Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide


Atlantic Centre for Green Chemistry and Department of Chemistry, Saint Mary’s University, 923 Robie St., Halifax, NS B3H 3C3, Canada
*
Author to whom correspondence should be addressed.
Molbank 2017, 2017(3), M957; https://doi.org/10.3390/M957
Submission received: 5 September 2017
/
Revised: 15 September 2017
/
Accepted: 16 September 2017
/
Published: 19 September 2017
(This article belongs to the Section Structure Determination)
{kind=link}
{kind=link}
Abstract
:The single crystal structure of bis(2,4,6-trimethylphenyl)phosphine oxide has been determined. All interatomic distances and angles can be considered normal. The aryl substituents adopt an intermediate configuration when compared to both sterically unhindered (e.g., diphenylphosphine oxide) and congested (e.g., bis(2,4,6-tri-tert-butylphenyl)phosphine oxide) secondary phosphine oxides, illustrating the influence of steric congestion on the molecular structure.
1. Introduction
The importance of organophosphorus compounds is illustrated by their widespread applications in areas of organic [1,2], inorganic [3,4], medicinal [5,6,7,8], and material [9] chemistries. Over the past decade, the synthetic potential of secondary phosphine oxides has become increasingly well-established. These compounds are precursors to phosphinoyl radicals; they are effective