Fluorescent Silicate Materials for the Detection of Paraoxon

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
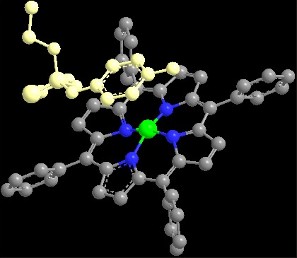
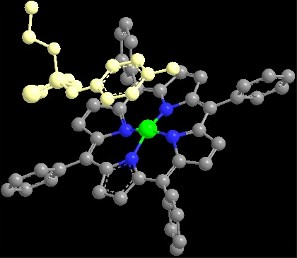
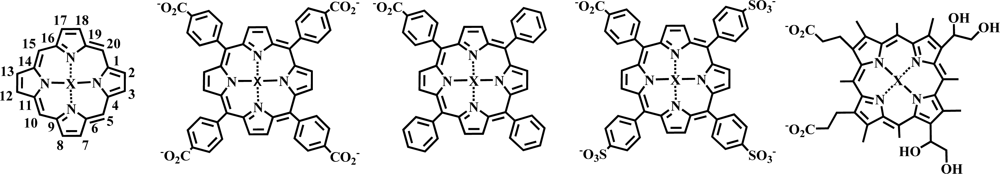
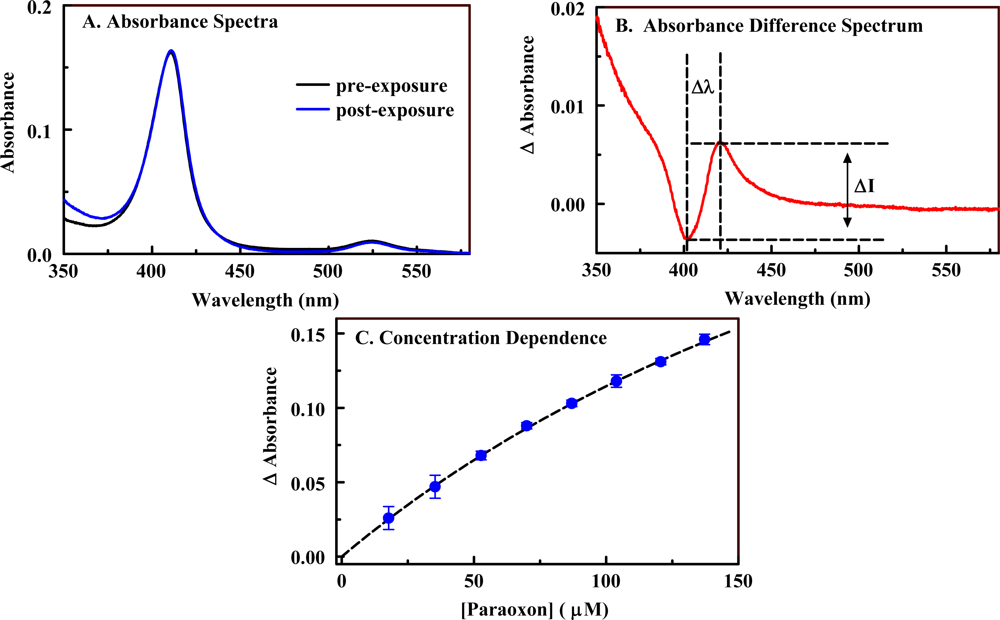
2.1. Porphyrin Indicator
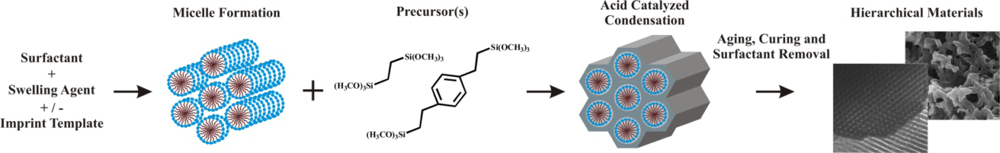
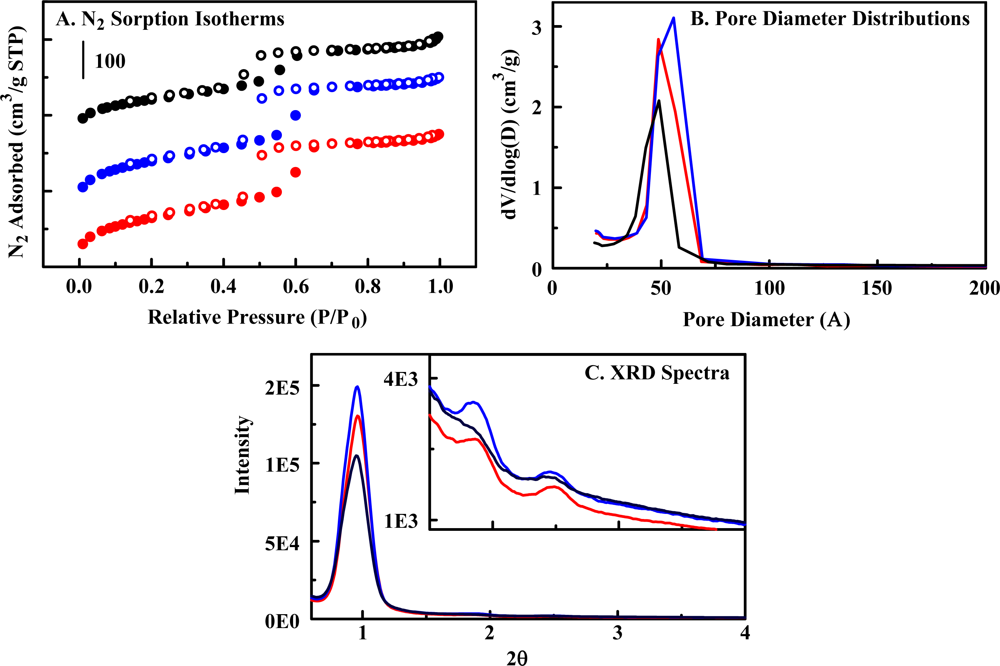
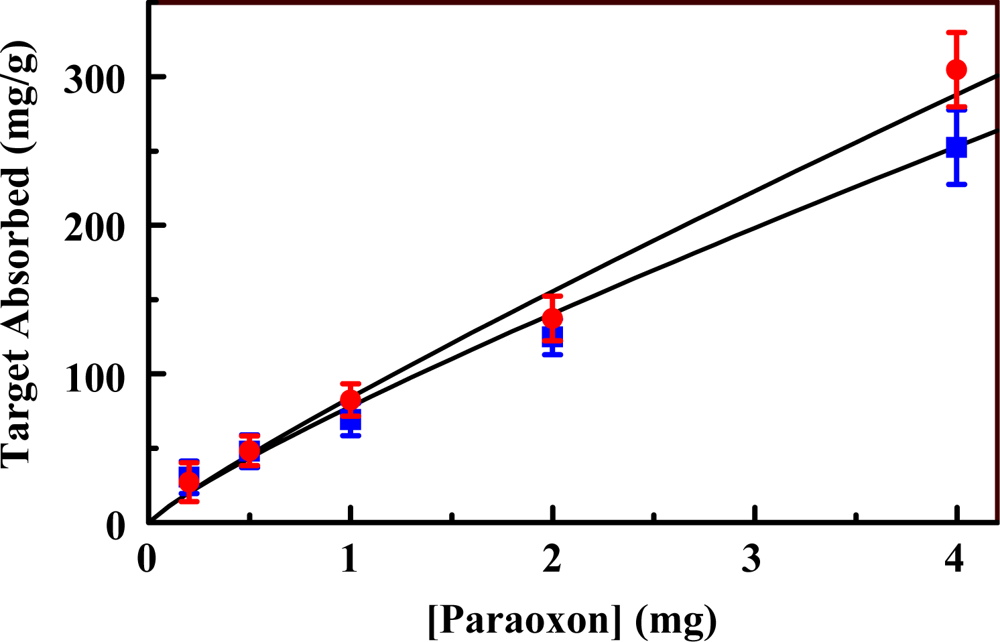
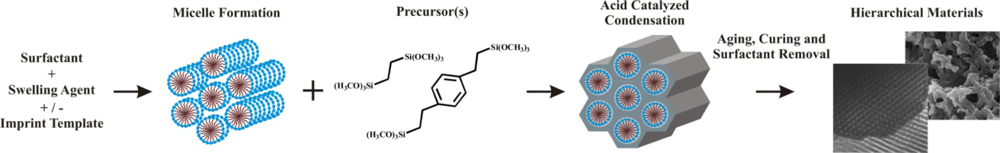
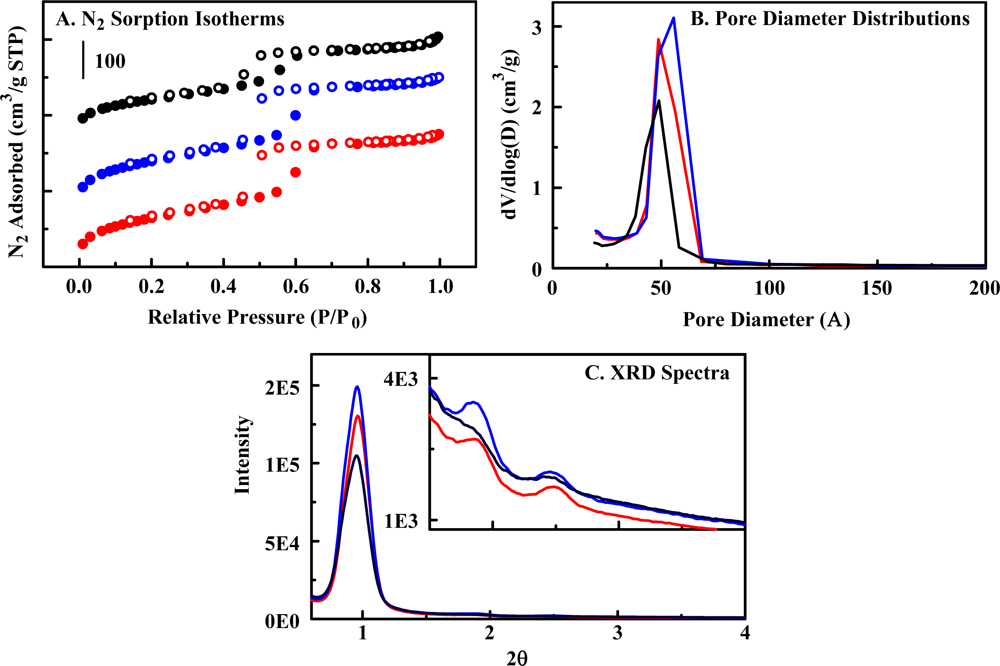
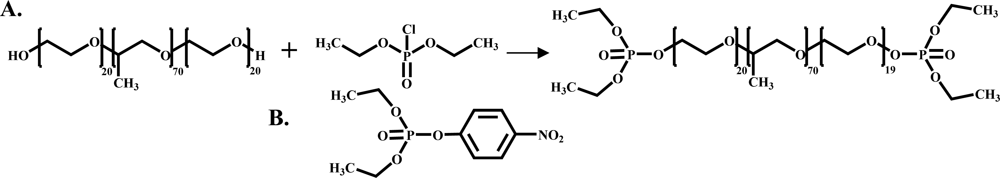
2.2. The Organosilicate Scaffold
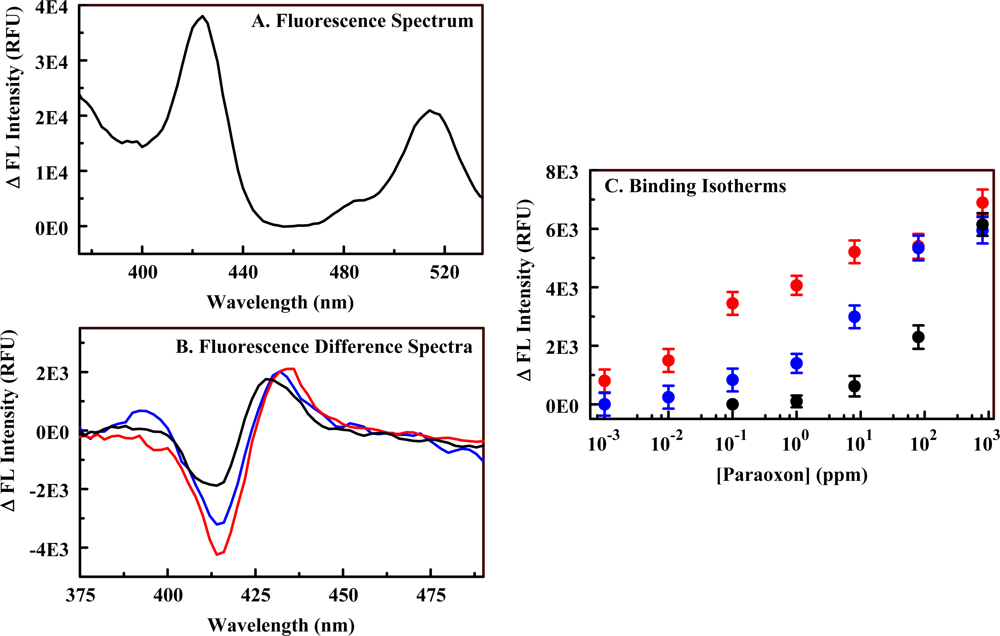
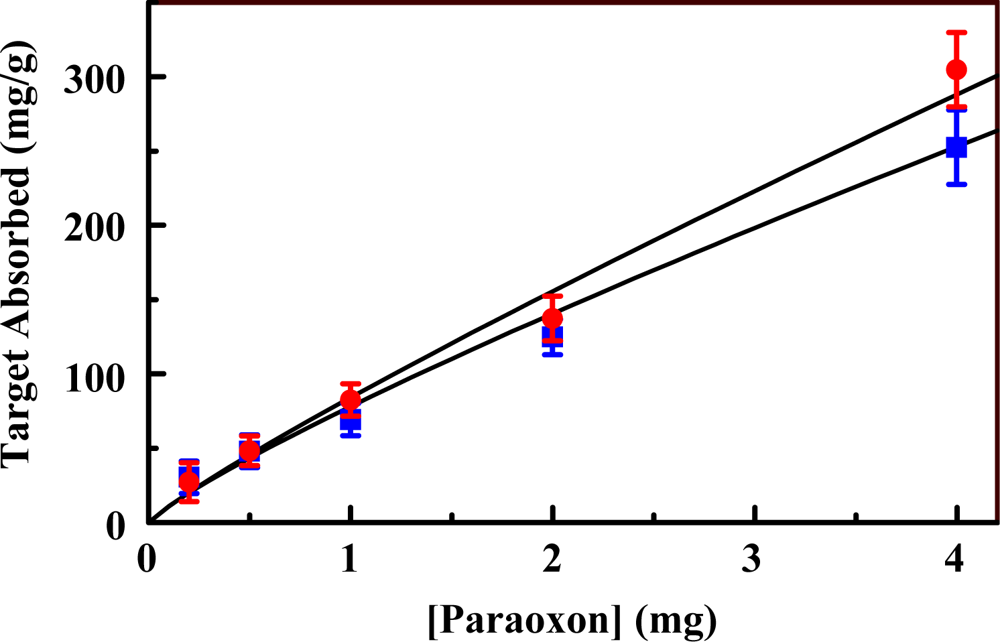
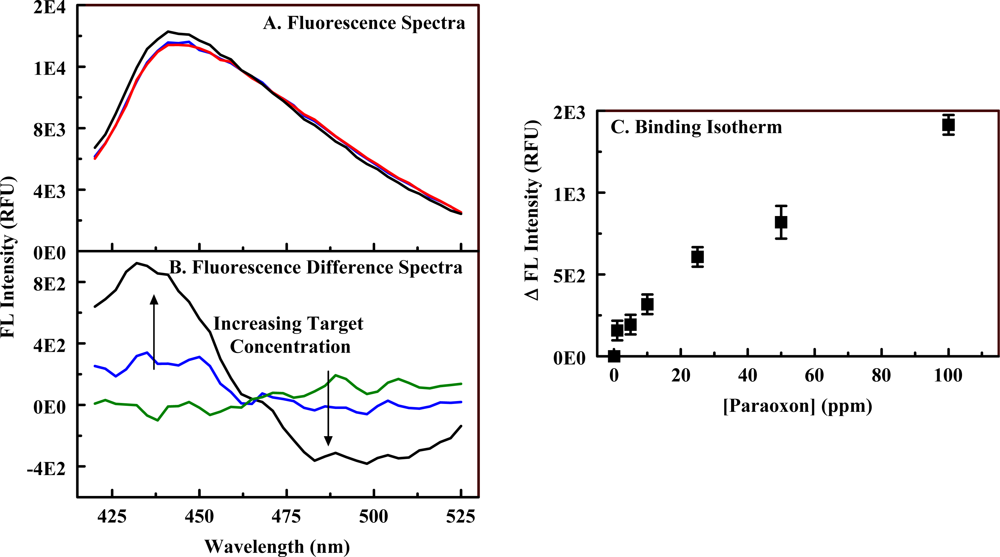
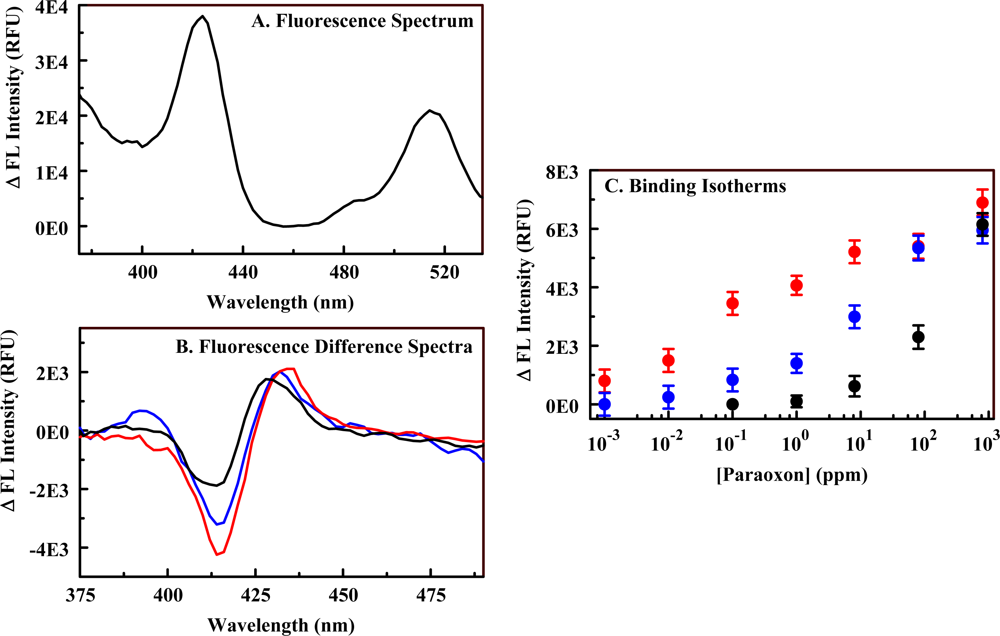
2.3. The Fluorescent Construct
3. Experimental Section
4. Conclusions
Acknowledgments
References and Notes
- Amao, Y. Probes and Polymers for Optical Sensing of Oxygen. Microchim. Acta 2003, 143, 1–12. [Google Scholar]
- Feng, Y.; Pilbrow, J.R. Porphyrin Intercalation and Non-specific ‘Edge on’ Outside Binding to Natural DNA. Biophys. Chem 1990, 36, 117–131. [Google Scholar]
- Malinski, T. Applications: Past, Present, Future. In The Porphyrin Handbook, 1st ed; Kadish, K. M., Smith, K.M., Guilard, R., Eds.; Academic Press: New York, NY, USA, 2000; Volume 6, p. 231. [Google Scholar]
- Ogoshi, H.; Mizutani, T.; Hayashi, T.; Kuroda, Y. Porphyris and Metalloporphyrins as Receptor Models in Molecular Recognition. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: New York, NY, USA, 2000; Volume 6, p. 279. [Google Scholar]
