The SPASIBA Force Field for Studying Iron-Tannins Interactions : Application to Fe3+ /Fe2+ Catechol Complexe

Abstract
:1. Introduction
2. Theoretical Calculations
2.1. Density Functional Theory
2.2 Empirical force field
3. Results and Discussion
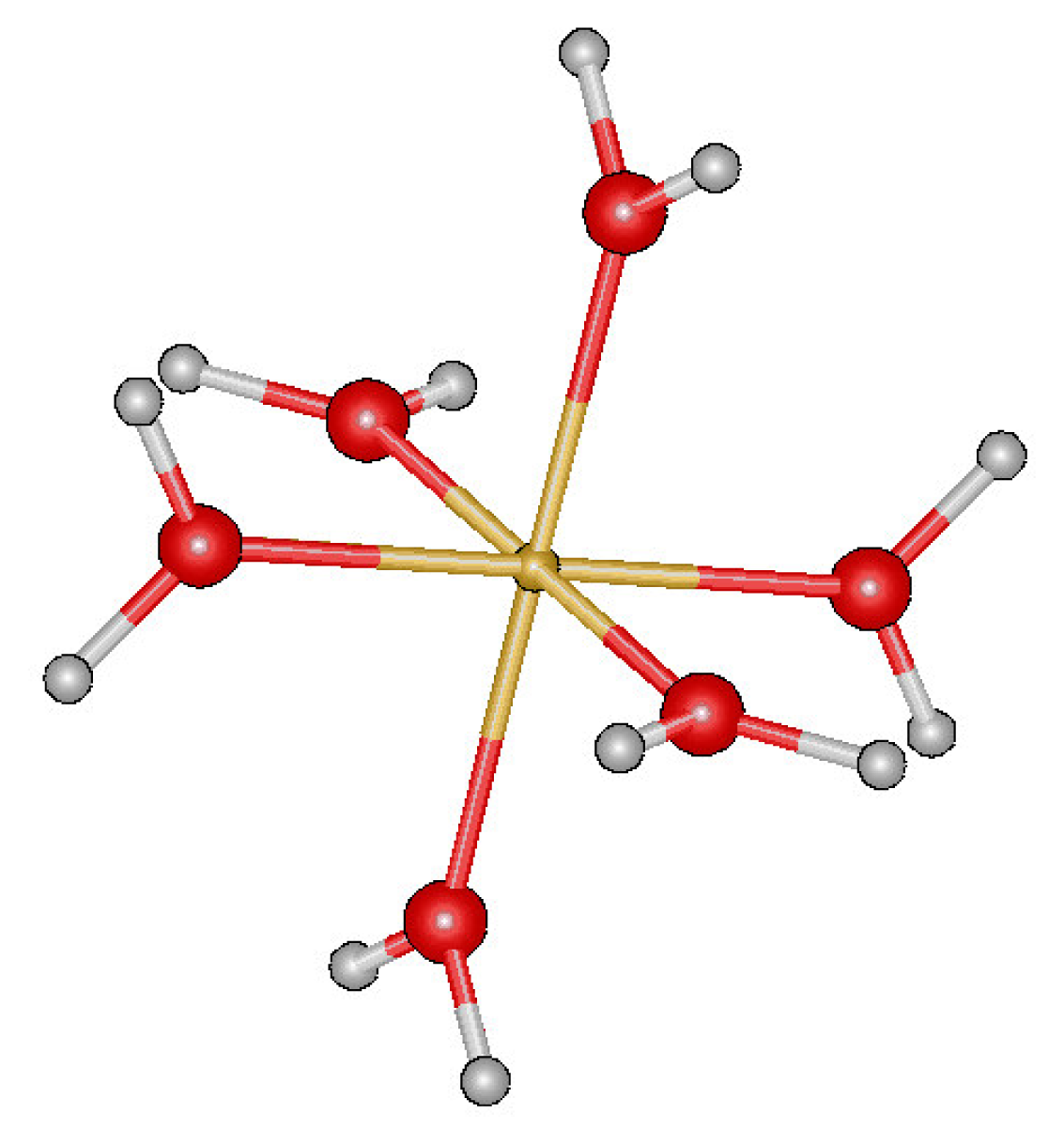
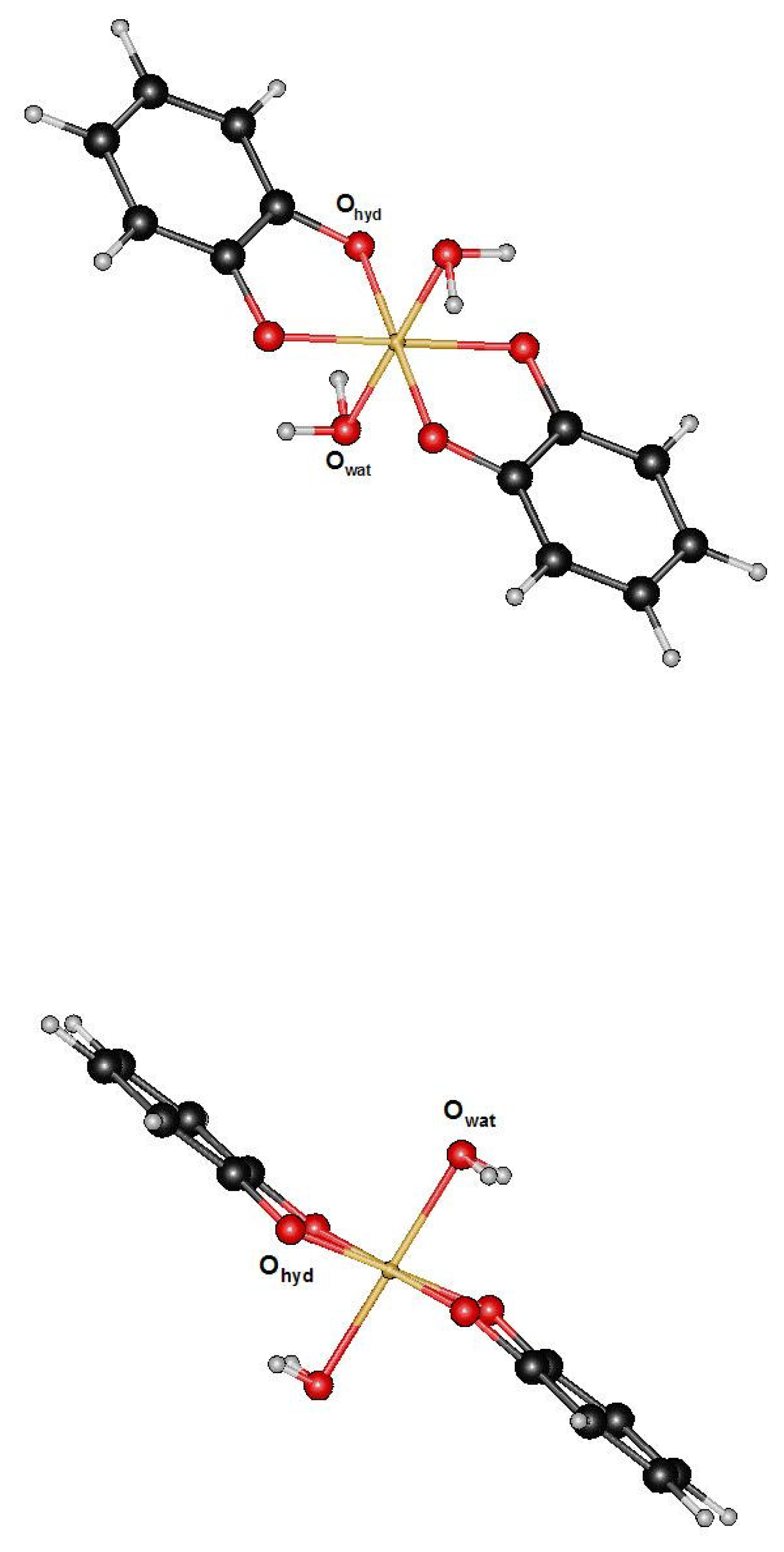
3.1. Fe(III) complexes [Fe(H2O)6]3+ and [Fe(Cat)2(H2O)2]−1
3.1.1. DFT calculations
3.1.2. SPASIBA parameters for Fe[(H2O)6]3+ -[Fe(Cat)2(H2O)2]−1
3.1.3. Normal modes analysis
3.2. [Fe(H2O)6]2+ -[Fe(Cat)2(H2O)2]−2 Fe(II)complexes
3.2.1. DFT calculations
3.2.2. SPASIBA parameters for the Ferrous complexes
3.2.3. Normal modes
Conclusion



{kind=link}
{kind=link}
| Complex | Method | Basis Set | Multiplicity | Global charge |
|---|---|---|---|---|
| [Fe(H2O)6]3+ | B3LYP | 6-31G(tm)**+/LAVCP** | 6 (High Spin) | +3 |
| [Fe(Cat)2(H2O)2]−1 | B3LYP | LAVCP** | 6 (High Spin) | −1 |
| [Fe(H2O)6]2+ | B3LYP | 6-31G(tm)**/6-31G(tm)**+ | 5 (High Spin) | +2 |
| [Fe(Cat)2(H20)2]−2 | B3LYP | LAVCP** | 5 (High Spin) | −2 |
| Parameters | Experimental | DFT | SPASIBA |
|---|---|---|---|
| Fe-O | 1.994–2.002(a),2.039(b) | 2.052(0.0) | 1.957–2.003(0.001) |
| O-H | 0.995–1.002(a),0.973(b) | 0.978(0.0) | 0.996(0.0) |
| O-Fe-O (direct angle) | 90.5–90.9(a),90.0(b) | 90.0(0.041) | 90.0(1.098) |
| O-Fe-O | - | 179.95(0.024) | 179.51(0.021) |
| Fe-O-H | - | 126.4(0.036) | 129.94(0.372) |
| H-O-H | 108.0–110.4(b,c) | 107.2(0.02) | 100.08(0.178) |
| Parameters | Experimental(a) | DFT | SPASIBA |
|---|---|---|---|
| Fe-Owat | 2.0 | 2.038 (0.105) | 1.957(0.029) |
| O-H | 0.96 | 0.95 (0.0) | 0.977 |
| C-C | 1.398 | 1.405 (0.011) | 1.404 |
| C-O | 1.364 | 1.340 (0.001) | 1.413 |
| C-H | 1.086 | 1.088 (0.0) | 1.100 |
| C-O-Fe | - | 109.51 (0.036) | 107.54 |
| C-C-C | 120.0 | 120.12 | 120.0 |
| C-C-O | 120.0 | 120.13 | 119.63 |
| O-Fe-O (direct angle) | 90.5–90.9 | 90.0 (0.041) | 90.0 |
| O-Fe-O | 180.0 | 179.97 (0.024) | 179.51 |
| Fe-O-C | - | 109.51 | 107.54 |
| Fe-O-H | - | 102.33 (0.014) | 110.0 |
| H-O-H | 104.5 | 104.2 (0.014) | 107.03 |
| Interplanar distance(in Å) | 0.60 | 0.13 |