reagents in phosphorylation reactions [10,11,12] due to the accessible P–H bond dissociation energies (ca. 80 kcal mol−1) [13], and also participate in olefin addition reactions [14,15]. Given the utility of these compounds, it seemed pertinent to report the single crystal structure of the title compound bis(2,4,6-trimethylphenyl)phosphine oxide, 1.
2. Results and Discussion
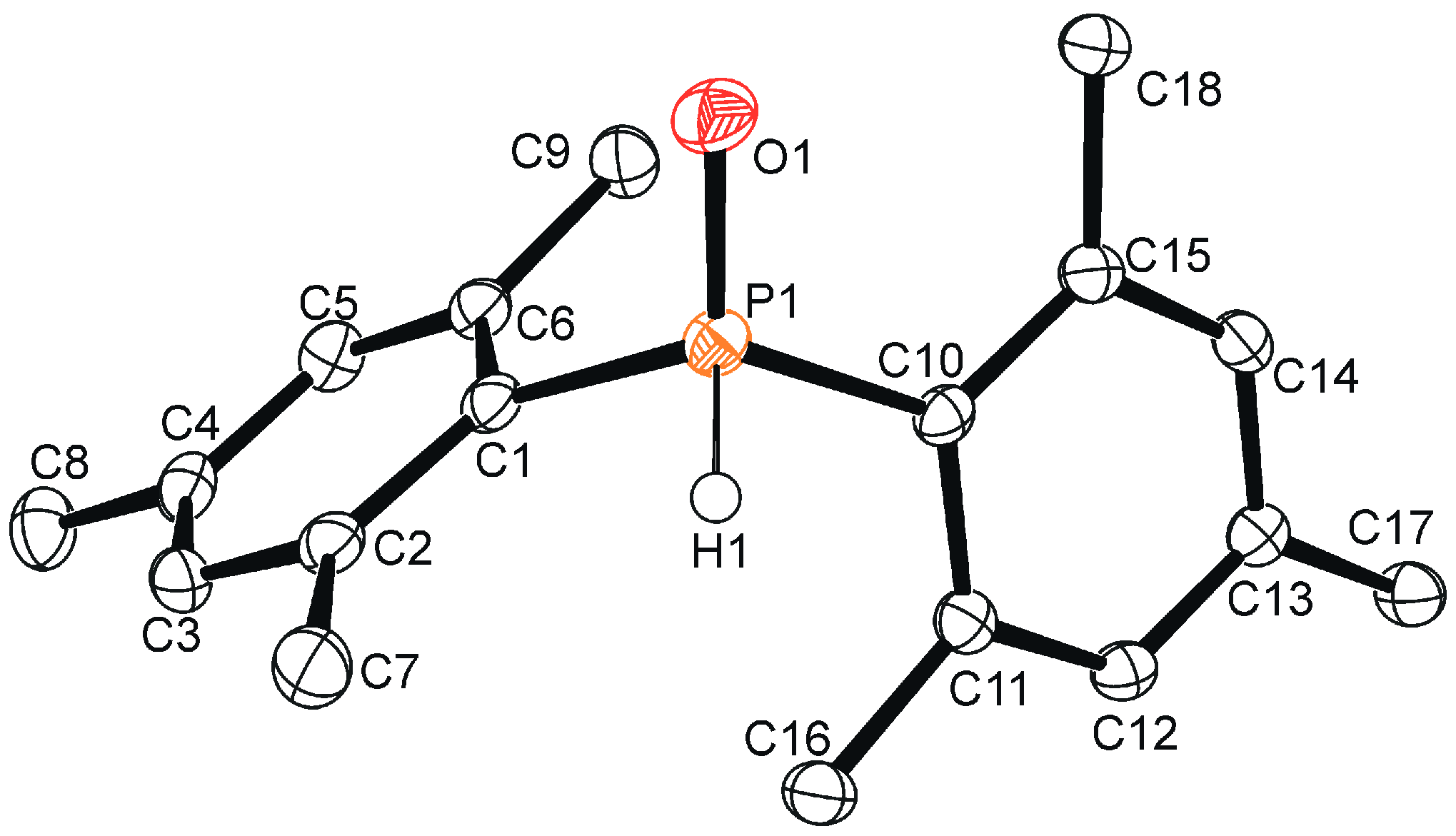
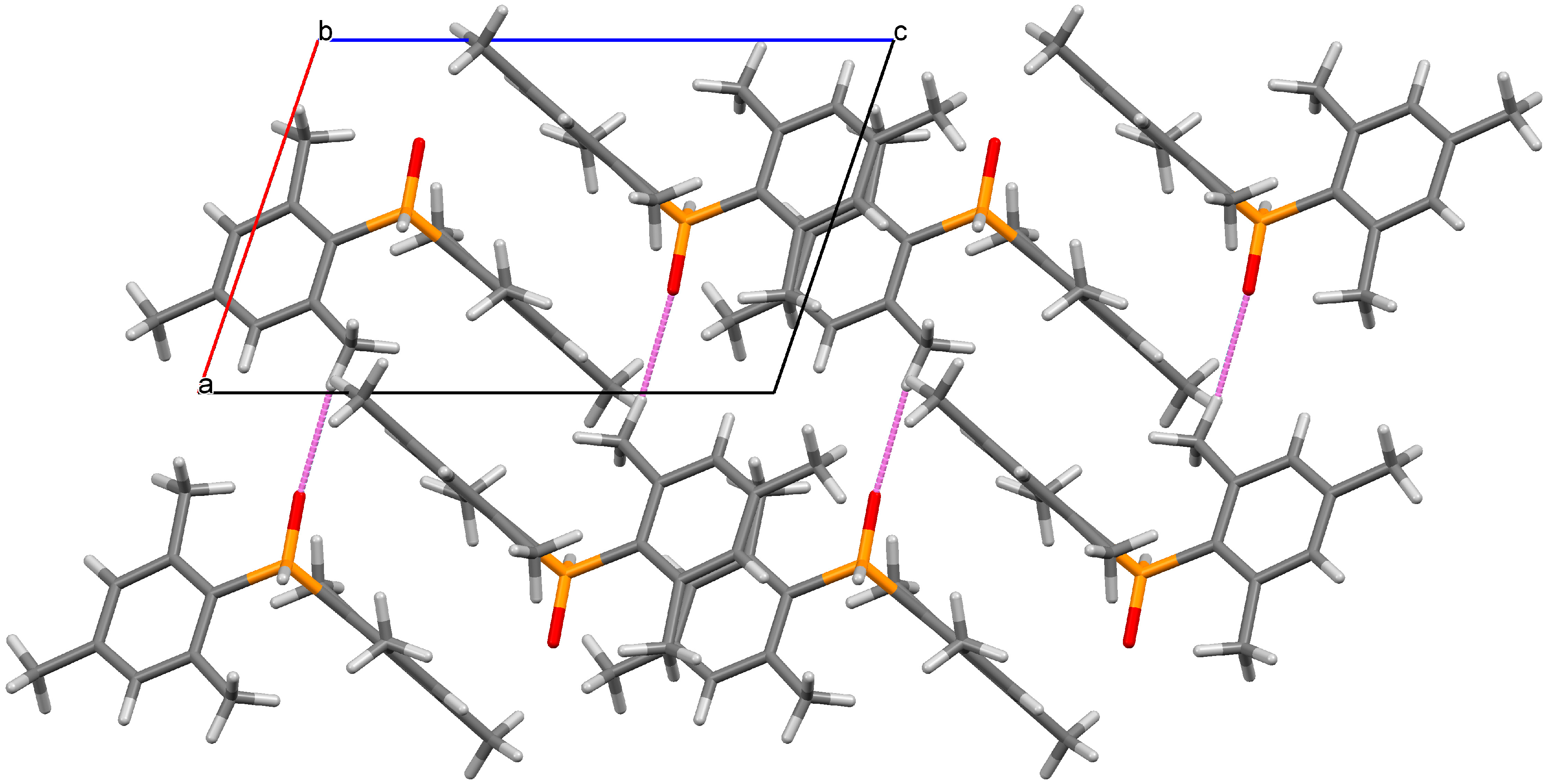
Crystals of the title compound were grown by pentane evaporation, and a crystal structure was obtained (Figure 1). As expected based on spectroscopic data [16] and related secondary phosphine oxides [16,17,18,19], the organophosphorus compound 1 exhibits a tetracoordinate geometry. The hydrogen atom H1 was located in the difference map and allowed to freely refine giving a P1–H1 distance of 1.319(17) Å. The P1–O1 distance measured 1.4854(13) Å and the P1–C1 and P1–C10 distances were 1.8151(18) Å and 1.8162(18) Å, respectively. The mesityl rings exhibit a twist of 31.93(16)° (measured as the dihedral angle between C1–C6–C10–C15) that is intermediate in magnitude when compared to diphenylphosphine oxide [17] (1.2°, relatively unhindered) and bis(2,4,6-tri-tert-butylphenyl)phosphine oxide [19] (52.4°, extremely congested), illustrating the influence of steric congestion on the molecular structure of the compound. In