Synthesis and Characterization of Some New Tetraaldehyde and Tetraketone Derivatives and X-ray Structure of 1,1'-(4,4'-(2- (1,3-bis(4-Acetylphenoxy)propan-2-ylidene)propane-1,3-diyl) bis(oxy)bis(4,1-phenylene))diethanone

Abstract
:Introduction
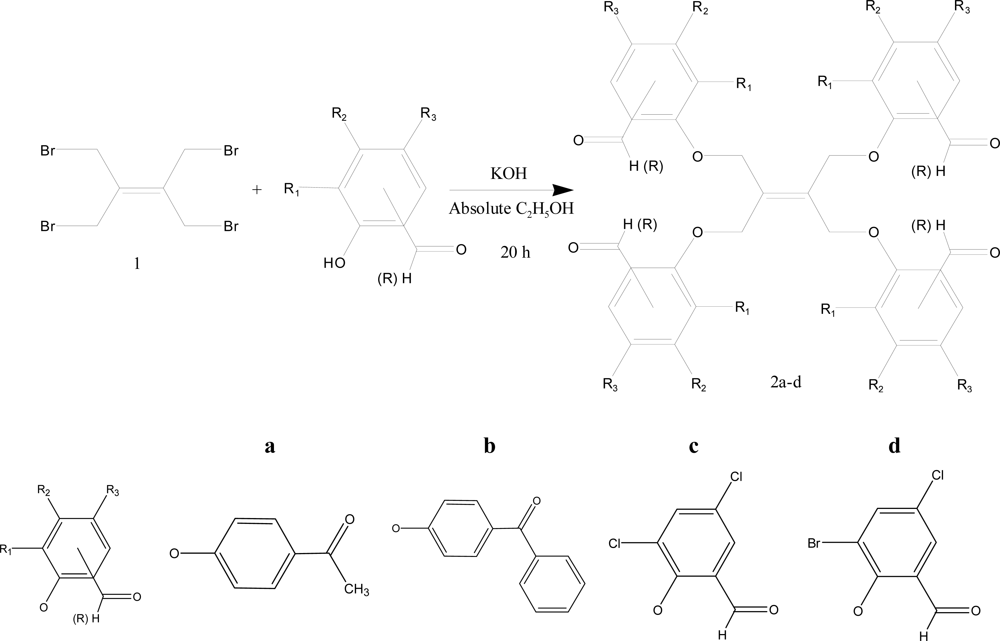
2. Experimental
2.1. Materials
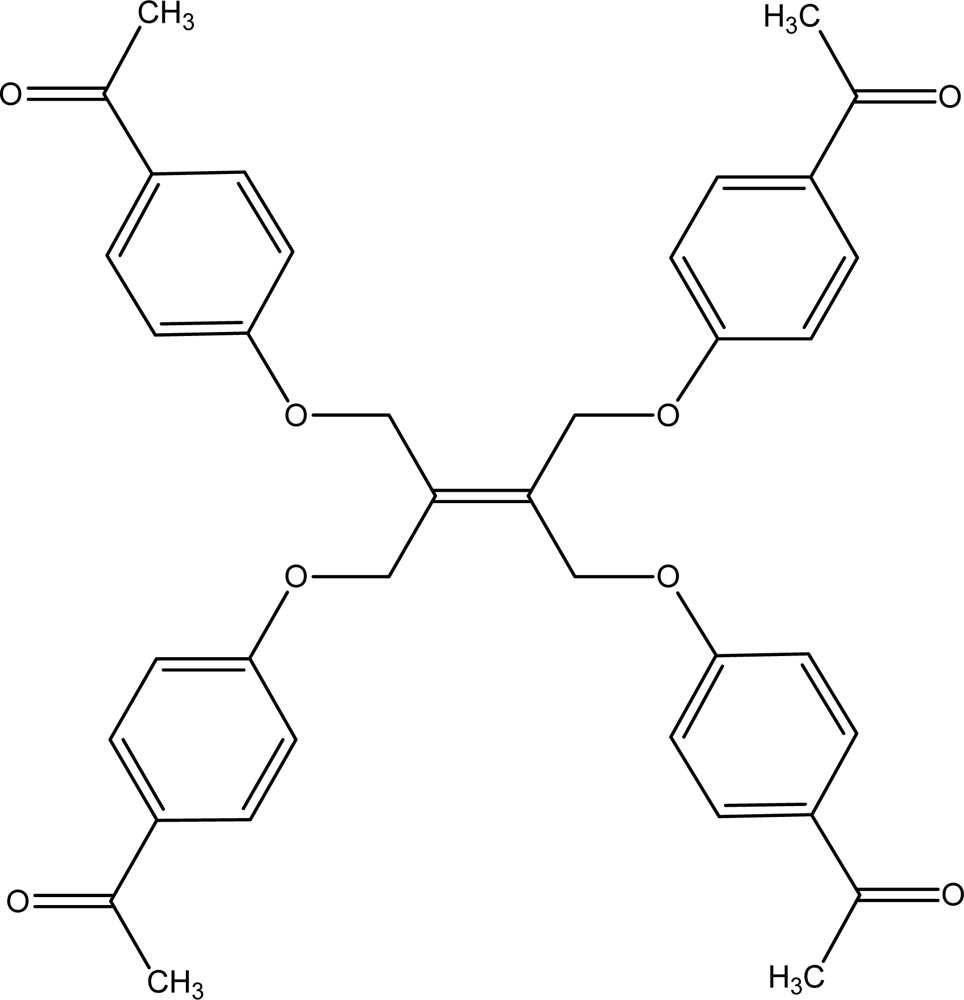
2.2. Synthesis of 1,1′-(4,4′-(2-(1,3-bis(4-acetylphenoxy)propan-2-ylidene)propane-1,3-di-yl)bis(oxy)bis (4,1-phenylene))diethanone (2a)
2.3. Synthesis of (4,4′-(2-(1,3-bis(4-benzoylphenoxy)propan-2-ylidene)propane-1,3-diyl)bis(oxy)bis (4,1-phenylene))bis(phenylmethanone) (2b)
2.4. Synthesis of 2,2′-(2-(1,3-bis(2,4-dichloro-6-formylphenoxy)propan-2-ylidene)propane-1,3-diyl)bis (oxy)bis(3,5-dichlorobenzaldehyde) (2c)
2.5. Synthesis of 2,2′-(2-(1,3-bis(2-bromo-4-chloro-6-formylphenoxy)propan-2-ylidene)propane-1,3-diyl)bis (oxy)bis(3-chloro-5-bromo benzaldehyde) (2d)
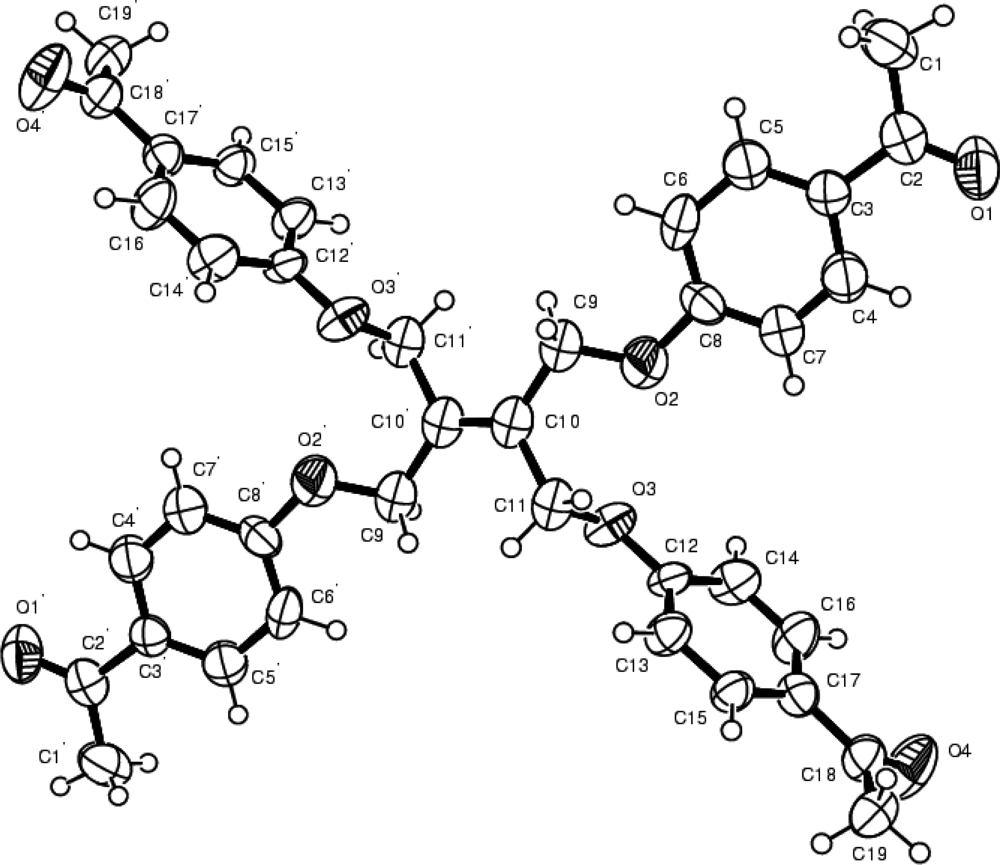
2.6. Crystallographic structure determination compound 2a
3. Results and Discussion
4. Conclusions
Acknowledgments
References and Notes
- Singh, BK; Jetley, UK; Sharma, RK; Garg, BS. Synthesis, characterization and biological activity of complexes of 2-hydroxy-3,5-dimethylacetophenoneoxime (HDMAOX) with copper(II), cobalt(II), nickel(II) and palladium(II). Spectrochimica Acta Part A 2007, 68, 63–73. [Google Scholar]
- Rajabi, L; Courreges, C; Montoya, J; Aguilera, RJ; Primm, TP. Acetophenones with selective antimycobacterial activity. Lett. Appl. Microbiol. 2005, 40, 212. [Google Scholar]