| Bonds | K (Kcal.mol−1) | R0 (Å) | |
|---|---|---|---|
| Fe-Ohyd | 117.0 | 2.00 | |
| Fe-Owat | 117.0 | 2.00 | |
| O-H | 511.0 | 0.98 | |
| C-O | 315. | 1.41 | |
| C-C | 2.90 | 1.40 | |
| Valence angle | H (Kcal.mol−1.rad−2) | Θ0 (degrees) | F(Kcal.mol−1. Å2) |
| Ohyd-Fe-Ohyd* | 10.0 | 91.0 | 9.0 |
| Ohyd-Fe-Ohyd** | 10.0 | 91.0 | 9.0 |
| Ohyd-Fe-Ohyd** | 10.0 | 179.0 | 9.0 |
| Ohyd-Fe-Owat | 10.0 | 91.0 | 9.0 |
| C-O-Fe | 41.0 | 109.0 | 19.0 |
| Fe-O-H | 20.0 | 120.0 | 0. |
| H-O-H | 33.0 | 107.0 | 91.0 |
| C-C-O | 34.0 | 120.0 | 85.0 |
| C-C-H | 32.0 | 120.0 | 0. |
| C-C-C | 45.0 | 120.0 | 30. |
| C-O-Fe | 41.0 | 109.0 | 19. |
| Torsions | Vn/2(Kcal.mol−1) | Phase (degrees) | n (order) |
| X-Fe-O-X | 0.045 | 0.0 | 3 |
| Assignments | Exp.(a) | DFT | SPASIBA |
|---|---|---|---|
| τ Fe-O | 59.1–258.8 | 32.9–274.7 | |
| δ O-Fe-O | 306–332 | 275.6–357.3 | 247.7–330.1 |
| ν Fe-O | 475–523 | 349.8–493 | 371–495.6 |
| δ Fe-O-C | - | 746.1–774.8 | 736.2–736.7 |
| 404.7–517.4(1) | 433.7–511.4(1) | ||
| δ Fe-O-H | 721–746(2) | 641.5–646.1(2) | 672–690.6(2) |
| 867.8–914.2(3) | 889.7–937.7(3) |
| Parameters | Exp. (a) | DFT | SPASIBA |
|---|---|---|---|
| Fe-O | 2.098–2.143 | 2.085–2.143 | 2.02–2.077 |
| O-Fe-O | 89.25–91.02 | 90.0 | 89.96–90.0 |
| O-Fe-O | - | 179.79 | 179.35 |
| Fe-O-H | - | 124.81 | 127.92 |
| H-O-H | 110.38 | 103.93 |
| Parameters | Exp. (b) | DFT | SPASIBA |
|---|---|---|---|
| Fe-O | 2.06–2.19 | 2.085 | 2.02 |
| O-H | 0.96 | 0.98 | 0.981 |
| C-C | 1.398 | 1.411 | 1.406 |
| C-O | 1.364 | 1.327 | 1.337 |
| C-H | 1.086 | 1.091 | 1.089 |
| C-O-Fe | 120.0 | 111.97 | 110.52 |
| C-C-C | 120.0 | 120.0 | 120.0 |
| C-C-H | 120.0 | 119.69 | 120.1 |
| C-C-O | 120.0 | 120.61 | 119.69 |
| O-Fe-O | 90.0 | 90.0 | 90.0 |
| O-Fe-O | 180.0 | 180.0 | 179.37 |
| Fe-O-H | - | 106.6 | 107.69 |
| H-O-H | 104.5 | 100.15 | 105.02 |
| Interplanar distance (Å) | 0.49 | 0.20 |
| Bonds | K(kcal.mol−1Å) | Ro(Å) | |
|---|---|---|---|
| Fe-O | 65.0 | 2.13 | |
| O-H | 511.0 | 0.98 | |
| C-O | 315.0 | 1.41 | |
| C-C | 290. | 1.40 | |
| Bendings | H(kcal.mol−1.rad−2) | θo(degrees) | F(kcal.mol−1.Å−2) |