the crystal, intermolecular O∙∙∙H–C contacts were identified between O1, H16C, and H17B with interatomic distances of 2.561(2) Å and 2.653(2) Å, respectively (Figure 2). In the crystal, C–H∙∙∙π interactions were also observed between C6–H8B (2.879(3) Å) and C12–H18C (2.842(3) Å), which resulted in a stacking of the mesityl rings (Figure 2).
The NMR spectroscopic data in CDCl3 for the title compound were consistent with previous reports [16]. The 1H NMR spectrum contained resonances at 6.86 (d, 4JHP = 3.7 Hz), 2.39, and 2.28 ppm for the meta-aromatic, ortho-methyl, and para-methyl positions of the mesityl groups, respectively. The P-H resonance was identified at 8.55 ppm as a doublet with a coupling constant of 476 Hz, which is typical for P(V) 1JHP constants [18]. Additional evidence was given by the coalescence of this doublet into a singlet at 8.55 ppm in the 1H{31P} NMR spectrum. The 31P{1H} NMR spectrum contained one resonance at 10.1 ppm. All resonances in the 13C{1H} NMR spectrum were observed as doublets. The aromatic region contained resonances at 141.9 (4JCP = 2.7 Hz), 141.7 (2JCP = 10.4 Hz), 130.5 (3JCP = 11.0 Hz), and 126.4 ppm (1JCP = 100.1 Hz) for the para, ortho, meta, and ipso carbon environments, respectively. Methyl environments were identified at 21.1 (5JCP = 1.0 Hz) and 20.8 ppm (3JCP = 7.8 Hz) for the ortho and para positions, respectively.
