(2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one

Institut für Organische Chemie der Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
*
Author to whom correspondence should be addressed.
Molbank 2005, 2005(2), M405; https://doi.org/10.3390/M405
Submission received: 8 March 2004
/
Accepted: 18 March 2004
/
Published: 3 October 2005
{kind=link}

1. Procedure
(2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (2) is synthesized via [4+3] cycloaddition of an oxyallyl intermediate generated from 1,3-dichloro-3-methylbutan-2-one with lithium perchlorate / diethylether / triethylamine.
A. 1,3-Dichloro-3-methylbutan-2-one (Note 1). Caution: The operation must be carried out in a well-vented hood. The evolving HCl and SO2 gas should be absorbed by passing over water.
A 2-L, three-necked, round-bottomed flask, fitted with a dropping funnel, immersion thermometer, drying tube (calcium chloride) and an efficient Teflon-coated magnetic stirring bar ("Circulus"), is charged with 3-methylbutan-2-one (isopropyl methyl ketone) (430 mL, ca. 346 g, 4.0 mol) (Note 2) and chilled in an ice bath. With magnetic stirring, freshly distilled sulfuryl chloride (800 mL, ca. 1334 g, 9.9 mol) is added dropwise. The internal temperature is controlled to be between 10 and 15°C; this takes approximately 3 hours. Stirring is continued overnight, while the ice-bath is allowed to melt.
Ice water (500 mL) is added to the reaction mixture, and the layers separated by means of a separatory funnel. The aqueous layer is extracted with diethyl ether (3 ´ 200 mL). The ether phases are combined with the organic layer and washed neutral with saturated aqueous NaHCO3 solution. Then the organic layer is dried overnight with anhydrous magnesium sulfate (Note 3).
Most of the solvent is removed by gentle heating in a rotary evaporator. The concentrate is transferred into a distillation flask that containes a teflon-coated magnetic stirring bar, and mixed with a small amount of calcium carbonate (Note 4). The flask is fitted with a 110 cm vacuum-jacketed Vigreux column (effective length 100 cm), a vacuum distillation head and a vacuum controler. The rectification – with continuous magnetic stirring – is started, using a pressure of ca. 40 Torr. The first fraction collected at bp 40-50°C/40 Torr (73 g) containes mainly the monochlorination product 3-chloro-3-methylbutan-2-one and a minor amount of 1,3-dichloro-3-methylbutan-2-one (1), the dichlorination product, as shown by GLC analysis (Note 5). The main product is collected as a fraction with bp 68–70°C/15 Torr, that consists of 1,3-dichloro-3-methylbutan-2-one (1), contaminated with traces of 1,1,3-trichloro-3-methylbutan-2-one. Yield is 469 g (76%) of NMR-pure substance (Note 6).
B. (2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (2). A 1-L three-necked flask, fitted with a dropping funnel and a reflux condenser with drying tube (KOH pellets), is charged with dry lithium perchlorate (21.2 g, 0.20 mol) and dry diethyl ether (120 mL) (Note 7). The mixture is stirred with the aid of an efficient Teflon-coated stirring bar (“Circulus”). When the salt is dissolved, dry 2-methylfuran (180 mL), followed by dry triethylamine (20.3 g, 0.20 mol) is added rapidly through the dropping funnel (Note 8).
To this well-stirred solution, a mixture of 1,3-dichloro-3-methylbutan-2-one (1, 15.5 g, 0.10 mol), diethyl ether (38 mL) and 2-methylfuran (38 mL) is added dropwise at room temperature within 1 hr. Gradually, a yellow mass with honey-like consistence deposits, and the color of the solution turnes orange to dark. Progress of the reaction may be monitored by GLC (see below).