| O-Fe-O | 4.0 | 89.0 | 2.0 |
| O-Fe-O | 4.0 | 179.0 | 2.0 |
| C-O-Fe | 46.0 | 112.0 | 21.0 |
| Fe-O-H | 18.0 | 120.0 | 0.0 |
| C-C-O | 34.0 | 120.0 | 85.0 |
| C-C-H | 32.0 | 120.0 | 0. |
| C-C-C | 45.0 | 120.0 | 30. |
| C-O-Fe | 46.0 | 112.0 | 21.0 |
| Torsions | Vn/2(kcalmol−1) | Phase(degrees) | n (order) |
| X-Fe-O-X | 0.043 | 0.0 | 3. |
| X-CA-CA-X | 3.925 | 180.0 | 2. |
| X-CA-O-X | 1.80 | 180.0 | 2. |
| Exp. (a) | DFT | SPASIBA | |
|---|---|---|---|
| τ Fe-O | - | 33–117.3 | 39.2–137.5 |
| δ O-Fe-O | 210–231 | 81.3–258.7 | 97.3–228.8 |
| ν Fe-O | 296–379 | 242.1–515.9 | 269 –493.7 |
| δ Fe-O-C | - | 712.7–714.2 | 725.4–726.5 |
| δ Fe-O-H | - | 524.4–688(1) | 509.1–674.5(1) |
| 857.6–882.5(2) | 871.3–906.3(2) |
References
- Spencer, C. M.; Cai, Y.; Martin, R.; Gaffney, S. H.; Goulding, P. N.; Magnolato, D.; Lilley, T. H.; Haslam, E. Polyphenol complexation –Some thoughts and observations. Phytochemistry 1988, 27, 2397–2409. [Google Scholar]
- Horton, S.; Levin, C. Commentaries on “Evidence that Iron deficiency Anemia causes reduced work capacity”. J. Nutr 2001, 131, 691S–696S. [Google Scholar]
- Cook, J. D.; Reddy, M. B.; Hurrel, R. F. The effect of read and white wines on nonheme-iron absorption in Humans. Am. J. Clin .Nutr 1995, 61, 800–804. [Google Scholar]
- Frankel, E. N.; German, J. B.; Kinsella, J. E.; Parks, E.; Kanner, J. Inhibition of oxidation of human low-density lipoprotein by phenolic substance in red wine. The Lancet 1993, 341, 454–457. [Google Scholar]
- Lokman, K.; Haslam, E.; Williamson, M. P. Structure and conformation of the procyanidin B2-dimer. Magnetic Resonance in Chemistry 1997, 35, 854–858. [Google Scholar]
- Hemingway, R. W.; Tobiason, F. L.; Wayne Mc Graw, G.; Steynberg, J. P. Conformation and complexation of tannins: NMR spectra and Molecular Search Modeling of Flavan-3-cis Magnetic Resonance in chemistry. Magnetic Resonance in Chemistry 1996, 34, 424–433. [Google Scholar]
- Lambrinidis, G.; Halabalaki, M.; Katsanou, E. S.; Skaltsounis, A. L.; Alexis, M. N.; Mikros, E. The estrogen receptor and polyphenols: molecular simulation studies of their interactions, a review. Environnmental Chemistry Letters 2006, 4, 159–174. [Google Scholar]