3. Materials and Methods
3.1. Materials
All reagents and non-deuterated solvents were purchased from Sigma-Aldrich (Oakville, ON, Canada) and used without further purification. NMR solvents were purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, USA) and stored over 4 Å molecular sieves for a minimum of one day prior to use.
3.2. Instrumentation
3.2.1. General Remarks
NMR spectra were recorded at 298 K on a 300 MHz spectrometer (Bruker, Milton, ON, Canada) and are reported in ppm. 1H-NMR spectra were collected in deuterated solvents and referenced internally to tetramethylsilane (TMS, δ = 0 ppm). The 13C{1H} NMR spectrum was referenced internally to CDCl3 relative to TMS (δ = 0 ppm). 31P NMR chemical shifts were referenced to an external standard of 85% phosphoric acid (δ = 0 ppm). Coupling constants are reported in Hz and given as absolute values. A sample of 1 was prepared as a KBr pellet for IR spectroscopy, and measured using a Bruker ALPHA FT-IR spectrometer. High-resolution mass spectrometry was performed on a micrOTOF from Bruker Daltonics with sample preparation as follows: A sample of 1 was dissolved in dichloromethane (HPLC grade) and diluted (>10,000 times) with methanol (HPLC grade). The sample was introduced to the mass spectrometer via syringe pump at a flow rate of 2 μL min−1 and ionized (electrospray ionization, ESI, positive mode). ESI conditions: spray voltage applied to the ESI needle = 4.0 kV; dry gas flow rate = 4 L min−1; nebulizer gas pressure = 1 Bar; source temperature = 180 °C. The melting point of 1 was recorded on an Electrothermal MEL-Temp 3.0 using a glass capillary sealed under inert conditions and is uncorrected.
3.2.2. X-ray Crystallography
Under inert conditions, crystals were prepared for mounting by suspending them in paratone-N oil on a microscope slide. A single crystal was attached to the tip of an appropriately sized MiTeGen loop with paratone-N oil and cooled to 125 K. Measurements were made on a Bruker APEX2 CCD-equipped diffractometer (30 mA, 50 mV) using monochromated Mo Kα radiation (λ = 0.71073 Å) at 125 K. The initial orientation and unit cell were indexed [20] using a least-squares analysis of a random set of reflections collected from three series of 0.5° wide scans, 10 seconds per frame, and 12 frames per series that were well distributed in reciprocal space. For data collection, the frame length was adjusted to 5 seconds per frame to give a predicted resolution of 0.65 Å, and five ω-scan frame series were collected with 0.5° wide scans and 366 frames per series at varying 2θ, ω, and φ angles (2θ = −28°, ω = −28°, φ = 0°, 90°, 180°, 270°; 2θ = 28° ω = 28°, φ = 0°) and a φ-scan with 0.5° wide scans and 720 frames (2θ, ω, and φ = 0°). Cell refinement and data reduction were performed with the Bruker SAINT software [21] which corrects for beam inhomogeneity, possible crystal decay, and Lorentz and polarization effects. Data processing and a multi-scan absorption correction was applied using APEX3 software package [21]. The structure was solved using direct methods [22] and all non-hydrogen atoms were refined anisotropically using the ShelXLE [23] graphical user interface and SHELXL [24]. Hydrogen atoms (excluding H1) were included at geometrically idealized positions and were fixed (Ar-H) or in the case of methyl groups, the dihedral angle of the idealized tetrahedral CH3 fragment was allowed to refine. Hydrogen atoms were placed in calculated positions using an appropriate riding model and coupled isotropic temperature factors. H1 was located in the difference map and allowed to freely refine its position, with a coupled isotropic temperature factor of −1.2 to P1. Figures were made using Ortep-3 for Windows [25] and Mercury [26].