- Mauzerall, D. Spectra of Molecular Complexes of Porphyrins in Aqueous Solution. Biochemistry 1965, 4, 1801–1810. [Google Scholar]
- Shelnutt, J.A. Molecular Complexes of Copper Uroporphyrin with Aromatic Acceptors. J. Phys. Chem 1983, 87, 605–616. [Google Scholar]
- Schneider, H.-J.; Wang, M. Ligand-Porphyrin Complexes: Quantitative Evaluation of Stacking and Ionic Contributions. J. Org. Chem 1994, 59, 7464–7472. [Google Scholar]
- Balaji, T.; Sasidharan, M.; Matsunaga, H. Optical Sensor for the Visual Detection of Mercury Using Mesoporous Silica Anchoring Porphyrin Moiety. Analyst 2005, 130, 1162–1167. [Google Scholar]
- Balaji, T.; El-Safty, S.A.; Matsunaga, H.; Hanaoka, T.; Mizukami, F. Optical Sensors Based on Nanostructured Cage Materials for the Detection of Toxic Metal Ions. Angew. Chem. Int. Ed 2006, 45, 7202–7208. [Google Scholar]
- Han, B.-H.; Manners, I.; Winnik, M.A. Oxygen Sensors Based on Mesoporous Silica Particles on Layer-by-Layer Self-assembled Films. Chem. Mater 2005, 17, 3160–3171. [Google Scholar]
- Zhang, H.; Sun, Y.; Zhang, P.; Wang, Y. Oxygen Sensing Materials Based on Mesoporous Silica MCM-41 and Pt(II)-porphyrin Complexes. J. Mater. Chem 2005, 15, 3181–3186. [Google Scholar]
- Huo, C.; Zhang, H.; Zhang, H.; Zhang, H.; Yang, B.; Zhang, P.; Wang, Y. Synthesis and Assembly with Mesoporous Silica MCM-48 of Platinum(II) Porphyrin Complexes Bearing Carbazyl Groups: Spectroscopic and Oxygen Sensing Properties. Inorg. Chem 2006, 45, 4735–4742. [Google Scholar]