- Jarzecki, A. A.; Anbar, A. D.; Spiro, T. G. DFT Analysis of Fe(H20)63+ and Fe(H20)62+. Structure and vibrations; Implications for Isotope Fractionation. J. Phys. Chem. A 2004, 108, 2726–2732. [Google Scholar]
- Yamahara, R.; Ogo, S.; Masuda, H.; Watanabe, Y. J. (Catecholato) iron(III) complexes: Structural and functional models for the catechol-bound iron(III) form of catechol dioxygenases. Inorg. Biochem. 2002, 88, 284–294. [Google Scholar]
- Wong, M. W. Vibrational frequency prediction using density functional theory. Chem. Phys. Letters 1996, 256, 391–399. [Google Scholar]
- Jaguar Program, 4.0; Schrödinger, Inc.: Portland, OR, 1999.
- Derreumaux, P.; Vergoten, G. A new spectroscopic molecular mechanics force field. Parameters for proteins. J. Chem. Phys 1995, 102, 8586–8605. [Google Scholar]
- Vergoten, G.; Mazur, I.; Lagant, P.; Michalski, J. C.; Zanetta, J. P. The SPASIBA force field as an essential tool for studying the structure and dynamics of saccharides. Biochimie 2003, 85, 65–73. [Google Scholar]
- Lagant, P.; Nolde, D.; Stote, R.; Vergoten, G.; Karplus, M. Increasing normal mode analysis accuracy: The SPASIBA spectroscopic force field introduced into the CHARMM Program. J. Phys. Chem. A 2004, 108, 4019–4029. [Google Scholar]
- Orville, A. M.; Lipscomb, J. D.; Ohlendorf, D. H. Binding of isotopically labeled substrates inhibitors, and cyanide by protocatechuate 3,4-dioxygenase. J. Biol. Chem 1989, 264, 8791–8801. [Google Scholar]
- Whittaker, J. W.; Lipscomb, J. D.; Kent, T. A.; Münck, E. Brevibacterium fuscum protocatechuate 3,4-dioxygenase. Purification, crystallization and characterization. J. Biol. Chem. 1984, 259, 4466–4475. [Google Scholar]
- Best, S. P.; Forsyth, J. B. Stereochemistry of tervalent aqua ions: Low-temperature Neutron Diffraction. Structure of CsFe(SO4)2.12H2O and CsFe(SeO4)2.12H2O. J. Chem. Soc. Dalton Trans. 1990, 395–400. [Google Scholar]