3.3. Synthesis
The title compound was prepared from the hydrolysis of bis(2,4,6-trimethylphenyl)phosphorus chloride according to a previously published literature procedure [16]. 31P-NMR (CDCl3): δ 10.1 ppm (d, 1JPH = 476 Hz). 1H-NMR (CDCl3): δ 8.55 (d, 1JHP = 476 Hz, 1H, P-H), 6.86 (d, 4JHP = 3.7 Hz, 4H, m-Ar), 2.39 (s, 12H, o-CH3), 2.28 ppm (s, 6H, p-CH3). 13C{1H} NMR (CDCl3): δ 141.9 (d, 4JCP = 2.7 Hz, para-Ar), 141.7 (d, 2JCP = 10.4 Hz, ortho-Ar), 130.5 (d, 3JCP = 11.0 Hz, meta-Ar), 126.4 (d, 1JCP = 100.1 Hz, ipso-Ar), 21.1 (d, 5JCP = 1.0 Hz, para-CH3), 20.8 ppm (d, 3JCP = 7.8 Hz, ortho-CH3). IR (KBr pellet): ν 3024 (m), 2957 (vs), 2917 (vs), 2853 (vs), 2737 (m), 2361 (vs, P-H), 2175 (w), 2149 (w), 1950 (w), 1920 (w), 1890 (w), 1764 (w), 1730 (w), 1605 (vs), 1558 (s) 1454 (vs) 1411 (vs), 1378 (vs), 1290 (m), 1274 (w), 1246 (s), 1190 (vs), 1170 (vs), 1082 (vs), 1034 (vs), 1003 (vs), 980 (vs), 959 (m), 914 (s), 850 (vs), 804 (w), 716 (m), 639 (vs), 612 (s), 572 (s), 562 (s), 545 (m), 517 (m), 505 (m), 440 (vs), 426 cm−1 (vs). m.p.: 135.5–141.3 °C. HRMS (ESI TOF): [M + Na+]+ found 309.1391. [C18H23PO + Na+]+ requires 309.1384. Crystal data for C18H23OP (M = 286.33 g mol−1): triclinic, space group P-1 (no. 2), a = 7.9123(6) Å, b = 8.2998(6) Å, c = 12.4165(9) Å, α = 99.613(4)°, β = 108.264(4)°, γ = 91.343(4)°, V = 760.94(10) Å3, Z = 2, T = 125(2) K, μ(MoKα) = 0.174 mm−1, Dcalc = 1.250 g cm−3, 9674 reflections measured (1.757° ≤ 2Θ ≤ 29.210°), 3786 unique (Rint = 0.0373, Rsigma = 0.0553) which were used in all calculations. The final R1 was 0.0467 (I > 2σ(I)) and wR2 was 0.1176 (all data). CCDC 1571849 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Supplementary Materials
The following are available online https://www.mdpi.com/1422-8599/2017/3/M957, Figure S1: 1H NMR spectrum of 1, Figure S2: 1H{31P} NMR spectrum of 1, Figure S3: 13C{1H} NMR spectrum of 1, Figure S4: 31P{1H} NMR spectrum of 1, Figure S5: 31P NMR spectrum of 1, Figure S6: IR spectrum of 1, Table S1: Atomic coordinates and equivalent isotropic displacement parameters of 1, Table S2: Bond lengths [Å] and angles [°] for 1, Table S3: Anisotropic displacement parameters for 1, Table S4: Hydrogen coordinates and isotropic displacement parameters for 1.
