Ab initio post-HF CCSD(T) Calculations for Triplet and Singlet Methylene in Four consecutive Dunning Basis Sets with Extrapolations to Infinite Limits for Various Molecular Properties

Abstract
:Introduction
- 1)
- The Hartree-Fock (HF) self-consistent-field (SCF) method uses one Slater determinant of LCAO-MO´s describing a single configuration of electrons which may serve as a reference state for most of the following post-HF models.
- 2)
- Configuration-interaction (CI) theory is based on a linear superposition of Slater determinants describing excitations of electrons from a reference state with variational determination of the expansion coefficients. Truncated configurations may use single (one-electron) excitations (S), double (two-electron) excitations (D) or similarly triple (T) or quadruple (Q) excited configurations. Treatment of all possible excited configurations are termed full CI (FCI).
- 3)
- The multireference CI (MRCI) method uses several determinants as reference configurations and generates all excitations up to a given level from each reference configuration, i. e. if all single and double excitations are included results the MRSDCI model. Alternatively this may be termed second order CI (SOCI) for single and double excitations out of a CASSCF reference function.
- 4)
- The multiconfigurational self-consistent field (MCSCF) method uses CI determinants which are variationally optimised simultaneously with the expansion coefficients. The MO space may be partitioned into three subspaces containing inactive (doubly occupied), virtual (unoccupied) and active orbitals (with variable occupancies of 0, 1 or 2 electrons). A MCSCF expansion distributing the active electrons in all possible ways among the active orbitals which is leading to non-integer occupancies is termed complete active space (CAS) method.
- 5)
- The coupled-cluster (CC) model treats excitations between pair-wise correlated electrons (pair clusters) in a non-linear way via a cluster operator acting on a single-determinantal reference state. The cluster operator is partitioned into classes of all single (S), double (D) or triple (T) excitations, In the CCSD(T) method [12, 13] are contributions from triple excitations estimated by a perturbative treatment. The CC methods account well for dynamical electron correlation.
- 6)
- Perturbation theory is applied as Møller-Plesset (MP) perturbation of second, third or fourth order (MP2, MP3 or MP4) related to HF SCF as the unperturbed reference state
Calculational Procedure
Results and Discussion
Total Energies

{kind=link}
{kind=link}
| 3B1 CH2 1 | CCSD(T) | cc-pVDZ | -39.041816 | -39.041248 | -39.041564 | -39.046371e | [4] |