When the reaction is completed (ca. 1 hr), water (250 mL) is added and the mixture transferred to a separatory funnel. The layers are separated, and the aqeous layer is extracted with diethyl ether (4 ´ 100 mL). The combined organic phases are washed with saturated brine (250 mL) and dried overnight with anhydrous magnesium sulfate. The solvent is removed in a rotary evaporator and the residue distilled in a kugelrohr at 70°C/0.01 Torr. The pale yellow, waxy product (12.8 g, ca. 63% yield) (Note 9) is dissolved in boiling n-heptane (15 mL) and allowed to cool slowly. The 2endo-isomer (2) separates as a white solid (8.3 g, 41%) with mp 101–103°C. (Note 10).
2. Notes
1. 1,3-Dichloro-3-methylbutan-2-one (1) may be prepared by vapor.phase chlorination of 3-methylbutan-2-one (methyl isopropyl ketone).1 Since this preparation requires a special equipment, we prepared dichloroketone 1 by reaction of 3-methylbutan-2-one with sulfuryl chloride (SO2Cl2), in analogy to the procedure of Wyman and Kaufman.2
2. 3-Methylbutan-2-on was purchased from Aldrich Chemical Company, Inc.
3. For drying of other chloroketones, the authors prefer a mixture of anhydrous calcium chloride and a smaller amount of calcium carbonate, see Note 4.
4. During the rather long distillation time, traces of hydrogen chloride may evolve. They are removed by neutralization with the calcium carbonate.
5. For gas chromatography, the we used a 2.3 m glass column, packed with 5% OV 101 on Gaschrom Q, using a temperature program from 80 to 220°C, 8 K/min, with FID detector; the carrier gas was nitrogen. Retention times: tR = 0.63 min (3-chloro-3-methylbutan-2-one), tR = 2.36 min (1,3-dichloro-3-methylbutan-2-one), tR = 3.31 min (1,1,3-trichloro-3-methylbutan-2-one).
6. The colorless liquid shows the following signals in the 1H NMR spectrum (60 MHz, CDCl3) d: 1.75 (s, 4-H and 3-CH3), 4.70 (s, 1-H). The purity of this product is sufficient for preparative work. In order to obtain a product with >99% purity, distillation in vacuo may be repeated. The authors used a 50 cm Fischer Spaltrohr column.
7. Lithium perchlorate (pro analysi or purum) was purchased from Fluka Feinchemikalien GmbH, Neu-Ulm, Germany or Janssen, Beerse, Belgium, respectively. It is dried in vacuo (oil pump) at 220–230°C. Diethyl ether is dried over sodium wire, distilled, and stored over molecular sieve 3Å.
8. 2-Methylfuran was purchased from Fluka or Janssen. It is shaken with 5 percent aqueous potassium hydroxide solution until the aqueous layer remains colorless, dried over calcium chloride and distilled before use from potassium hydroxide pellets. Triethylamine is distilled from calcium hydride before use.
9. Before crystallization, the crude distilled product contains, besides 2, the (2exo)-2-chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one epimer (3), (4endo)-4-chloro-1,2,2-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (4), and traces of the 4exo-isomer, (4exo)-4-chloro-1,2,2-trimethyl-8-oxabicyclo[3.2.1]-oct-6-en-3-one (5). According to a capillary GLC (21 m glass column, coated with Emulphor), the proportions of 2, 3, 4 and 5 were 85 : 5 : 9 : 0.8, i.e the regioselectivity of the cycloaddition is [2 + 3] : [4 + 5] = ca. 9:1 (GLC). The endo/exo selectivity is [2 + 4] : [3 + 5] = ca. 13:1.