- Gündoğdu-Karaburun, N; Benkli, K; Tunalı, Y; Uçucu, Ü; Demirayak, Ş. Synthesis and antifungal activities of some aryl [3-(imidazol-1-yl/triazol-1-ylmethyl)benzofuran-2-yl] ketoximes. Eur. J. Med. Chem. 2006, 41, 651–656. [Google Scholar]
- Jung, KY; Kim, SK; Gao, ZG; Gross, AS; Melman, N; Jacobson, KA; Kim, YC. Structure-activity relationships of thiazole and thiadiazole derivatives as potent and selective human adenosine A[3] receptor antagonists. Bioorg. Med. Chem. 2004, 12, 613–623. [Google Scholar]
- Gul, HI; Denizci, AA; Erciyas, E. Antimicrobial evaluation of some Mannish bases of acetophenones and representative quaternary derivatives. Arzneimittel Forschung. 2002, 52, 773. [Google Scholar]
- Teruyuki, M; Yoshiharu, H; Ka, Y. Oxime derivative thereof, Process for preparing thereof, Herbicidal composition and methods for the destruction of undesirable weeds. Asahi Chemical Ind., Japan 1986. [Google Scholar]
- Miyazawa, M; Shimamura, H; Nakamura, S. Suppression of furylfuramide-induced SOS response by acetophenones using Salmonella typhimurium TA1535/pSK1002 umu test. J. Agric. Food Chem. 2000, 48, 4377. [Google Scholar]
- Guyot, C; Bouseta, A; Scheirman, VV. Floral origin markers of chestnut and lime tree honeys. J. Agric. Food Chem. 1998, 46, 625. [Google Scholar]
- Wilkins, CK; Scholl, S. Volatile metabolites of some barley storage molds. Int. J. Food Microbiol. 1989, 8, 11. [Google Scholar]