| bent | cc-pVTZ | -39.077854 | -39.077802 | -39.077501 | -39.066184f | [29] | |
| cc-pVTZ | -39.094104 | [22] | |||||
| triplet GS | cc-pVQZ | -39.087331 | -39.087285 | -39.086958 | |||
| cc-pV5Z | -39.090075 | -39.090029 | -39.089024d | ||||
| cc-pV6Z | - | -39.090903 | - | ||||
| CBS limit | -39.0912 | -39.092197j | -39.09174 | ||||
| CCSDT | cc-pVDZ | -39.041670 | |||||
| cc-pVTZ | -39.078279 | ||||||
| cc-pVQZ | -39.087741 | ||||||
| FCI | cc-pVDZ | -39.041695 | -39.046816e | [4] | |||
| FCI | ANO | -39.046260 | [23] | ||||
| FCI | cc-pVTZ | -39.078346 | -39.066738f | [29] | |||
| CISD | cc-pVDZ | -39.036887 | -39.045381 | [52] | |||
| cc-pVTZ | -39.070465 | -39.080444 | [52] | ||||
| cc-pVQZ | -39.079271 | ||||||
| cc-pV5Z/QZ | -39.081170d | ||||||
| MP4 | cc-pVDZ | -39.040477 | |||||
| cc-pVTZ | -39.076541 | ||||||
| cc-pVQZ | -39.085976 | ||||||
| cc-pV5Z/QZ | -39.088060d | ||||||
| CAS+1+2 | cc-pVDZ | -39.040273 | |||||
| cc-pVTZ | -39.074544 | ||||||
| cc-pVQZ | -39.083477 | ||||||
| cc-pV5Z/QZ | -39.085397d | ||||||
| CMRCIg | cc-pCVDZ | -39.041579 | [27] | ||||
| cc-pCVTZ | -39.075452 | [27] | |||||
| cc-pCVQZ | -39.083886 | [27] | |||||
| cc-pCV5Z | -39.086206 | [27] | |||||
| CBS limit | -39.08796 | [27] | |||||
| MCI CAS | cc-pCVTZ | -39.07989 | [49] | ||||
| MRCI ISh | ANO QZ | -39.083084 | [21] | ||||
| MRCI SOi | ANO QZ | -39.083222 | [21] | ||||
| MR SOCI | ANO | -39.084972 | [25] | ||||
| 3Σ- CH2 2 | CCSD(T) | cc-pVDZ | -39.030906 | ||||
| Linear | cc-pVTZ | -39.068449 | MCI CAS: | -39.07895 | [49] | ||
| cc-pVQZ | -39.078196 | ||||||
| cc-pV5Z | -39.081045 | ||||||
| C BS limit | -39.0822 |
| 1A1 CH2 3 | CCSD(T) | cc-pVDZ | -39.022584 | -39.022029 | -39.024154 | -39.025791e | [4] |
| bent | cc-pVTZ | -39.061384 | -39.061367 | -39.062456 | -39.048005f | [29] | |
| singlet | cc-pVQZ | -39.071913 | -39.071909 | -39.072413 | |||
| GS | cc-pV5Z | -39.075039 | -39.075037 | -39.075244 | |||
| cc-pV6Z | - | -39.076081j | - | ||||
| CBS limit | -39.0764 | -39.076408k | -39.07733 | ||||
| CCSDT | cc-pVDZ | -39.022748 | |||||
| cc-pVTZ | -39.062180 | ||||||
| cc-pVQZ | -39.072695 | ||||||
| FCI | cc-pVDZ | - | -39.022937 | -39.026635e | [4] | ||
| FCI | ANO | -39.027183 | [24] | ||||
| FCI | cc-pVTZ | -39.062405 | -39.048984f | [29] | |||
| CMRCIg | cc-pCVDZ | -39.023177 | [27] | ||||
| cc-pCVTZ | -39.059919 | [27] | |||||
| cc-pCVQZ | -39.069396 | [27] | |||||
| cc-pCV5Z | -39.072067 | [27] | |||||
| CBS limit | -37.07405 | [27] | |||||
| MRCI ISh | ANO QZ | -39.068284 | [21] | ||||
| MRCI SOi | ANO QZ | -39.068284 | [21] | ||||
| SOCI | ANO | -39.070250 | [25] | ||||
| 1Δg CH2 4 | CCSD(T) | cc-pVDZ | -38.967818 | ||||
| linear | cc-pVTZ | -39.010679 | |||||