The physical properties are as follows:
2: 1H NMR (60 MHz, CDCl3) d: 1.05 (s), 1.37 (s), 1.65 (s), 4.49 (d, J = 1.8 Hz), 4.53 (s), AB sub-spectrum centered at 6.28, with dA = 6.36, dB = 6.19, JAB = 6 Hz; the lines of the A part are split into doublets with 1.8 Hz; 13C NMR (75.47 MHz, CDCl3) d: 20.3, 21.3, 24.5, 53.3, 68.4, 86.75, 88.5, 134.9, 203.3; CIMS (CH4, 70 eV) m/z (%): 203 (2), 201 (6), 165 (3), 149 (4), 147 (13), 123 (11), 121 (30), 119 (100), 95 (78), 83 (7); see also Ref.9
3: 1H NMR (60 MHz, CDCl3) d: 1.00 (s), 1.50 (s), 1.57 (s), 3.77 (s), 4.49 (d, J = 1.8 Hz), AB sub-spectrum centered at 6.26, with dA = 6.46, dB = 6.06, JAB = 6 Hz; the lines of the A part are split into doublets with 1.8 Hz; 13C NMR (75.47 MHz, CDCl3) d: 19.8, 20.5, 26.8, 51.7, 61.3, 85.4, 86.7, 134.7, 136.6, 205.8; CIMS (CH4, 70 eV) m/z (%): 203 (2), 201 (5), 167 (6), 157 (2), 155 (6), 149 (8), 147 (16), 123 (81), 121 (36), 119 (100), 95 (96), 85 (15), 83 (10), 71 (11); mp 48–50°C (petroleum ether).
4: 1H NMR (60 MHz, CDCl3) d: 1.10 (s), 1.30 (s), 1.40 (s); AB sub-spectrum centered at 4.90, with dA = 5.02, dB = 4.79, JAB = 6 Hz; the lines of the A part are split into doublets with ca. 1 Hz; 6.30 (s); 13C NMR (75.47 MHz, CDCl3) d: 16.75, 20.1, 21.4, 57.3, 62.4, 82.4, 89.6, 131.2, 139.7, 203.4; CIMS (CH4, 70 eV) m/z (%): 203 (2), 201 (5), 165 (7), 149 (3), 147 (8), 137 (9), 121 (33), 119 (100), 95 (76), 85 (4), 83 (7); mp 91–93°C (petroleum ether).
5: CIMS (CH4, 70 eV) m/z (%): 203 (3), 201 (10), 185 (13), 183 (38), 165 (8), 155 (12), 143 (21), 137 (22), 121 (21), 119 (63), 95 (100), 85 (17).
10. The substance is pure enough for further synthetic work, according to GLC (5% OV 101 on Merck Volaspher A 2, particle size 125-150 mm, 100–120 mesh ASTM, 2 m glass column, 80 ® 250°C, 8 K/min, tR = 10.2 min), and TLC (petroleum ether/ethyl acetate 4:1, Rf = 0.40). The mother liquor from the heptane crystallization were worked-up by medium pressure liquid chromatography (MPLC) on Lichroprep Si 60 (Merck, Germany), using petroleum ether/ethyl acetate (24:1) for elution. Isomers 2, 3 and 4 could be separated; however the trace compound 5 escaped isolation.
3. Discussion
A major progress in the synthesis of seven-membered ring ketones has been achieved by the development of the [4 + 3] cycloaddition reaction of furans and other 1,3-dienes with oxyallyl intermediates.3 For the generation of these intermediates, the reduction of a,a'-dibromo- and polybromoketones has been the preferred method over the last few years.
Dehydrohalogenation of a-halogenoketones is an older approach to generate oxyallyls (“Favorskii intermediates”) which has several advantages, as discussed elsewere.3d,4,5 However, in order to obtain synthetically useful yields of [4 + 3] cycloadducts, the crucial point is the choice of the appropriate basic reaction medium. Inter alia, lithium perchlorate/diethylether6 and 2,2,2-trifluoroethanol7 have been found to be useful for these [4 + 3] cycloaddition reactions. Whereas for the reductive methods of oxyallyl generation, di- or polybromoketones are needed, the dehydrohalogenation methods ("base-mediated conditions"3d) can be effected with a-chloroketones. The latter are more shelf-stable and less lacrymatory than the bromo analogues.
