
A Palladium-Tin Modified Microband Electrode Array for Nitrate Determination


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Chemicals
2.2. Apparatus
2.3. Working Electrode Modification
3. Results and Discussion
3.1. Preparation of Pd-Sn Electrode
3.1.1. Electrodeposition of Palladium


3.1.2. Electrodeposition of Tin
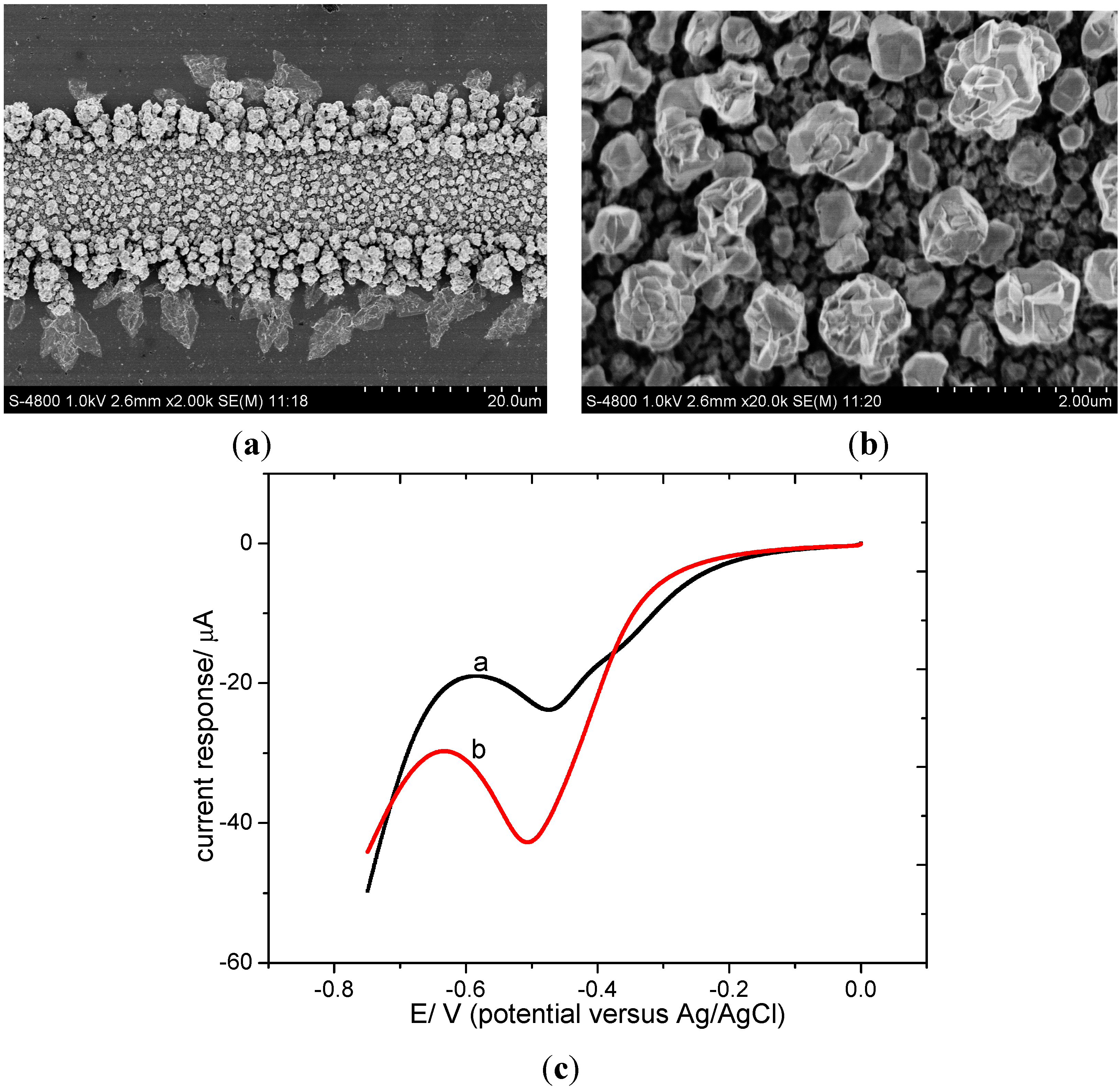
3.1.3. Characterization of Pd-Sn Electrode