| singlet | cc-pVQZ | -39.022215 | |||||
| cc-pV5Z | -39.025820 |
Core-Valence Correlation
Equilibrium CH Bond Lengths
| 3B1 CH2 1 | CCSD(T) | cc-pVDZ | 1.0957 | 1.0955 | 1.0947 | 1.0875e | [4] |
| bent triplet | cc-pVTZ | 1.0785 | 1.0783 | 1.0782 | 1.0772f | [29] | |
| GS | cc-pVQZ | 1.0773 | 1.0771 | 1.0771 | 1.07793 | [21] | |
| cc-pV5Z | 1.0771 | 1.0772d | 1.0767 | 1.07114 | [22] | ||
| ANO | 1.082 | [23] | |||||
| eqn. 1 | CBS limit | 1.07706 | 1.0767 | 1.079 | [25] | ||
| eqn. 2 | Prediction | 1.07526 | 1.07598g | [30] | |||
| experiment | 1.0748 | ± 0.0004 | [8] | ||||
| experiment | 1.0753 | ± 0.0003 | [9] | ||||
| CMRCI | cc-pCVDZ | 1.0951 | [27] | ||||
| cc-pCVTZ | 1.0784 | ||||||
| cc-pCVQZ | 1.0773 | ||||||
| cc-pCV5Z | 1.0769 | ||||||
| CBS limit | 1.0769 | ||||||
| CISD | cc-pVDZ | 1.0932 | |||||
| cc-pVTZ | 1.0758 | ||||||
| cc-pVQZ | 1.0744 | ||||||
| cc-pV5Z/QZ | 1.0744 | ||||||
| MP4 | cc-pVDZ | 1.9044 | |||||
| cc-pVTZ | 1.0774 | ||||||
| cc-pVQZ | 1.0762 | ||||||
| cc-pV5Z/QZ | 1.0762 | ||||||
| CAS+1+2 | cc-pVDZ | 1.0959 | |||||
| cc-pVTZ | 1.0786 | ||||||
| cc-pVQZ | 1.0773 | ||||||
| cc-pV5Z/QZ | 1.0773 | ||||||
| 3Σ- CH2 2 | CCSD(T) | cc-pVDZ | 1.0837 | ||||
| Linear | cc-pVTZ | 1.0676 | MCI CAS: | 1.068 | [49] | ||
| Triplet | cc-pVQZ | 1.0665 | |||||
| cc-pV5Z | 1.0663 | ||||||
| eqn. 1 | CBS limit | 1.0663 | |||||
| eqn. 2 | Prediction | 1.0645 | |||||
| experiment | 1.060 | ±0.005 | [8] |
| 1A1 CH2 3 | cc-pVDZ | 1.1291 | 1.1286 | 1.1199e | [4] | ||
| Bent | cc-pVTZ | 1.1105 | 1.1103 | 1.1089f | [29] | ||
| Singlet GS | cc-pVQZ | 1.1086 | 1.1086 | 1.10863 | [21] | ||
| cc-pV5Z | 1.1080 | 1.1080 | 1.117 | [23] | |||
| 1.112 | [26] | ||||||
| 1.110 | [25] | ||||||
| 1.10691 | [30] | ||||||
| eqn. 1 | CBS limit | 1.1077 | 1.1079 | ||||
| eqn. 2 | Prediction | 1.1059 | |||||
| experiment | 1.1112 | [9] | |||||
| experiment | 1.1070 | ±0.002 | [10] | ||||
| CMRCIg | cc-pCVDZ | 1.1290 | [27] | ||||
| cc-pCVTZ | 1.1107 | ||||||
| cc-pCVQZ | 1.1088 | ||||||
| cc-pCV5Z | 1.1083 | ||||||
| CBS limit | 1.1081 | ||||||
| 1Δg CH2 ‘4 | cc-pVDZ | 1.0789 | |||||
| Linear | cc-pVTZ | 1.0645 | |||||
| Singlet | cc-pVQZ | 1.0640 | |||||
| cc-pV5Z | 1.0640 | ||||||
| eqn. 1 | CBS limit | 1.0640 | |||||
| eqn. 2 | Prediction | 1.0622 | Experiment: | 1.070 | [51] |
HCH Valence Angles
| 3B1 CH2 1 | CCSD(T) | cc-pVDZ | 132.198 | 132.10 | 132.13 | 132.15 | [4] |
| Bent | cc-pVTZ | 133.565 | 133.33 | 133.33 | 133.29 | [29] | |
| Triplet GS | cc-pVQZ | 133.648 | 133.51 | 133.51 | 132.900 | [21] | |
| cc-pV5Z | 133.539 | 133.57 | 133.56 | 134.127 | [22] | ||
| 132.4 | [23] | ||||||
| 133.6 | [25] | ||||||
| 132.7 | [26] | ||||||