- Cardoso, W.S.; Francisco, M.S.P.; Landers, R.; Gushikem, Y. Co(II) porphyrin Adsorbed on SiO2/SnO2/phosphate Prepared by the Sol-gel Method. Application in Electroreduction of Dissolved Dioxygen. Electrochim. Acta 2005, 50, 4378–4384. [Google Scholar]
- Cardoso, W.S.; Gushikem, Y. Electrocatalytic Oxidation of Nitrite on a Carbon Paste Electrode Modified with Co(II) Porphyrin Adsorbed on SiO2/SnO2/Phosphate Prepared by the Sol-gel Method. J. Electroanal. Chem 2005, 583, 300–306. [Google Scholar]
- Tao, S.; Li, G. Porphyrin-doped Mesoporous Silica Films for Rapid TNT Detection. Colloid Polym. Sci 2007, 285, 721–728. [Google Scholar]
- Tao, S.; Li, G.; Zhu, H. Metalloporphyrins as Sensing Elements for the Rapid Detection of Trace TNT Vapor. J. Mater. Chem 2006, 16, 4521–4528. [Google Scholar]
- Tao, S.; Shi, Z.; Li, G.; Li, P. Hierarchically Structured Nanocomposite Films as Highly Sensitive Chemosensory Materials for TNT Detection. Chem. Phys. Chem 2006, 7, 1902–1905. [Google Scholar]
- Johnson-White, B.; Zeinali, M.; Shaffer, K.M.; Patterson, J.; Charles, P.T.; Markowitz, M.A. Detection of Organics Using Porphyrin Embedded Nanoporous Organosilicas. Biosens. Bioelect 2007, 22, 1154–1162. [Google Scholar]
- Kosuge, K.; Murakami, T.; Kikukawa, N.; Takemori, M. Direct Synthesis of Porous Pure and Thiol-Functional Silica Spheres through the S+X-I+ Assembly Pathway. Chem. Mater 2003, 15, 3184–3189. [Google Scholar]
