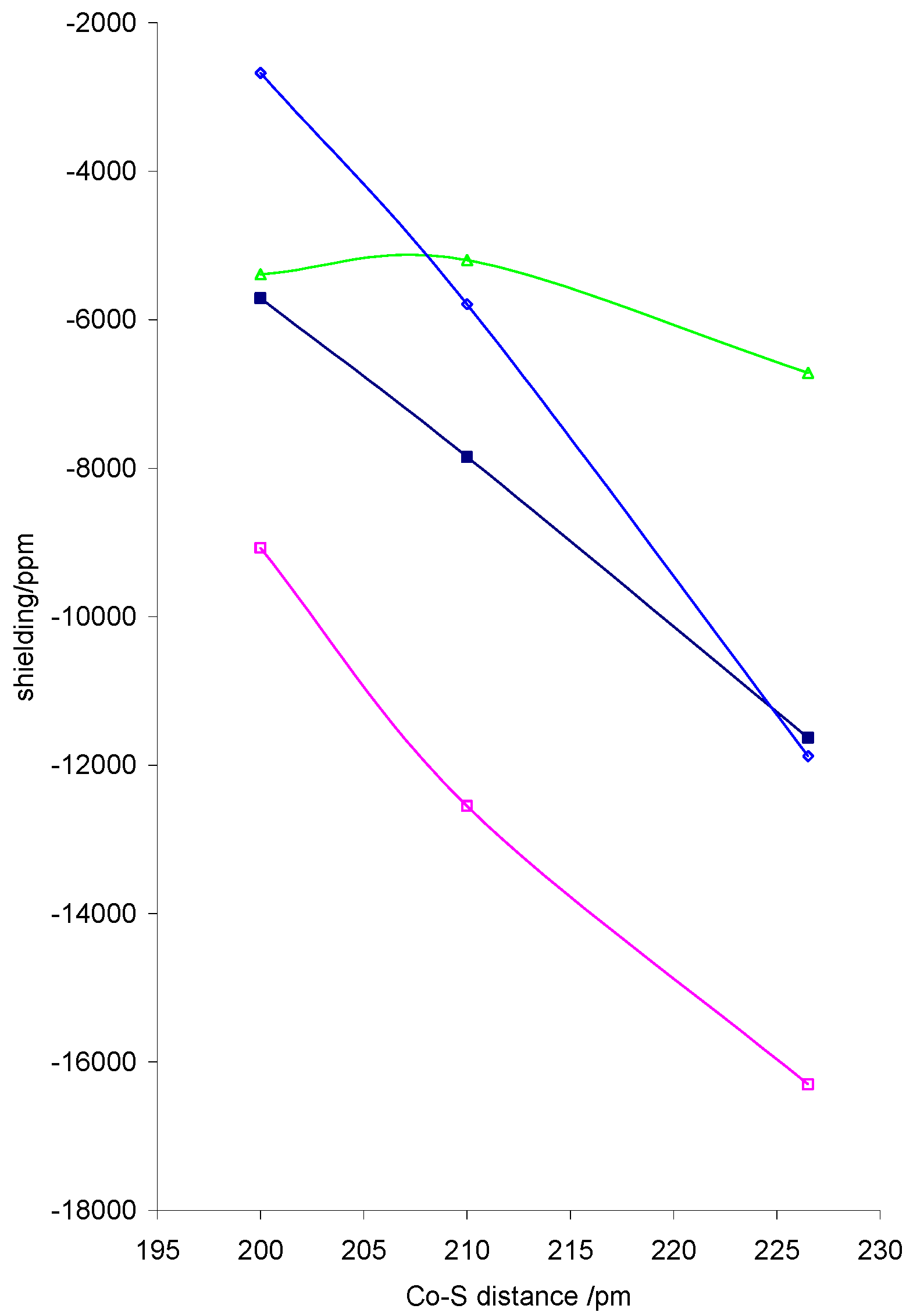
Figure 3.
Variation of calculated Co shielding with NO distance for a model porphin complex with r(CoN) = 1.74Å isotropic shielding, σ11, σ22 and ◊ σ33
Figure 3.