| CBS limit | – | 133.59 | 133.848 | [30] | |||
| experiment | 133.84±0.05 | [8] | |||||
| experiment | 133.93±0.01 | [9] | |||||
| 1A1 CH2 3 | CCSD(T) | cc-pVDZ | 100.542 | 100.54 | 101.28 | [4] | |
| Bent GS | cc-pVTZ | 101.617 | 101.63 | 101.72 | [29] | ||
| cc-pVQZ | 101.929 | 101.90 | 102.137 | [30] | |||
| cc-pV5Z | 101.914 | 102.01 | |||||
| CBS limit | – | 102.05 | |||||
| CMRCIg | cc-pCVDZ | 100.54 | |||||
| cc-pCVTZ | 101.63 | ||||||
| cc-pCVQZ | 101.90 | ||||||
| cc-pCV5Z | 102.01 | ||||||
| CBS limit | 102.08 | ||||||
| experiment | 101.95 | [9] | |||||
| experiment | 102.4 ±0.4 | [10] |
Singlet-Triplet Energy Gap
| 12.068 | 10.335 | 9.675 | 9.435 | - | 9.287 | CCSD(T) VEc | |
| 12.060 | 10.313 | 9.649 | 9.408 | 9.301 | (9.299)d | CCSD(T) VEe,f | [30] |
| 12.019 | 10.527 | 9.998 | 9.799 | 9.661 | - | CCSD(T) AEe,f | [30] |
| 11.791 | 10.054 | 9.408 | 9.184 | - | 9.042 | CCSD(T) VEe | [27] |
| 11.862 | 10.330 | 9.792 | 9.590 | - | 9.475 | CCSD(T) AEe | [27] |
| 11.547 | 9.747 | 9.093 | 9.661 | 8.872 | 8.729 | CMRI VEe | [27] |
| 11.652 | 10.051 | 9.502 | 9.307 | - | 9.193 | CMRI AEe | [27] |
| 11.496 | 9.620 | 8.931 | 8.699 | - | 8.553 | CMRI+Q VEe,f | [27] |
| 11.590 | 9.901 | 9.315 | 9.106 | - | 8.980 | CMRI+Q AEe,f | [27] |
| 11.771 | 10.003 | FCI VEe,f | [30] | ||||
| 11.729 | FCI AEe,f | [30] | |||||
| 12.664 [4] | 11.141 [29] | FCI VE | |||||
| 11.97 [23] | FCI VE | [23] | |||||
| 9.287 (IS) | 9.373 (SO) | MRCI VEg | [21] | ||||
| 9.592 (IS) | 9.740 (SO) | MRCI AEg | [21] | ||||
| 9.238 SOCI | 9.111 (SO+Q) | MRCI VEg | [25] | ||||
| 10.608 | MCPF AEg | [26] | |||||
| 9.436 | CISD | [52] |
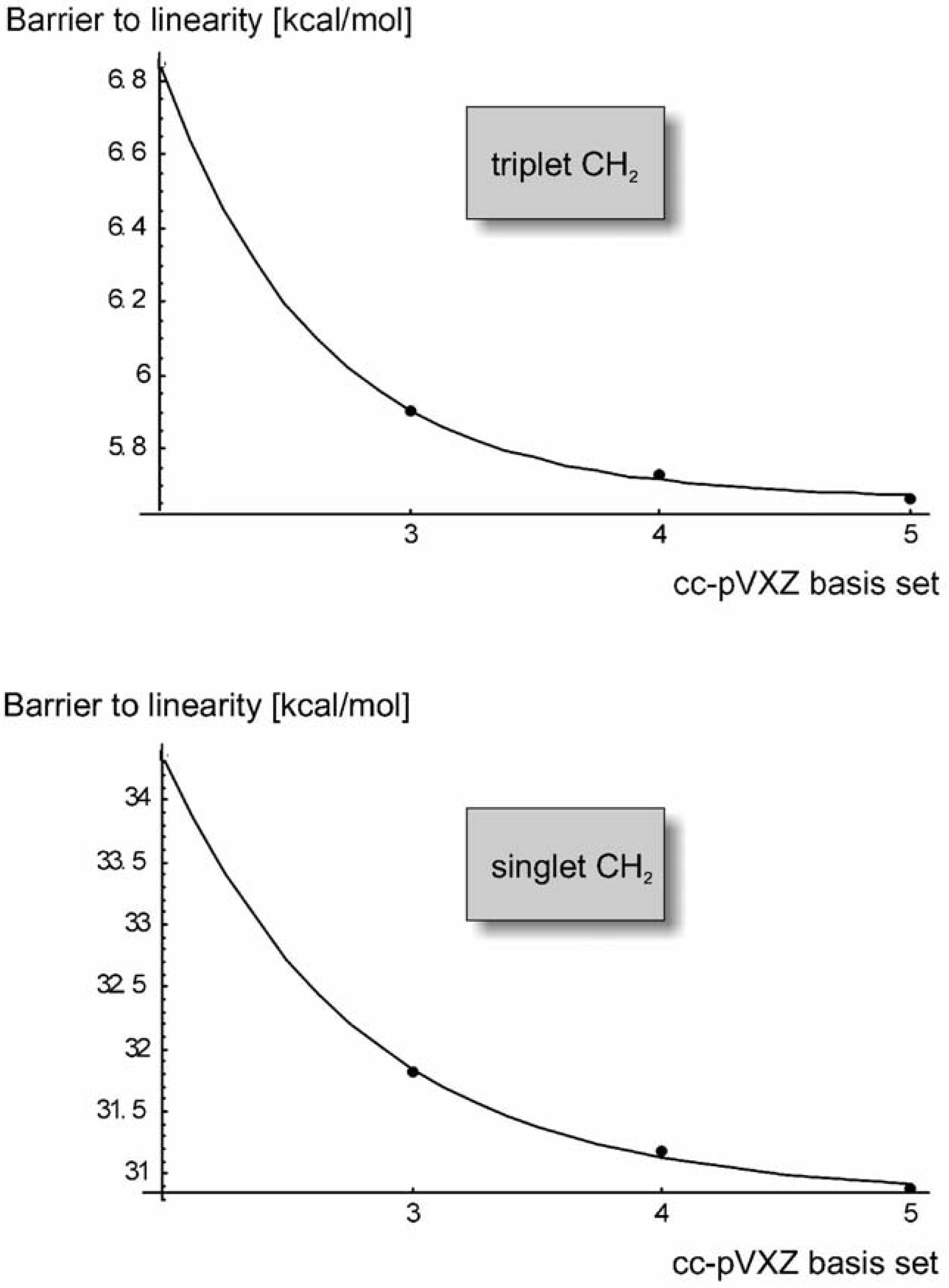
Barriers to Linearity

| cc-pVDZ | 6.8460 | 34.3662 |
| cc-pVTZ | 5.9017 | 31.8179 |
| cc-pVQZ | 5.7323 | 31.1860 |
| cc-pV5Z | 5.6660 | 30.8850 |
| CBS limit | 5.6234 | 30.8422 |
| MCI CAS AE: | 5.9 [49] | CMCI: 25.160 [48] |
| Experiment | 5.54 ± 0.23 [8] | 24.59 ± 1.14 [50] |
| Experiment | 5.478 [9] | 26.88 [51] |
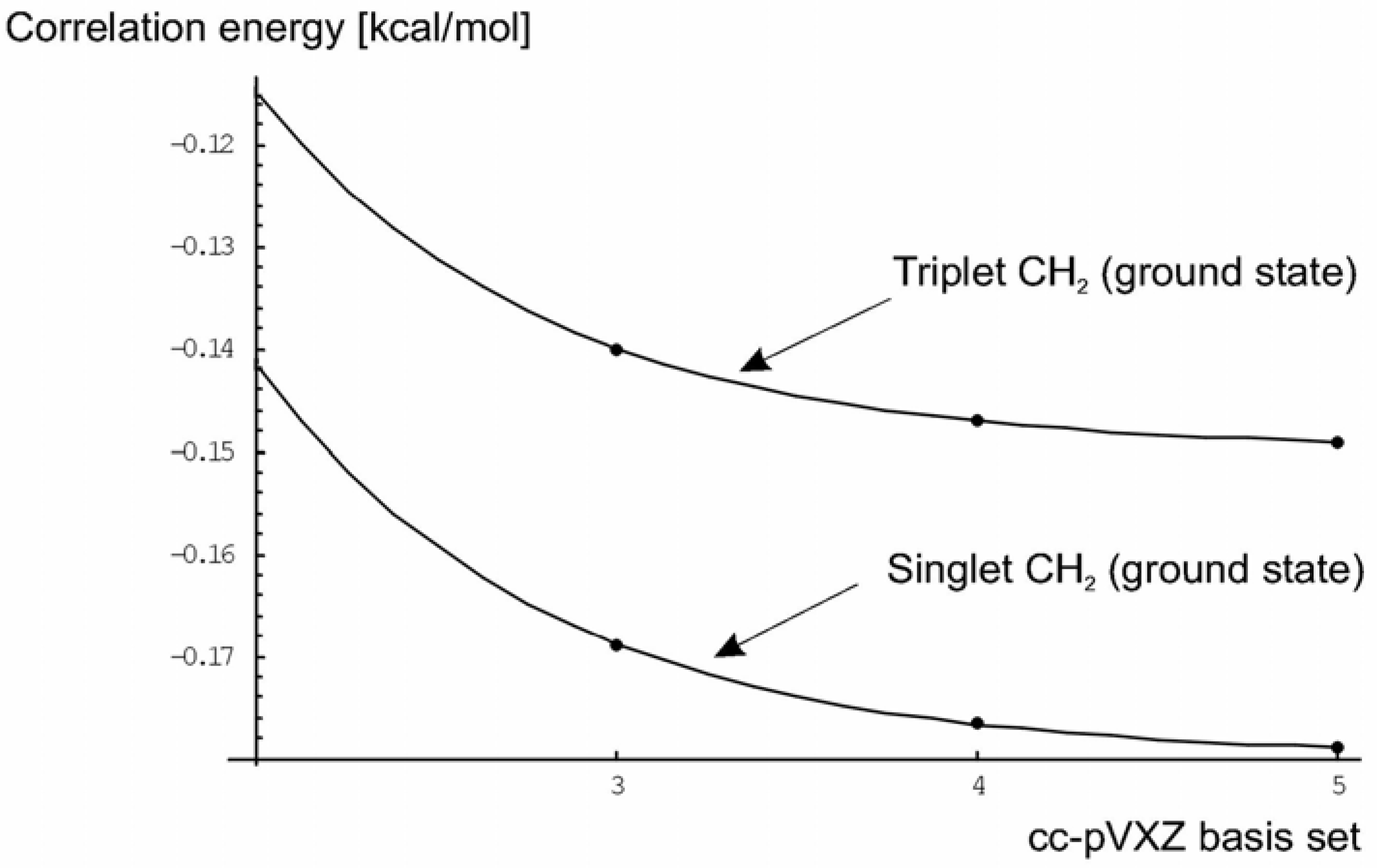
Correlation Energies
| 3B1 CH2 bent 1 | cc-pVDZ | -0.114976 |
| triplet | cc-pVTZ | -0.139995 |
| (ground state) | cc-pVQZ | -0.146980 |
| cc-pV5Z | -0.149101 | |
| CBS limit | -0.150026 | |
| 3Σ- CH2 2 | cc-pVDZ | -0.116565 |
| triplet | cc-pVTZ | -0.142558 |
| (linear) | cc-pVQZ | -0.149845 |
| cc-pV5Z | -0.152062 | |
| CBS limit | -0.153032 | |
| 1A1 CH2 bent 3 | cc-pVDZ | -0.141485 |
| singlet | cc-pVTZ | -0.168839 |
| (ground state) | cc-pVQZ | -0.176555 |
| cc-pV5Z | -0.178935 | |
| CBS limit | -0.179997 | |
| 1Δg CH2 4 | cc-pVDZ | -0.144840 |
| Singlet | cc-pVTZ | -0.173029 |
| (linear) | cc-pVQZ | -0.181233 |
| cc-pV5Z | -0.183755 | |
| CBS limit | -0.184875 |