- Matsumoto, A.; Misran, H.; Tsutsumi, K. Adsorption Characteristics of Organosilica Based Mesoporous Materials. Langmuir 2004, 20, 7139–7145. [Google Scholar]
- Palaniappan, A.; Su, X.; Tay, F.E.H. Functionalized Mesoporous Silica Films for Gas Sensing Applications. J. Electroceram 2006, 16, 503–505. [Google Scholar]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered Mesoporous Molecular Sieves Synthesized by a Liquid-crystal Template Mechanism. Nature 1992, 359, 710–712. [Google Scholar]
- Burleigh, M.C.; Markowitz, M.A.; Wong, E.M.; Lin, J.S.; Gaber, B.P. Synthesis of Periodic Mesoporous Organosilicas with Block Copolymer Templates. Chem. Mater 2001, 13, 4411–4412. [Google Scholar]
- Goto, Y.; Inagaki, S. Synthesis of Large-pore Phenylene-bridged Mesoporous Organosilica Using Triblock Copolymer Surfactant. Chem. Commun 2002, 20, 2410–2411. [Google Scholar]
- Melde, B.J.; Johnson, B.J.; Dinderman, M.A.; Deschamps, J.R. Macroporous periodic mesoporous organosilicas with diethylbenzene bridging groups. Microp. Mesop. Mater 2009, 130, 180–188. [Google Scholar]
- Nakanishi, K. Pore Structure Control of Silica Gels Based on Phase Separation. J. Porous Mater 1997, 4, 67–112. [Google Scholar]
- Nakanishi, K.; Kobayashi, Y.; Amatani, T.; Hirao, K.; Kodaira, T. Spontaneous Formation of Hierarchical Macro-Mesoporous Ethane-Silica Monolith. Chem. Mater 2004, 16, 3652–3658. [Google Scholar]
- Nakanishi, K.; Kanamori, K. Organic-Inorganic Hybrid Poly(silsesquioxane) Monoliths with Controlled Macro- and Mesopores. J. Mater. Chem 2005, 15, 3776–3786. [Google Scholar]