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Acknowledgments
We thank the Natural Sciences and Engineering Research Council of Canada (through the Discovery Grants Program to J.D.M.). J.D.M. also acknowledges support from the Canadian Foundation for Innovation, the Nova Scotia Research and Innovation Trust Fund and Saint Mary’s University.
Author Contributions
A.J.V.: X-ray crystallography, literature search, manuscript preparation. K.R.: Crystallization of title compound, collection and interpretation of spectroscopic data, manuscript preparation. N.A.G.: Synthesis of title compound. J.D.M.: Synthesis planning, literature research, manuscript preparation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boutagy, J.; Thomas, R. Olefin synthesis with organic phosphonate carbanions. Chem. Rev. 1974, 74, 87–99. [Google Scholar] [CrossRef]
- Kitamura, M.; Tokunaga, M.; Noyori, R. Asymmetric Hydrogenation of β-Keto Phosphonates: A Practical Way to Fosfomycin. J. Am. Chem. Soc. 1995, 117, 2931–2932. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (Nobel lecture 2001). Adv. Synth. Catal. 2003, 345, 3–13. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]
- Sawa, M.; Kiyoi, T.; Kurokawa, K.; Kumihara, H.; Yamamoto, M.; Miyasaka, T.; Ito, Y.; Hirayama, R.; Inoue, T.; Kirii, Y. New type of metalloproteinase inhibitor: Design and synthesis of new phosphonamide-based hydroxamic acids. J. Med. Chem. 2002, 45, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Zhang, P.; Murphy, M.E.; Marmorstein, R.; George, D.L. Structural basis for the inhibition of HSP70 and dnak chaperones by small-molecule targeting of a c-terminal allosteric pocket. ACS Chem. Biol. 2014, 9, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.K.; Budina-Kolomets, A.; Murphy, M.E.; Nefedova, Y. Efficacy of the HSP70 inhibitor PET-16 in multiple myeloma. Cancer Biol. Ther. 2015, 16, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Stetz, G.; Verkhivker, G.M. Probing allosteric inhibition mechanisms of the Hsp70 chaperone proteins using molecular dynamics simulations and analysis of the residue interaction networks. J. Chem. Inf. Model. 2016, 56, 1490–1517. [Google Scholar] [CrossRef] [PubMed]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface modification using phosphonic acids and esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, P.; Gao, Y.; Chen, Y.; Tang, G.; Zhao, Y. Copper-catalyzed P-arylation via direct coupling of diaryliodonium salts with phosphorus nucleophiles at room temperature. J. Org. Chem. 2013, 78, 8176–8183. [Google Scholar] [CrossRef] [PubMed]
- Ke, J.; Tang, Y.; Yi, H.; Li, Y.; Cheng, Y.; Liu, C.; Lei, A. Copper-catalyzed radical/radical Csp3–H/P–H cross-coupling: α-Phosphorylation of Aryl Ketone O-Acetyloximes. Angew. Chem. Int. Ed. 2015, 54, 6604–6607. [Google Scholar] [CrossRef] [PubMed]
- Noël-Duchesneau, L.; Lagadic, E.; Morlet-Savary, F.; Lohier, J.; Chataigner, I.; Breugst, M.; Lalevée, J.; Gaumont, A.; Lakhdar, S. Metal-Free Synthesis of 6-phosphorylated phenanthridines: Synthetic and mechanistic insights. Org. Lett. 2016, 18, 5900–5903. [Google Scholar] [CrossRef] [PubMed]
- Jessop, C.M.; Parsons, A.F.; Routledge, A.; Irvine, D.J. Radical addition reactions of phosphorus hydrides: tuning the reactivity of phosphorus hydrides, the use of microwaves and horner–wadsworth–emmons-type reactions. Eur. J. Org. Chem. 2006, 2006, 1547–1554. [Google Scholar] [CrossRef]