Conclusions
References
- Shavitt, I. Geometry and Singlet-Triplet Energy Gap in Methylene: A Critical Review of Experimental and Theoretical Determinations. Tetrahedron 1985, 41, 1531–1542. [Google Scholar] [CrossRef]
- Goddard III, W. A. Theoretical Chemistry Comes Alive: Full Partner with Experiment. Science 1985, 227, 917–923. [Google Scholar]
- Schaefer III, H. F. Methylene: A Paradigm for Computational Quantum Chemistry. Science 1986, 231, 1100–1107. [Google Scholar]
- Sherill, C. D.; Van Huis, T. J.; Yamaguchi, Y.; Schaefer III, H. F. Full Configuration Interaction Benchmarks for the 3B1, 1A1, 1B1 and 1A1 States of Methylene. J. Mol. Struct. (Theochem) 1997, 400, 130–156. [Google Scholar]
- Foster, J. M.; Boys, S. F. Quantum Variational Calculations for a Range of CH2 Configurations. Rev. Mol. Phys. 1960, 32, 305–307. [Google Scholar] [CrossRef]
- Herzberg, G.; Shoesmith, J. Spectrum and Structure of the Free Methylene Radical. Nature 1959, 183, 1801–1802. [Google Scholar] [CrossRef]
- Herzberg, G.; Johns, J. W. C. On the Structure of CH2 in its Triplet State. J. Chem. Phys. 1971, 54, 2276–2278. [Google Scholar] [CrossRef]
- Bunker, P. R.; Jensen, P. A Refined Potential Surface for the 3B1 Electronic State of Methylene CH2. J. Chem. Phys. 1983, 79, 1224–1228. [Google Scholar] [CrossRef]
- Jensen, P.; Bunker, P. R. The Potential Surface and Stretching Frequencies of X 3B1 Methylene (CH2) Determined from Experiment Using the Morse Oscillator-Rigid Bender Internal Dynamic Hamiltonian. J. Chem. Phys. 1988, 89, 1327–1332. [Google Scholar] [CrossRef]
- Petek, H.; Nesbitt, D. J.; Darwin, D. C.; Ogilby, P. R.; Moore, C. B. Analysis of CH2 1A1 (1,0,0) and (0,0,1) Coriolis-Coupled States, 1A1 – 3B1 Spin-Orbit Coupling, and the Equilibrium Structure of CH2 1A1 State. J. Chem,. Phys. 1989, 91, 6566–6578. [Google Scholar] [CrossRef]
- Helgaker, T.; Joergensen, P.; Olsen, J. Molecular Electronic Structure Theory; Wiley: Chichester, New York, 2000. [Google Scholar]
- Purvis III, G. D.; Bartlett, R. J. A Full Coupled-Cluster Singles and Doubles Model: The Inclusion of Disconnected Triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G. W.; Pople, J. A.; Head-Gordon, M. A Fifth Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Dunning, T. H., Jr. Gaussian Basis Functions for Use in Molecular Calculations I. Contraction of (9s,5p) Atomic Basis Sets for the First Row Atoms. J. Chem. Phys. 1970, 53, 2823–2833. [Google Scholar] [CrossRef]
- Dunning, T. H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Almlöf, J.; Taylor, P. R. General Contraction of Gaussian Basis Sets. I. Atomic Natural Orbitals for First- and Second-Row Atoms. J. Chem. Phys. 1987, 86, 4070–4077. [Google Scholar] [CrossRef]