Variation of calculated Co shielding with NO distance for a model porphin complex with r(CoN) = 1.74Å isotropic shielding, σ11, σ22 and ◊ σ33
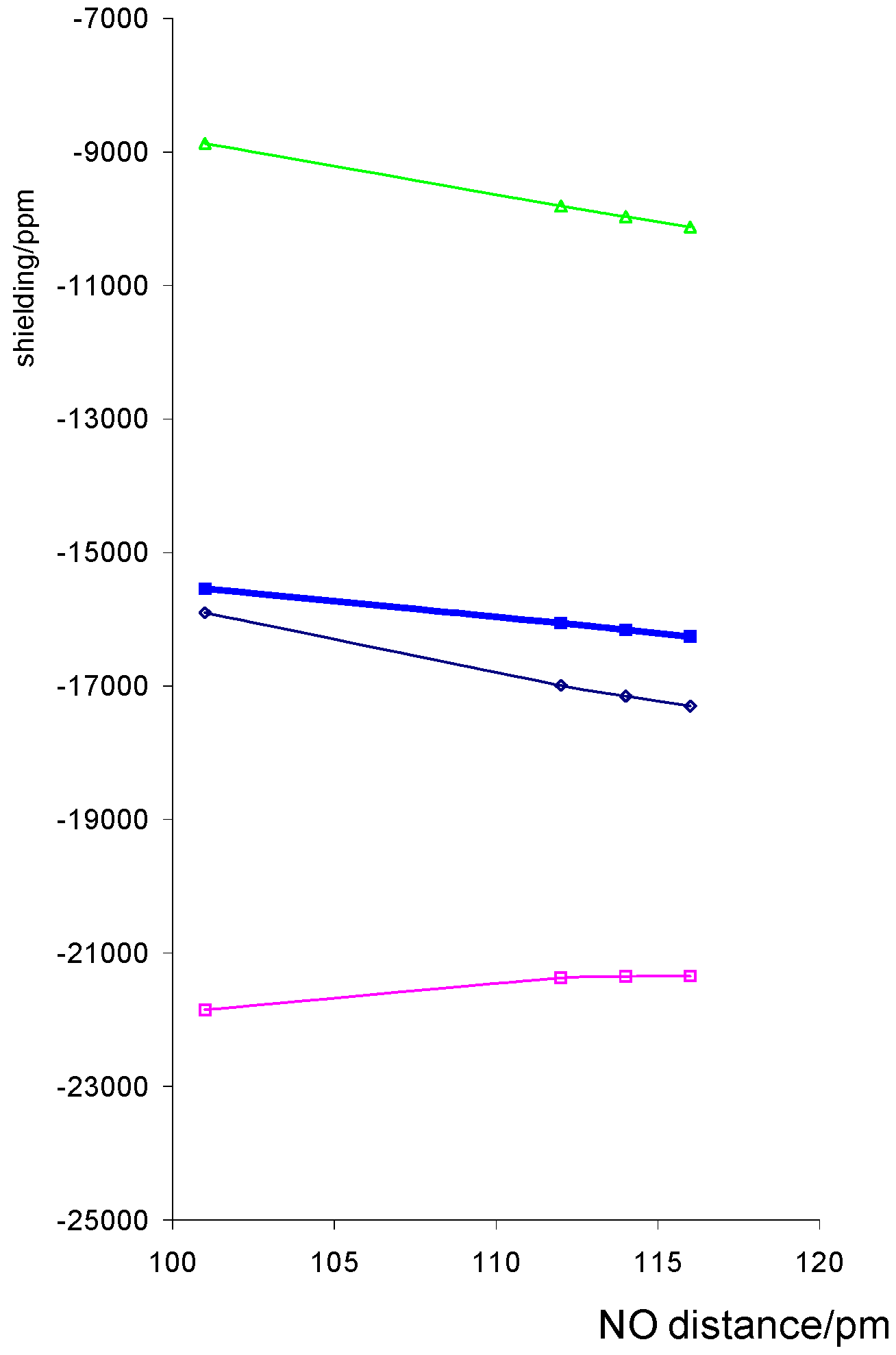
Figure 4.
Variation of calculated 59Co shielding with CoNO angle for model nitrosyl complexes: dithiocarbamato complex with optimised CoNO geometry (solid line), model Schiff base complex with r(CoN) = 1.738Å and r(NO) = 1.12Å (dotted line) isotropic shielding, σ11, σ22 and ◊ σ33
Figure 4.