- Yasuda, T; Kon, R; Nakazawa, T; Ohsawa, K. Metabolism of Paeonol in Rats. J. Nat. Product 1999, 62, 1142. [Google Scholar]
- Soykan, C; Erol, İ. Synthesis, Characterization, and Biologicaj Activity of N-(4-Acetylphenyl) maleimide and Its Oxime, Carbazone, Thiosemicarbazone Derivatives and Their Polymers. J.Polymer Science, Polymer Chem. 2003, 41, 1942–1951. [Google Scholar]
- Poddar, SN. Ortho-hydroxy acetophenone oxime as an analytical reagent. Part I. Analyt. Bioanalyt. Chem. 1957, 154, 254. [Google Scholar]
- Yadav, JS; Reddy, PT; Nanda, S; Rao, AB. Stereoselective synthesis of (R)-(–)-denopamine, (R)-(–)-tembamide and (R)-(–)-aegeline via asymmetric reduction of azidoketones by Daucus carota in aqueous medium. Tetrahedron:Asymmetry 2001, 12, 3381–3385. [Google Scholar]
- Cope, AC; Kagan, F. Cyclic Polyolefins. J. Am. Chem. Soc. 1958, 80, 5499–5502. [Google Scholar]
- Siemens: SMART and SAINT. Area Detector Control and Integration Software; Siemens Analytical X-ray Systems Inc.: Madison, Wisconsin, USA, 1996. [Google Scholar]
- Sheldrick, GM. SADABS. Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Germany, 1996. [Google Scholar]
- Karle, J; Karle, I. The symbolic addition procedure for phase determination for centrosymmetric and non-centrosymmetric crystals. Acta Crystallogr. 1996, 21, 849–859. [Google Scholar]
- International Tables for X-ray Crystallography; Kynoch; Birmingham, UK, 1974; Vol. IV, (present distributor: Reidel: Dordrecht, Netherlands).