- Amatani, T.; Nakanishi, K.; Hirao, K.; Kodaira, T. Monolithic Periodic Mesoporous Silica with Well-Defined Macropores. Chem. Mater 2005, 17, 2114–2119. [Google Scholar]
- Nakanishi, K.; Amatani, T.; Yano, S.; Kodaira, T. Multiscale Templating of Siloxane Gels via Polymerization-Induced Phase Separation. Chem. Mater 2008, 20, 1108–1115. [Google Scholar]
- Brandhuber, D.; Peterlik, H.; Huesing, N. Facile Self-Assembly Processes to Phenylene-Bridged Silica Monoliths with Four Levels of Hierarchy. Small 2006, 2, 503–506. [Google Scholar]
- Zhong, H.; Zhu, G.; Yang, J.; Wang, P.; Yang, Q. Periodic Mesoporous Hybrid Monolith with Hierarchical Macro-Mesopores. Micropor. Mesopor. Mater 2007, 100, 259–267. [Google Scholar]
- Jayasundera, S.; Burleigh, M.C.; Zeinali, M.; Spector, M.S.; Miller, J.B.; Yan, W.; Dai, S.; Markowitz, M.A. Organosilica Copolymers for the Adsorption and Separation of Multiple Pollutants. J. Phys. Chem. B 2005, 109, 9198–9201. [Google Scholar]
- Loy, D.A.; Shea, K.J. Bridged Polysilsesquioxanes. Highly Porous Hybrid Organic-Inorganic Materials. Chem. Rev 1995, 95, 1431–1442. [Google Scholar]
- Johnson, B.J.; Melde, B.J.; Charles, P.T.; Cardona, D.C.; Dinderman, M.A.; Malanoski, A.P.; Qadri, S.B. Imprinted Nanoporous Organosilicas for Selective Adsorption of Nitroenergetic Targets. Langmuir 2008, 24, 9024–9029. [Google Scholar]
- Johnson-White, B.; Zeinali, M.; Malanoski, A.P.; Dinderman, M. Sunlight Catalyzed Conversion of Cyclic Organics with Novel Mesoporous Organosilicas. Catalysis Comm 2007, 8, 1052–1056. [Google Scholar]