- Stockland, R.A.; Taylor, R.I.; Thompson, L.E.; Patel, P.B. Microwave-Assisted Regioselective Addition of P (O)−H bonds to alkenes without added solvent or catalyst. Org. Lett. 2005, 7, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Lenker, H.K.; Richard, M.E.; Reese, K.P.; Carter, A.F.; Zawisky, J.D.; Winter, E.F.; Bergeron, T.W.; Guydon, K.S.; Stockland, R.A., Jr. Phospha-michael additions to activated internal alkenes: Steric and electronic effects. J. Org. Chem. 2012, 77, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.W.; Fawcett, J.; Hanton, M.J. Rigid N-phosphino guanidine P, N ligands and their use in nickel-catalyzed ethylene oligomerization. Organometallics 2008, 27, 5082–5087. [Google Scholar] [CrossRef]
- Härling, S.; Greiser, J.; Al-Shboul, T.M.; Görls, H.; Krieck, S.; Westerhausen, M. Calcium-mediated hydrophosphorylation of organic isocyanates with diphenylphosphane oxide. Aust. J. Chem. 2013, 66, 1264–1273. [Google Scholar] [CrossRef]
- Christiansen, A.; Li, C.; Garland, M.; Selent, D.; Ludwig, R.; Spannenberg, A.; Baumann, W.; Franke, R.; Börner, A. On the tautomerism of secondary phosphane oxides. Eur. J. Org. Chem. 2010, 2010, 2733–2741. [Google Scholar] [CrossRef]
- Fleming, C.G.; Slawin, A.M.; Arachchige, K.S.A.; Randall, R.; Bühl, M.; Kilian, P. Synthetic and computational study of geminally bis (supermesityl) substituted phosphorus compounds. Dalton Trans. 2013, 42, 1437–1450. [Google Scholar] [CrossRef] [PubMed]
- APEX 3, v. 2016.1-0; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SAINT, v. 8. 37A; Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Single crystal structure of 1. Ellipsoids are shown at the 50% probability level. Hydrogen atoms (excluding H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): P1–H1 1.319(17), P1–O1 1.4854(13), P1–C1 1.8151(18), P1–C10 1.8162(18), O1–P1–H1 111.7(7), C1–P1–H1 103.9(7), C10–P1–H1 101.2(7), C1–P1–C10 108.07(8), O1–P1–C1 113.94(8), O1–P1–C10 116.64(8), C1–C6–C10–C15 31.93(16)°.
Figure 1.
Single crystal structure of 1. Ellipsoids are shown at the 50% probability level. Hydrogen atoms (excluding H1) have been omitted for clarity. Selected bond lengths (Å) and angles (°): P1–H1 1.319(17), P1–O1 1.4854(13), P1–C1 1.8151(18), P1–C10 1.8162(18), O1–P1–H1 111.7(7), C1–P1–H1 103.9(7), C10–P1–H1 101.2(7), C1–P1–C10 108.07(8), O1–P1–C1 113.94(8), O1–P1–C10 116.64(8), C1–C6–C10–C15 31.93(16)°.

Figure 2.
Packing diagram showing the close contacts in the crystal of 1 when viewed along the b axis. Selected distance (Å): O1–H16C 2.561(2).
Figure 2.
Packing diagram showing the close contacts in the crystal of 1 when viewed along the b axis. Selected distance (Å): O1–H16C 2.561(2).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Veinot, A.J.; Ramgoolam, K.; Giffin, N.A.; Masuda, J.D. Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide. Molbank 2017, 2017, M957. https://doi.org/10.3390/M957
AMA Style
Veinot AJ, Ramgoolam K, Giffin NA, Masuda JD. Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide. Molbank. 2017; 2017(3):M957. https://doi.org/10.3390/M957
Chicago/Turabian StyleVeinot, Alex J., Ketnavi Ramgoolam, Nick A. Giffin, and Jason D. Masuda. 2017. "Crystal Structure of Bis(2,4,6-trimethylphenyl)-phosphine Oxide" Molbank 2017, no. 3: M957. https://doi.org/10.3390/M957
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.