- Durier, V.; Tristram, F.; Vergoten, G. Molecular force field development for saccharides using the SPASIBA spectroscopic potential. Force field parameters for α-D-Glucose. J. Mol. Struct. (Theochem) 1997, 395–396, 81–90. [Google Scholar]
- Funabiki, T.; Yamazaki, T. Mechanism of oxygenative cleavage of catechols by nonheme iron complexs in relevance to catechol dioxygenases studied by quantum chemical calculations. Journal of Molecular Catalysis. A: Chemical 1999, 150, 37–47. [Google Scholar]
- Torrens, F. Nature of FeIII-O2, FeII-CO and FeIII-CN complexes of hemoprotein models. Polyhedron 2003, 22, 1091–1098. [Google Scholar]
- Allouche, A.; Pourcin, B. Ab initio calculation of vibrational force fields: Determination of non-redundant symmetry coordinates by least-square component analysis. Spectrochim. Acta 1993, 49A, 571–580. [Google Scholar]
- Pulay, P. Possibilities and limitations of ab initio calculatio of vibrational spectra. J. Mol. Struct 1995, 347, 293–308. [Google Scholar]
- Best, S. P.; Armstrong, R. S.; Beattie, J. K. Single-crystal Raman spectroscopy of the α-Alums CsM(SO4)2.12H2O (M=Co or Ir) between 275 and 1200 cm−1). J. Chem. Soc., Dalton Trans. 1992, 299–304. [Google Scholar]
- Cotton, F.A.; Daniels, L. M.; Murillo, C. A.; Quesada, J. F. Hexaaqua dispositive ions of the first transition series: new and accurate structures; expected and unexpected trends. Inorg. Chem 1993, 32, 486. [Google Scholar]
- Jenkins, T. E.; Lewis, J. A Raman investigation of some metal(II) hexafluorosilicate(IV) and hexafluorotitanate(IV) salts. Spectrochim. Acta. PartA: Mol. Spectrosc 1981, 37A, 47–50. [Google Scholar]
- Merlel, M.; Muller, V.; Krebs, B. Novel iron(III) comolexes with phenolate containing tetradentate ligands as model systems for catechol 1,2-dioxygenase. Inorganica Chimica Acta 2002, 337, 308–316. [Google Scholar]
- Joseph, M.; Suni, V.; Nayar, C. R.; Kurup, M. R. P.; Hoong-Kun-Fun. Synthesis, spectral characterization and crystal structure of 2-benzylpyridine N(4) cyclohexylthiosemicarbazone. J. Mol. Struct. 2004, 705, 63–70. [Google Scholar]
- Chis, V. Molecular and vibrational structure of 2,4 dinitrophenol: FT-IR, FT-Raman and quantum chemical calculations. Chem. Phys 2004, 300, 1–11. [Google Scholar]