Variation of calculated 59Co shielding with CoNO angle for model nitrosyl complexes: dithiocarbamato complex with optimised CoNO geometry (solid line), model Schiff base complex with r(CoN) = 1.738Å and r(NO) = 1.12Å (dotted line) isotropic shielding, σ11, σ22 and ◊ σ33
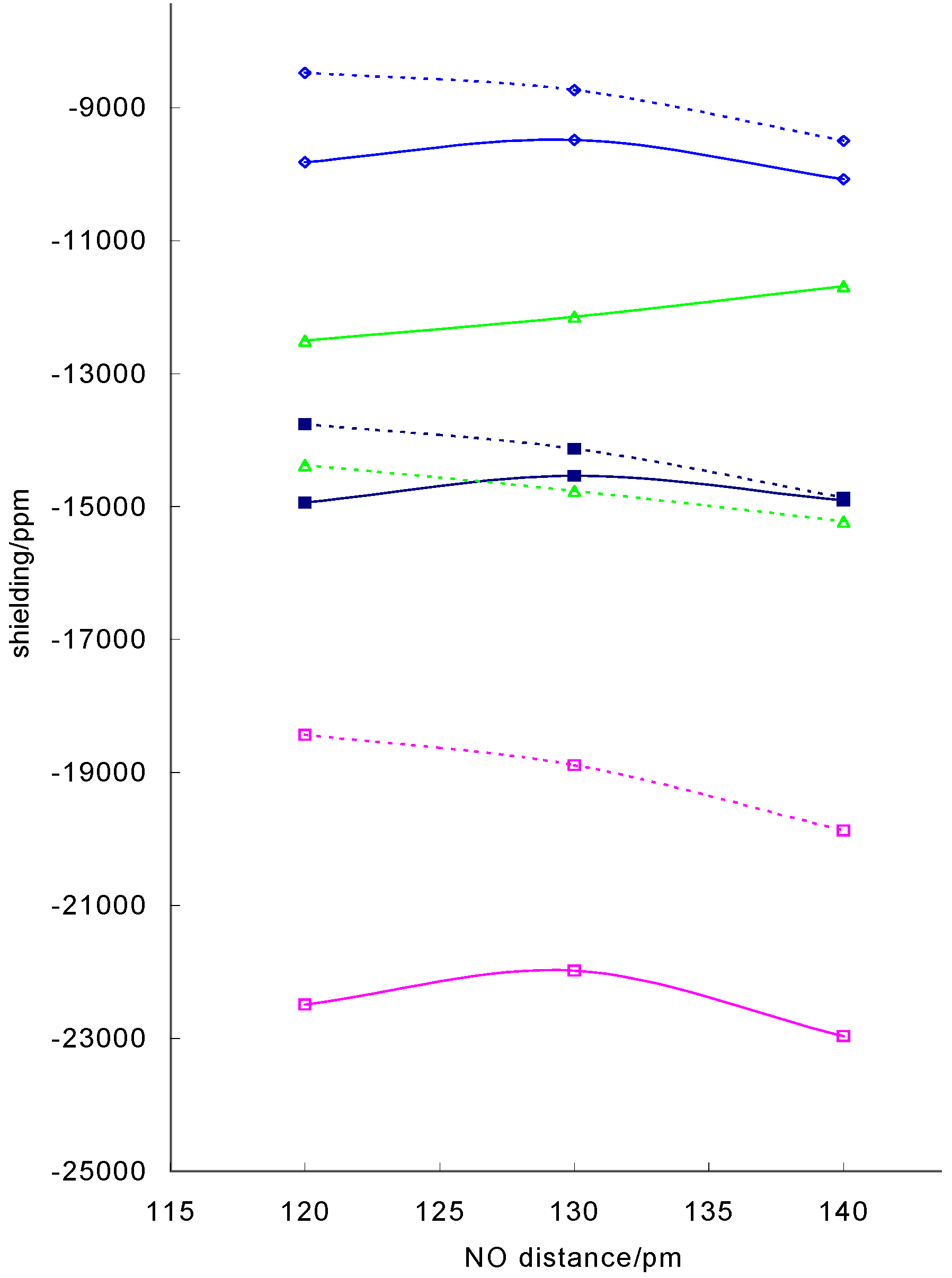
In this case, the partially optimised structure displays a different pattern to the series with fixed CoNO geometry with the shielding going through a maximum. In these structure the Co-N distance decreases with increasing angle and as shown in figure 1 this will lead to an increase in shielding, offsetting the decrease due to increase in angle.