- Almlöf, J.; Taylor, P. R. Atomic Natural Orbital (ANO) Basis Sets for Quantum Chemical Calculations. Adv. Quantum Chem. 1992, 22, 301–373. [Google Scholar]
- Helgaker, T.; Taylor, P. R. In Modern Electronic Structure Theory; Yarkony, D. R., Ed.; World Scientific: Singapore, 1995. [Google Scholar]
- O'Neil, S. V.; Schaefer III, H. F.; Bender, C. F. C2v Potential Energy Surfaces for Seven Low-Lying States of CH2. J. Chem. Phys. 1971, 55, 162–169. [Google Scholar] [CrossRef]
- Alexander, S. A.; McDonald, C.; Matsen, F. A. Ab Initio Surfaces of Singlet and Triplet Methylene. Int. J. Quantum Chem. 1983, 17, 407–414. [Google Scholar]
- Comeau, D. C.; Shavitt, I.; Jensen, P.; Bunker, P. R. Ab initio Determination of the Potential-Energy Surfaces and Rotation-Vibration Levels of Methylene in the Lowest Triplet and Singlet States and the Singlet-Triplet Splitting. J. Chem. Phys. 1989, 90, 6491–6500. [Google Scholar] [CrossRef]
- Yu, J.-S.; Chen, S.-Y.; Yu, C.-H. Analytical Fittings for the Global Potential Energy Surface of the Ground State of Methylene. J. Chem. Phys. 2003, 118, 582–594. [Google Scholar] [CrossRef]
- Bauschlicher, C. W., Jr.; Taylor, P. R. A Full CI Treatment of the 1A1-3B1 Separation in Methylene. J. Chem. Phys. 1986, 85, 6510–6512. [Google Scholar] [CrossRef]
- Bauschlicher, C. W., Jr.; Taylor, P. R. Full CI Benchmark Calculations for Several States of the Same Symmetry. J. Chem. Phys. 1987, 86, 2844–2848. [Google Scholar] [CrossRef]
- Bauschlicher, C. W., Jr.; Langhoff, S. R.; Taylor, P. R. 1A1-3B1 Separation in CH2 and SiH2. J. Chem. Phys. 1987, 87, 387–391. [Google Scholar] [CrossRef]
- Bauschlicher, C. W., Jr.; Langhoff, S. R.; Taylor, P. R. Core-Core and Core-Valence Correlation. J. Chem. Phys. 1988, 88, 2540–2546. [Google Scholar] [CrossRef]
- Woon, D. E.; Dunning, T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. V. Core-Valence Basis Sets for Boron through Neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K. A.; Dunning, T. H., Jr. Benchmark Calculations with Correlated Molecular Wave Functions. Bond Energies and Equilibrium Geometries of the CHn and C2Hn (n = 1-4) Series. J. Chem. Phys. 1997, 106, 4119–4140. [Google Scholar]
- Sherrill, C. D.; Leininger, M. L.; Van Huis, T. J.; Schaefer, H. F., III. Structures and Vibrational Frequencies in the Full Configuration Interaction Limit: Predictions of Four Electronic States of Methylene Using a Triple-Zeta Plus Double Polarzation (TZ2P) Basis. J Chem. Phys. 1998, 108, 1040–1049. [Google Scholar] [CrossRef]
- Császár, A. G.; Leininger, M. L.; Szalay, V. The Standard Enthalpy of Formation of CH2. J. Chem. Phys. 2003, 118, 10631–10641. [Google Scholar] [CrossRef]