- Kagawa, T.; Kawai, R.; Kashino, S.; Haisa, M. The crystal and molecular structure of 2,4 dinitrophenol. Acta. Cryst. B32 1976, 3171. [Google Scholar]
- Cabral, B. J. C.; Gil, R.; Fonseca, B.; Simoes, J. A. M. Density functional and density functional reaction field calculations of the molecular properties of phenol. Chem. Phys. Lett 1996, 258, 436–444. [Google Scholar]
- Hämäläinen, R.; Urpeinen, U. The structure, Thermal and Magnetic properties of hexaquaion(II) bis[bis(N-salicylideneglycinato) ferrata(III)] dihydrate. Acta. Chem. Scand 1989, 43, 15–18. [Google Scholar]
- Scott, R.; Wang, S.; Eidsness, M. K.; Kriauciunas, A.; Frolich, C. A.; Chen, V. J. X-ray absorption spectroscopic studies of the high-spin iron(II) active site of isopenicillin N-synthase: evidence for iron-sulfur interaction in the enzyme-substrate complex. Biochemistry 1992, 31, 4596–4601. [Google Scholar]
- Dimitrimova, Y. Ab initio and DFT studies of the vibrational spectra of hydrogen-bonded phOH (H2O)4 complexes. Spectochim. Acta, Part A 2004, 60, 3049–3057. [Google Scholar]
- Sanchez-Cortès, S.; Garcia-Ramos, J. V. Adsorption and chemical modification of phenols on a silver surface. Journal of Colloid and Interface Science 2000, 231, 98–106. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Yapo-Kicho, D.; Lagant, P.; Vergoten, G. The SPASIBA Force Field for Studying Iron-Tannins Interactions : Application to Fe3+ /Fe2+ Catechol Complexe. Int. J. Mol. Sci. 2007, 8, 259-272. https://doi.org/10.3390/i8030259
Yapo-Kicho D, Lagant P, Vergoten G. The SPASIBA Force Field for Studying Iron-Tannins Interactions : Application to Fe3+ /Fe2+ Catechol Complexe. International Journal of Molecular Sciences. 2007; 8(3):259-272. https://doi.org/10.3390/i8030259
Chicago/Turabian StyleYapo-Kicho, D., P. Lagant, and G. Vergoten. 2007. "The SPASIBA Force Field for Studying Iron-Tannins Interactions : Application to Fe3+ /Fe2+ Catechol Complexe" International Journal of Molecular Sciences 8, no. 3: 259-272. https://doi.org/10.3390/i8030259