The effect of ligand swinging was investigated by calculating the shielding with the nitrosyl oxygen in two positions, between two basal ligating atoms and over one basal atom. The changes in the tensor for a typical dithiocarbamato complex can be seen by comparing the starred entries with non-starred entries for the dithiocarbamato complex with Co-N = 1.728Å, N-O = 1.1Å and CoNO angle of 120° in Table 1.
These differences are not large but are noticeable. Averaging over these two positions would give a reduced span because the tensor principal components will not be exactly aligned for the two positions. For the complex with the O between the ligands, then one principal axis is along the y axis with the other two rotated by 12° from the x and z axes, whereas all three axes are rotated for the other complex leading to one axis lying almost over Co-S direction where S is the atom over which the nitrosyl oxygen lies.
Discussion and Conclusions
Density functional calculations of cobalt shielding in Co(III) complexes using DFT/GIAO methods with moderately-sized basis sets and hybrid density functionals have been found by several groups to give good agreement with experimental shielding values. Agreement is good not only for simple octahedral complexes but also for larger systems such as porphinato complexes. We have found that such calculations also give good results for square-pyramidal nitrosyl complexes of cobalt.
Analysis of the shielding tensors shows that the largest component arises from the dz2 to dyz transition. The paramagnetic part of this component increases rapidly with increasing cobalt -ligand distance. The principal component closest to zz, where the z axis is along the Co-N(NO) bond, shows a marked dependence on cobalt-basal ligating atom distance and the nature of the ligand as would be expected for a component arising primarily from orbitals in the basal plane. The third principal component by contrast is more sensitive to Co-N(NO) distance than to the basal ligands. The isotropic shielding displays large changes with Co-N and Co-basal ligating atom distance. The large changes in the shielding of the order of -103 - 104 ppm/Å with variation in cobalt ligating atom distance agree with experimental observations of temperature variation and isotope shifts which indicate that cobalt shielding is very sensitive to changes in geometry. The isotropic shielding decreases with NO distance but the derivative is smaller than that with respect to Co-ligating atom distances and the most negative component of the shielding tensor can decrease with increasing NO distance. Changes in the shielding tensor with CoNO angle are much smaller. For complexes with the basal ligands arranged at the experimental distances from the cobalt, then the shielding increases N< N,O < S.
A recent study on B12 model systems [40] shows that increase in axial Co-C bond distance due to substitution of different alkyl groups leads to an increased HOMO-LUMO gap via decrease of the HOMO energy. Our studies show that increase of the Co-N(NO) distance leads to a lowering in energy of both HOMO (Co dz2/NO σ/π) and LUMO(Co dyz/ NO π∗) with a greater lowering of the LUMO leading to a decrease in HOMO-LUMO energy gap.
Ideally, for comparison with experimental chemical shifts calculations should be corrected for temperature effects, although this is difficult except for simple cases such as [Co(CN)6]3- where most of the effect comes from the variation in one distance (CoC).
Acknowledgement
I should like to thank the UK computational chemistry working party for a grant of time on the Engineering and Physical Science Research Council (EPSRC)’s superscalar facility at the Rutherford Appleton Laboratory.
References
- Chan, C.C.; Au-Yeung, S. C. F. Annual Reports on NMR Spectroscopy; Webb, G. A., Ed.; Academic Press: London, SanDiego, 2000; vol.41, pp. 1–54. [Google Scholar]
- Ramsey, N. F. Magnetic Shielding of Nuclei in Molecules. Phys. Rev. 1950, 78, 699–703. [Google Scholar]
- Freeman, R.; Murray, G. R.; Richards, R. E. Cobalt nuclear resonance spectra. Proc. Roy. Soc. A 1957, 242, 455–466. [Google Scholar] [CrossRef]
- Juranić, N. Inorg. Chem. 1980, 19, 1093.