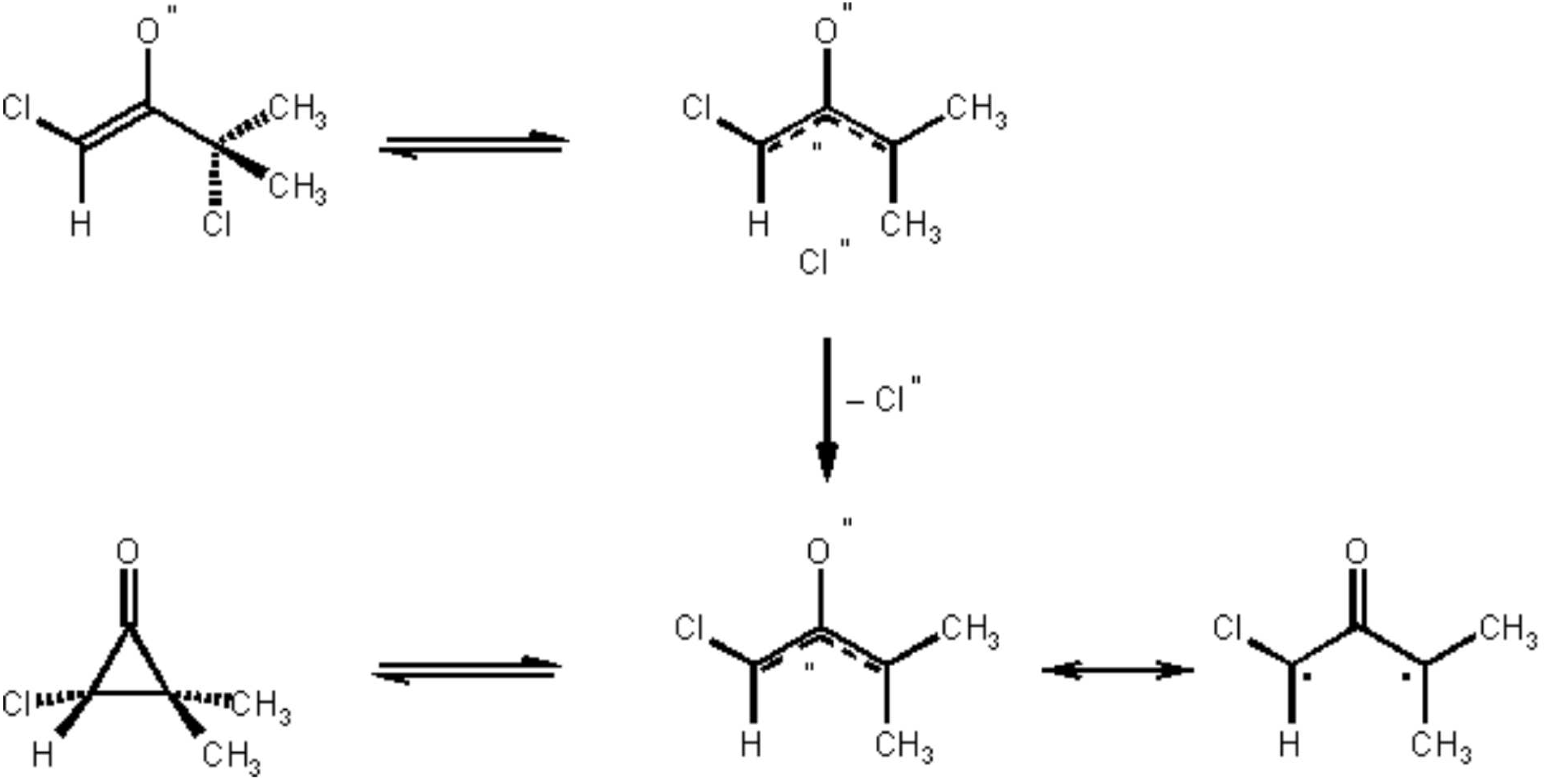
The procedure illustrates the [4+3] cycloaddition of oxyallyls generated from a-halogenoketones in the LiClO4/diethyl ether solvent system. Moreover it is an example for the generation of a monochloro oxyallyl derived from an a,a'-dichloro ketone (Scheme 1), and its regioselective cycloaddition to a 2-substituted furan.8 The major product of this [4 + 3] cycloaddition is 2, that can be separated easily from the minor products by crystallization (Note 9).