- White, B.J.; Harmon, H.J. Optical Determination of Bacterial Exosporium Sugars Using Immobilized Porphyrins. IEEE Sensors J 2005, 5, 726–732. [Google Scholar]
- Johnson, B.J.; Melde, B.J.; Charles, P.T.; Malanoski, A.P. Porphyrin-embedded organosilicas for detection and decontamination. Proceedings of 2009 SPIE International Defense, Security and Sensing Symposium, Orlando, FL, USA, April 2009; Kumar, V., Prabhakar, S., Ross, A.A., Halvorson, H.S., Southern, S.O., Eds.; SPIE: Orlando, FL, USA, 2009. [Google Scholar]
- Kim, H.J.; Guiochon, G. Comparison of the Thermodynamic Properties of Particulate and Monolithic Columns of Molecularly Imprinted Copolymers. Anal. Chem 2005, 77, 93–102. [Google Scholar]
- Umpleby, R.J.; Baxter, S.C.; Bode, M.; Berch, J.K.; Shah, R.N.; Shimizu, K.D. Application of the Freundlich Adsorption Isotherm in the Characterization of Molecularly Imprinted Polymers. Anal. Chim. Acta 2001, 435, 35–42. [Google Scholar]
- Nozawa, A.; Ohnuma, T. Improved High-performance Liquid-chromatographic Analysis of Ehtylene-oxide Condensates by Their Esterification with 2,5-Dinitrobenzoyl Chloride. J. Chromatogr 1980, 187, 261–263. [Google Scholar]
- Sun, C.; Baird, M.; Anderson, H.A.; Brydon, D.L. Separation and Determination of Oligomers and Homologues of Aliphatic Alcohol Ethoxylates in Textile Lubricants and Lubricant Emulsion by High-performance Liquid Chromatography. J. Chromatogr 1997, 771, 145–154. [Google Scholar]