- Feller, D. Application of Systematic Sequences of Wave Functions of the Water Dimer. J. Chem. Phys. 1992, 96, 6104–6114. [Google Scholar] [CrossRef]
- Neugebauer, A.; Häfelinger, G. Prediction of Experimentally Unknown re-Distances of Organic Molecules from Dunning Basis Set Extrapolations for ab initio Post-HF Calculations. J. Phys. Org. Chem. 2005. in print. [Google Scholar]
- Koch, W.; Holthausen, M. C. A Chemist´s Guide to Density Functional Theory; Wiley-VCH: Weinheim, New York, 1999. [Google Scholar]
- Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, J. A.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, D. J.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98, Revision A.7; Gaussian, Inc.: Pittsburgh, PA, 1998. [Google Scholar]
- Pauling, L.; Wilson, E. B. Introduction to Quantum Mechanics with Applications to Chemistry; McGraw-Hill: New York, 1935. [Google Scholar]
- Coulson, C. A. Valence, 2nd Ed. ed; Oxford University Press: Oxford, U.K, 1961. [Google Scholar]
- Langhoff, S. R.; Davidson, E. R. Configuration Interaction Calculations on the Nitrogen Molecule. Internat. J. Quantum. Chem. 1974, 8, 61–72. [Google Scholar] [CrossRef]
- Zittel, P. F.; Ellison, G. B.; ONeil, S. V.; Herbst, E.; Lineberger, W. C.; Reinhardt, W. P. Laser Photoelectron Spectrometry of CH2- . Singlet-Triplet Splitting and Electron Affinity of CH2. J. Am. Chem. Soc. 1976, 98, 3731–3732. [Google Scholar] [CrossRef]
- Hay, P. J.; Hunt, W. J.; Goddard III, W. A. Generalized Valence Bond Wave Functions for the Low Lying States of Methylene. Chem. Phys. Lett. 1972, 13, 30–35. [Google Scholar] [CrossRef]
- Bender, C. F.; Schaefer III, H. F.; Franceschetti, D. R.; Allen, L. C. Singlet-Triplet Energy Separation, Walsh-Mulliken Diagrams, and Singlet d-Polarization Effects in Methylene. J. Am. Chem. Soc. 1972, 94, 6888–6893. [Google Scholar] [CrossRef]
- Harding, L. B.; Goddard III, W. A. Methylene: ab initio Vibronic Analysis and Reinterpretation of the Spectroscopic and Negative Ion Photoelectron Experiments. Chem. Phys. Lett. 1978, 55, 217–220. [Google Scholar] [CrossRef]
- Leopold, D. G.; Murray, K. K.; Lineberger, W. C. Laser Photoelectron Spectroscopy of Vibrationally Relaxed CH2- : A Reinvestigation of the Singlet-Triplet Splitting in Methylene. J. Chem. Phys. 1984, 81, 1048–1050. [Google Scholar] [CrossRef]
- McKellar, A. R. W.; Bunker, P. R.; Sears, T. J.; Evenson, K. M.; Saykally, R. J.; Langhoff, S. R. Far Infrared Laser Magnetic Resonance of Singlet Methylene: Singlet-Triplet Perturbations, Singlet.Triplet Transitions, and the Singlet-Triplet Splitting. J. Chem. Phys. 1983, 79, 5251–5264. [Google Scholar] [CrossRef]