- Ustabaş, R; Çoruh, U; Er, M; Serbest, K; Vazquez-Lopez, EM. 2,2′-[2,3-Bis(1-formyl-2-naphthyloxymethyl)but-2-ene-1,4-diyldioxy]bis(naphthalene-1-carbaldehyde. Acta Cryst. 2006, E62, o5006–o5007. [Google Scholar]
- Zhang, HY; Sun, H; Mu, SC; Wang, JK; Chen, W. trans-1,4-Bis(pyrimidin-2-ylsulfanyl)but-2-ene. Acta Cryst. 2005, E61, o4142–o4143. [Google Scholar]
- Wang, J; Wang, W; Chi, HJ; Yang, QS. trans-2,2′-[(2-Butene-1,4-diyl)dithio]bis(4,5-dihydro-1,3-thiazine). Acta Cryst. 2006, E62, o4621–o4622. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Chemical formula | C38H36O8 | α(°) | 90 |
| Formula weight | 620.67 | β(°) | 95.875(3) |
| Crystal colour, habit | Colourless, prism | γ(°) | 90 |
| Crystal system | Monoclinic | V(Å3) | 1566.8(4) |
| Crystal dimensions | 0.30×021×0.21 | Z | 2 |
| Space group | P 21/n | Dcalc(g/cm3) | 1.316 |
| a(Å) | 9.0395(12) | μ(Mo Kα), cm−1 | 0.092 |
| b(Å) | 12.6114(17) 13.8166(18) | No unique reflections | 8396 |
| c(Å) | No of observations | 2430 | |
| R | 0.0481 | ||
| Rw | 0.0722 |
| C10-C10’ 1.332(6) C15-C17-C16 118.7(3) |
| O1-C2 1.218(3) C15-C17-C18 123.7(3) |
| O4-C18 1.226(3) C16-C17-C18 117.5(3) |
| C4-C3-C5 118.0(3) C8-O2-C9-C10 159.8(2) |
| C4-C3-C2 119.9(3) C12-O3-C11-C10 −173.0(2) |
| C5-C3-C2 122.0(3) |
Share and Cite
Er, M.; Ustabaş, R.; Çoruh, U.; Sancak, K.; Vázquez-López, E. Synthesis and Characterization of Some New Tetraaldehyde and Tetraketone Derivatives and X-ray Structure of 1,1'-(4,4'-(2- (1,3-bis(4-Acetylphenoxy)propan-2-ylidene)propane-1,3-diyl) bis(oxy)bis(4,1-phenylene))diethanone. Int. J. Mol. Sci. 2008, 9, 1000-1007. https://doi.org/10.3390/ijms9061000
Er M, Ustabaş R, Çoruh U, Sancak K, Vázquez-López E. Synthesis and Characterization of Some New Tetraaldehyde and Tetraketone Derivatives and X-ray Structure of 1,1'-(4,4'-(2- (1,3-bis(4-Acetylphenoxy)propan-2-ylidene)propane-1,3-diyl) bis(oxy)bis(4,1-phenylene))diethanone. International Journal of Molecular Sciences. 2008; 9(6):1000-1007. https://doi.org/10.3390/ijms9061000
Chicago/Turabian StyleEr, Mustafa, Reşat Ustabaş, Ufuk Çoruh, Kemal Sancak, and Ezequiel Vázquez-López. 2008. "Synthesis and Characterization of Some New Tetraaldehyde and Tetraketone Derivatives and X-ray Structure of 1,1'-(4,4'-(2- (1,3-bis(4-Acetylphenoxy)propan-2-ylidene)propane-1,3-diyl) bis(oxy)bis(4,1-phenylene))diethanone" International Journal of Molecular Sciences 9, no. 6: 1000-1007. https://doi.org/10.3390/ijms9061000