(2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (2) has been prepared previously by Shimizu et al.9 The japanese researchers transformed dichloroketone 1 into the trimethylsilylenol ether. The latter was reacted with 2-methylfuran using silver perchlorate and calcium carbonate in nitromethane for generation of the oxyallyl intermediate. For a 4.4 mmol scale, a 57% isolated yield of 2 was reported, after chromatographic separation from the by-product, 1-chloro-3-methyl-3-(5-methyl-2-furyl)-butan-2-one. The latter came from a competing electrophilic substitution of 2-methylfuran by the oxyallyl cation. The product ratio (cycloaddition vs. substitution) was 3 : 1.
(2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (2) may be used as a building block for cycloheptane systems with a 1,1,4-trimethyl substitution pattern. An approach to the terpenoid himachalane skeleton has been worked-out.10 Key steps are: (a) Catalytic hydrogenation of the C=C double bond of the oxabicycle without touching the chloro substituent. (b) Attachment of the 3-oxobutyl side chain at C-2 via Michael reaction of the saturated bicyclic a-chloro ketone with methyl vinyl ketone. The carbonyl function in 2 can be reduced by LiAlH4 with high endo-selectivity to give (2endo,3endo)-2-chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-ol.11
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Supplementary File 5Supplementary File 6References
- Rabjohn, N.; Rogier, E. R. J. Org. Chem. 1946, 11, 781.
- Wyman, D. P.; Kaufman, P. R. J. Org. Chem. 1964, 29, 1956.
- (a) Noyori, R.; Hayakawa, Y. Org. React. 1983, 29, 163. [Google Scholar] (b) Hoffmann, H. M. R. Angew. Chem., Int. Ed. Engl. 1984, 23, 1. [Google Scholar] [CrossRef] (c) Mann, J. Tetrahedron 1986, 42, 4611. (d) Rigby, J. H.; Pigge, F. C. Org. React. 1997, 51, 351. [Google Scholar] (e) Harmata, M. Tetrahedron 1997, 53, 6235.
- Sendelbach, S.; Schwetzler-Raschke, R.; Radl, A.; Kaiser, R.; Henle, G. H.; Korfant, H.; Reiner, S.; Föhlisch, B. J. Org. Chem. 1999, 64, 3398.
- Oh, J.; Ziani-Cherif, C.; Choi, J.-R.; Cha, J. K. Org. Synth. 2001, 78, ####. [Google Scholar]
- Herter, R.; Föhlisch, B. Synthesis 1982, 976.
- Föhlisch, B.; Gehrlach, E.; Herter, R. Angew. Angew. Chem 1982, 94, 144. [Google Scholar]; Angew. Chem. Suppl. 1982, 241.; Angew. Chem. Internat. Edit. Engl. 1982, 21, 137.
- Föhlisch, B.; Flogaus, R.; Oexle, J.; Schädel, A. Tetrahedron Lett. 1984, 35, 1773.
- Shimizu, N.; Tanaka, M.; Tsuno, Y. J. Am. Chem. Soc. 1982, 104, 1330.
- Oexle, J. Dissertation, Univ. Stuttgart, Germany, 1986. Details may be obtained from the senior author (B. F.).
- Föhlisch, B.; Kreiselmeier, G. Tetrahedron 2001, 57, 10077.
Scheme 1.
Share and Cite
MDPI and ACS Style
Oexle, J.; Buchinger, M.; Föhlisch, B. (2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one. Molbank 2005, 2005, M405. https://doi.org/10.3390/M405
AMA Style
Oexle J, Buchinger M, Föhlisch B. (2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one. Molbank. 2005; 2005(2):M405. https://doi.org/10.3390/M405
Chicago/Turabian StyleOexle, Jutta, Michael Buchinger, and Baldur Föhlisch. 2005. "(2endo)-2-Chloro-1,4,4-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one" Molbank 2005, no. 2: M405. https://doi.org/10.3390/M405
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.