- Sun, C.; Baird, M.; Simpson, J. Determination of Poly(ethylene glycol)s by Both Normal-phase and Reversed-phase Modes of High-performance Liquid. J. Chromatogr 1998, 800, 231–238. [Google Scholar]
- Gebreegzi, Y.T.; Foster, G.D.; Khan, S.U. Simulltaneous Determination of Carbaryl, Malathion, Fenitrothion, and Diazinon Residues in Sesame Seeds (Seasmum indicum L). J. Agric. Food Chem 2000, 48, 5165–5168. [Google Scholar]
- Tscharntke, T.; Hochberg, M.E.; Rand, T.A.; Resh, V.H.; Krauss, J. Author Sequence and Credit for Contributions in Multiauthored Publications. PLoS Biol 2007, 5, e18. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porphyrin | Δ λ | Δ I | K11 (1/mM) | Δε11 Peak (A/mM) | Δε11 Trough (A/mM) |
|---|---|---|---|---|---|
| Selected Candidates | |||||
| Ni(II) C1S3TPP | 19 | 0.010 | 0.66 | 57 | 4.3 |
| Fe(II) C4TPP | 12 | 0.010 | 0.97 | 160 | 19 |
| Ni(II) C4TPP | 11 | 0.006 | 3.1 | 43 | 11 |
| Cu(II) C4TPP | 14 | 0.019 | 3.1 | 120 | 38 |
| Ni(II) C1TPP | N/A | 0.432 | 4.1 | N/A | 220 |
| Rejected Candidates | |||||
| Co(II) C1TPP | N/A | 0.012 | 0.22 | N/A | 3.0 |
| Fe(II) C1S3TPP | 10 | 0.010 | 0.09 | 14 | 36 |
| Pt(IV) C4TPP | 8 | 0.002 | 0.06 | 5 | 63 |
| Material | Designation | BET Surface Area (m2/g) | BJH Pore Volume (cm3/g) | Average Pore Diameter (Å) |
|---|---|---|---|---|
| 50:50 DEB:BTE No Imprint | Material 1 | 445 | 0.476 | 49 |
| 50:50 DEB:BTE Imprinted | Material 2 | 478 | 0.527 | 56 |
| 100% DEB* | -- | 455 | 0.414 | 45 |
| 100% BTE* | -- | 800 | >1 | 75 |