- Sears, T. J.; Bunker, P. R. A Reinterpretation of the CH2- Photoelectron Spectrum. J. Chem. Phys. 1983, 79, 5265–5271. [Google Scholar] [CrossRef]
- McLean, A. D.; Bunker, P. R.; Escribano, R. M.; Jensen, P. An ab initio Calculation of ν1 and ν3 for Triplet Methylene ( 3B1 CH2) and the Determination of the Vibrationless Singlet-Triplet Splitting Te( 1A1). J. Chem. Phys. 1987, 87, 2166–2169. [Google Scholar] [CrossRef]
- Gu, J.-P.; Hirsch, G.; Buenker, R. J.; Brumm, M.; Osmann, G.; Bunker, P. R.; Jensen, P. A Theoretical Study of the Absorption Spectrum of Singlet CH2. J. Molec. Struct. 2000, 517+518, 247–264. [Google Scholar] [CrossRef]
- Green, W. H., Jr.; Handy, N. C.; Knowles, P. J.; Carter, S. Theoretical Assignment of the Visible Spectrum of Singlet Methylene. J. Chem. Phys. 1991, 94, 118–132. [Google Scholar] [CrossRef]
- Harding, L. B.; Guadagnini, R.; Schatz, G. C. Theoretical Study of the Reactions H + CH →C + H2 and C + H2 → CH2 Using an ab initio Global Ground-State Potential Surface for CH2. J. Phys. Chem. 1993, 97, 5472–5481. [Google Scholar] [CrossRef]
- Hartland, G. V.; Qin, D.; Dai, H,-L. Renner-Teller Effect on the Highly Excited Bending Levels of 1A1 CH2. J. Chem. Phys. 1995, 102, 6641–6645. [Google Scholar] [CrossRef]
- Duxbury, G.; Alijah, A.; McDonald, B. D.; Jungen, C. Stretch-Bender Calculations of the Effects of Orbital Angular Momentum and Vibrational Resonances in the Spectrum of Singlet Methylene. J. Chem. Phys. 1998, 108, 2351–2360. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Sherill, C. D.; Schaefer, H. F., III. The 3B1, 1A1, 1B1 and 1A1 Electronic States of CH2. J. Phys. Chem. 1996, 100, 7911–7918. [Google Scholar] [CrossRef]
© 2005 by MDPI (http://www.mdpi.org).
Share and Cite
Neugebauer, A.; Häfelinger, G. Ab initio post-HF CCSD(T) Calculations for Triplet and Singlet Methylene in Four consecutive Dunning Basis Sets with Extrapolations to Infinite Limits for Various Molecular Properties. Int. J. Mol. Sci. 2005, 6, 157-176. https://doi.org/10.3390/i6010157
Neugebauer A, Häfelinger G. Ab initio post-HF CCSD(T) Calculations for Triplet and Singlet Methylene in Four consecutive Dunning Basis Sets with Extrapolations to Infinite Limits for Various Molecular Properties. International Journal of Molecular Sciences. 2005; 6(1):157-176. https://doi.org/10.3390/i6010157
Chicago/Turabian StyleNeugebauer, Alexander, and Günter Häfelinger. 2005. "Ab initio post-HF CCSD(T) Calculations for Triplet and Singlet Methylene in Four consecutive Dunning Basis Sets with Extrapolations to Infinite Limits for Various Molecular Properties" International Journal of Molecular Sciences 6, no. 1: 157-176. https://doi.org/10.3390/i6010157