- Juranić, N. Nephelauxetic effect in paramagnetic shielding of transition-metal nuclei in octahedral d6 complexes. J. Am. Chem. Soc. 1988, 110, 8341–3. [Google Scholar] [CrossRef]
- Bramley, R.; Brorson, M.; Sargenson, A. M.; Schäffer, C. F. J. Am. Chem. Soc. 1985, 107, 2780. [CrossRef]
- Mason, J. Patterns of Nuclear Magnetic Shielding of Transition-Metal Nuclei. Che. Rev. 1987, 87, 1299–1312. [Google Scholar] [CrossRef]
- Chan, J. C. C.; Au-Yeung, C. F. Interpretation of 59Co NMR shielding using the hard and soft acid-base concept. J. Chem. Soc. Faraday Trans. 1996, 92, 1121–1128. [Google Scholar] [CrossRef]
- Gauss, J. J. Chem. Phys. 1993, 99, 3629.
- Bouman, T. D.; Hansen, Aa. E. NMR shielding calculations beyond coupled Hartree-Fock: second order correlation effects in localised-orbital/local-origin calculations of molecules containing phosphorus. Chem. Phys. Lett. 1990, 175, 292–298. [Google Scholar] [CrossRef]
- Van Wüllen, C.; Kutzelnigg, W. Calculation of nuclear magnetic resonance shieldings and magnetic susceptibilities using multiconfiguration Hartree-Fock wave functions and local gauge origins. J. Chem. Phys. 1996, 104, 2330–2337. [Google Scholar] [CrossRef]
- Chan, J. C. C.; Au-Yeung, S. C. F.; Wilson, P. J.; Webb, G. A. SOS-DFPT-IGLO calculations of 59Co NMR shielding parameters of hexacoordinated diamagnetic Co(III) complexes. J. Mol. Struct. (THEOCHEM) 1996, 365, 125–130. [Google Scholar] [CrossRef]
- Malkin, V. G.; Zhidomorov, G. M. Zh. Strukt. Khim. 1988, 29, 32.
- Malkin, V. G.; Malkina, O. L.; Salahub, D. R. Chem. Phys. Lett. 1993, 204, 80.
- Malkin, V. G.; Malkina, O. L.; Salahub, D. R. Chem. Phys. Lett. 1993, 204, 87.
- Malkin, V. G.; Malkina, O. L.; Casida, M. E.; Salahub, D. R. J. Am. Chem. Soc. 1994, 116, 5898.
- Perdew, J. P. Phys. Rev.B 1986, 33, 8822. [CrossRef]
- Schäfer, A. J. Chem. Phys. 1992, 97, 2571.
- Wachters, A. J. H. J. Chem. Phys. 1970, 52, 1033.
- Kutzelnigg, W.; Fleischer, U.; Schindler, M. NMR: Basic Principles and Progress 1990, 23, 165.
- Lamphun, B. N.; Webb, G. A. J. Mol. Struct. (THEOCHEM) 1983, 104, 191. [CrossRef]
- Chan, J. C. C.; Au-Yeung, S. C. F. A comparative study of the calculation of 59Co NMR shielding constants of hexacoordinated diamagnetic Co(III) complexes using DFT-IGLO and DFT-GIAO methods. J. Mol. Struct. (THEOCHEM) 1997, 393, 93–96. [Google Scholar]
- Gaussian 94. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Ciolowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P.Y.; Chen, W.; Womg, M.W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian, Inc.: Pittsburgh PA, 1995. [Google Scholar]
- Chan, J. C.; Au-Yeung, S. C. F. Density Functional Study of 59Co Chemical Shielding Tensors Using Gauge-Including Atomic Orbitals. J. Phys. Chem. A 1997, 101, 3637–3640. [Google Scholar] [CrossRef]
- Godbout, N.; Oldfield, E. Density Functional Study of Cobalt-59 Nuclear Magnetic resonance Chemical Shifts and Shielding Tensor Elements in Co(III) Complexes. J. Am. Chem. Soc. 1997, 119, 8065–8069. [Google Scholar] [CrossRef]
- Wachters, A. J. H. J. Chem. Phys. 1970, 52, 1033.