| 50:50 DEB:BTE No Imprint with amine functionality | Material 3 | 648 | 0.587 | 49 |
| 50:50 DEB:BTE Imprinted with amine functionality | Material 4 | 405 | 0.443 | 48 |
| 50:50 DEB:BTE Imprinted with high amine functionality | Material 5 | 343 | 0.374 | 49 |
| Scaffold | Porphyrin | Peak (nm) | Trough (nm) | LOD (ppm) |
|---|---|---|---|---|
| Material 3 | Fe(II) C4TPP | 430 | 410 | 90 |
| Ni(II) C4TPP | 416 | 438 | 50 | |
| Ni(II) C1TPP | N/A | N/A | N/A* | |
| Material 4 | Ni(II) C1S3TPP | 495 | 435 | 3 |
| Ni(II) C1TPP | 420 | 412 | 50 | |
| Ni(II) C4TPP | 417 | 433 | 10 | |
| Material 5 | Ni(II) C1S3TPP | 414 | 430 | 0.05 |
| Ni(II) C1TPP | 424 | 414 | 8 | |
| C4TPP | 420 | N/A | 50 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Johnson, B.J.; Melde, B.J.; Thomas, C.; Malanoski, A.P.; Leska, I.A.; Charles, P.T.; Parrish, D.A.; Deschamps, J.R. Fluorescent Silicate Materials for the Detection of Paraoxon. Sensors 2010, 10, 2315-2331. https://doi.org/10.3390/s100302315
Johnson BJ, Melde BJ, Thomas C, Malanoski AP, Leska IA, Charles PT, Parrish DA, Deschamps JR. Fluorescent Silicate Materials for the Detection of Paraoxon. Sensors. 2010; 10(3):2315-2331. https://doi.org/10.3390/s100302315
Chicago/Turabian StyleJohnson, Brandy J., Brian J. Melde, Cassandra Thomas, Anthony P. Malanoski, Iwona A. Leska, Paul T. Charles, Damon A. Parrish, and Jeffrey R. Deschamps. 2010. "Fluorescent Silicate Materials for the Detection of Paraoxon" Sensors 10, no. 3: 2315-2331. https://doi.org/10.3390/s100302315