- Wachters, A. J. H. IBM Tech. Rept. 1969, RJ584. Basis sets were obtained from the Extensible Computational Chemistry Environment Basis Set Database, Version 1.0, as developed and distributed by the Molecular Science Computing Facility, Environmental and Molecular Sciences Laboratory which is part of the Pacific Northwest Laboratory, P.O. Box 999, Richland, Washington 99352, USA, and funded by the U.S. Department of Energy. The Pacific Northwest Laboratory is a multi-program laboratory operated by Battelle Memorial Institue for the U.S. Department of Energy under contract DE-AC06 76RLO 1830. Contact David Feller or Karen Schuchardt for further information..
- Schrechenbach, G. J. Chem. Phys. 1990, 110, 11936.
- Xu, X-P.; Au-Yeung, S. C. F. A DFT and 59Co Solid-State NMR Study of the Chemical Shielding Property and Electronic Interaction in the Metalloporphyrin System. J. Am. Chem. Soc. 2000, 122, 6468–6475. [Google Scholar] [CrossRef]
- Maclean, A. D.; Chandler, G. S. J. J. Chem. Phys. 1993, 98, 5648.
- Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650.
- Hay, P. J. J. Chem. Phys. 1977, 66, 4377.
- Zhou, P.; Au-Yeung, S. C. F.; Xu, X-P. A DFT and 59Co Solid-State NMR study of the Second-Sphere Interaction in Polyammonium Macrocycles Cobalt Cyanide Supercomplexes. J. Am. Chem. Soc. 1999, 121, 1030–6. [Google Scholar] [CrossRef]
- Jameson, C. J.; Rehder, D.; Hoch, M. J. Am. Chem. Soc. 1987, 109, 2589–2594. [CrossRef]
- Groombridge, C. J.; Larkworthy, L.F.; Marécaux, A.; Povey, D. C.; Smith, G. W.; Mason, J. Synthesis and 15N Nuclear Magnetic Resonance Shift Tensors of Bent Nitrosyl Complexes with N-Substituted Salicylideneiminate Coligands; the Shift tensor as a Criterion of MNO Bond Angle. J. Chem. Soc. Dalton Trans. 1992, 3125–3131. [Google Scholar] [CrossRef]
- Gaussian 98, Revision A.7. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ciolowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Womg, M.W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian, Inc.: Pittsburgh PA, 1998. [Google Scholar]
- Schafer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571, Basis sets were obtained from the Extensible Computational Chemistry Environment Basis Set Database, Version 9/11/00, as developed and distributed by the Molecular Science Computing Facility, Environmental and Molecular Sciences Laboratory which is part of the Pacific Northwest Laboratory, P.O. Box 999, Richland, Washington 99352, USA, and funded by the U.S. Department of Energy. The Pacific Northwest Laboratory is a multi-program laboratory operated by Battelle Memorial Institue for the U.S. Department of Energy under contract DE-AC06 76RLO 1830. Contact David Feller or Karen Schuchardt for further information..
- Mingos, D. M. P. Inorg. Chem. 1973, 12, 1209.
- Duffin, P. A.; Larkworthy, L. F.; Mason, J.; Stephens, A. N.; Thompson, R. M. Ligand Field Effects in the Nuclear Magnetic Shielding of Nitrogen-15 and Cobalt-59 in Bent Nitrosyl Complexes of Cobalt(III). Inorg. Chem. 1987, 26, 2034–2040. [Google Scholar] [CrossRef]
- Jensen, K. P.; Sauer, S. P. A.; Liljefors, T.; Norrby, P-O. Theoretical Investigation of Steric and Electronic Effects in Coenzyme B12 Models. Organometallics 2001, 20, 550–6. [Google Scholar] [CrossRef]
© 2002 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Moore, E.A. Ab Initio Calculations of Co Shielding in Model Complexes. Int. J. Mol. Sci. 2002, 3, 873-887. https://doi.org/10.3390/i3080873
AMA Style
Moore EA. Ab Initio Calculations of Co Shielding in Model Complexes. International Journal of Molecular Sciences. 2002; 3(8):873-887. https://doi.org/10.3390/i3080873
Chicago/Turabian StyleMoore, Elaine A. 2002. "Ab Initio Calculations of Co Shielding in Model Complexes" International Journal of Molecular Sciences 3, no. 8: 873-887. https://doi.org/10.3390/i3080873