3.2. Nitrate Determination
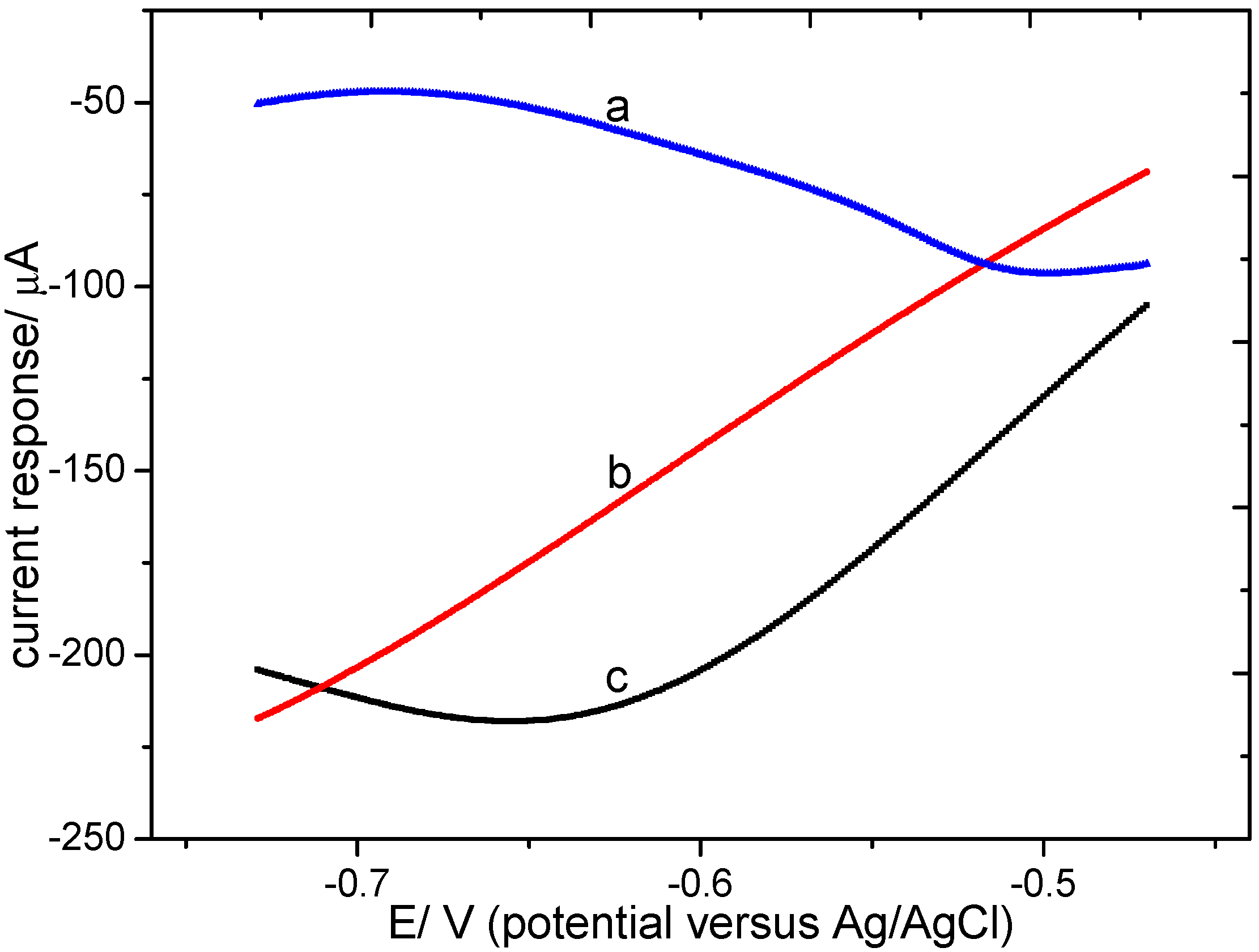
3.2.1. Electrochemical Measurement with Different Modified Electrodes



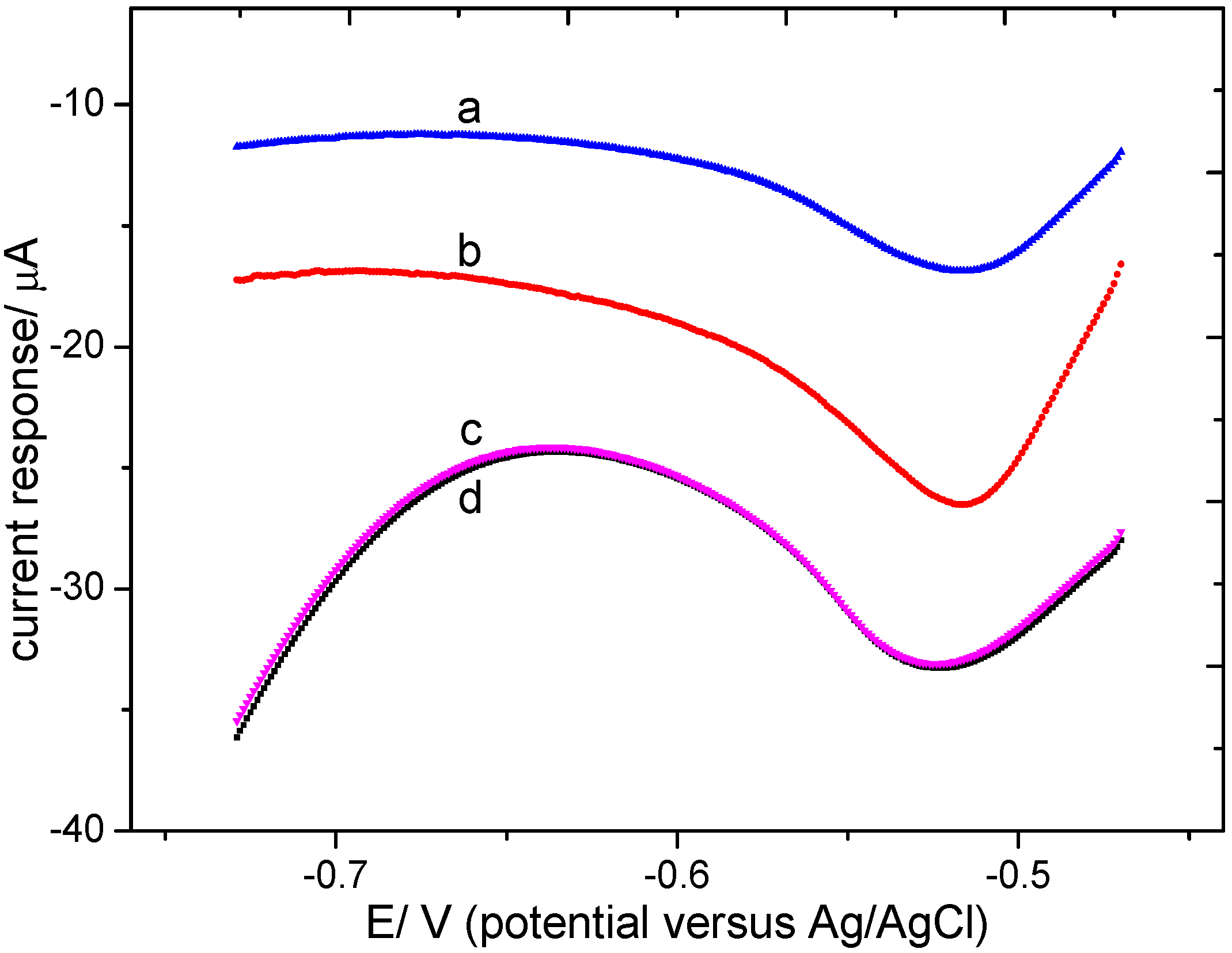
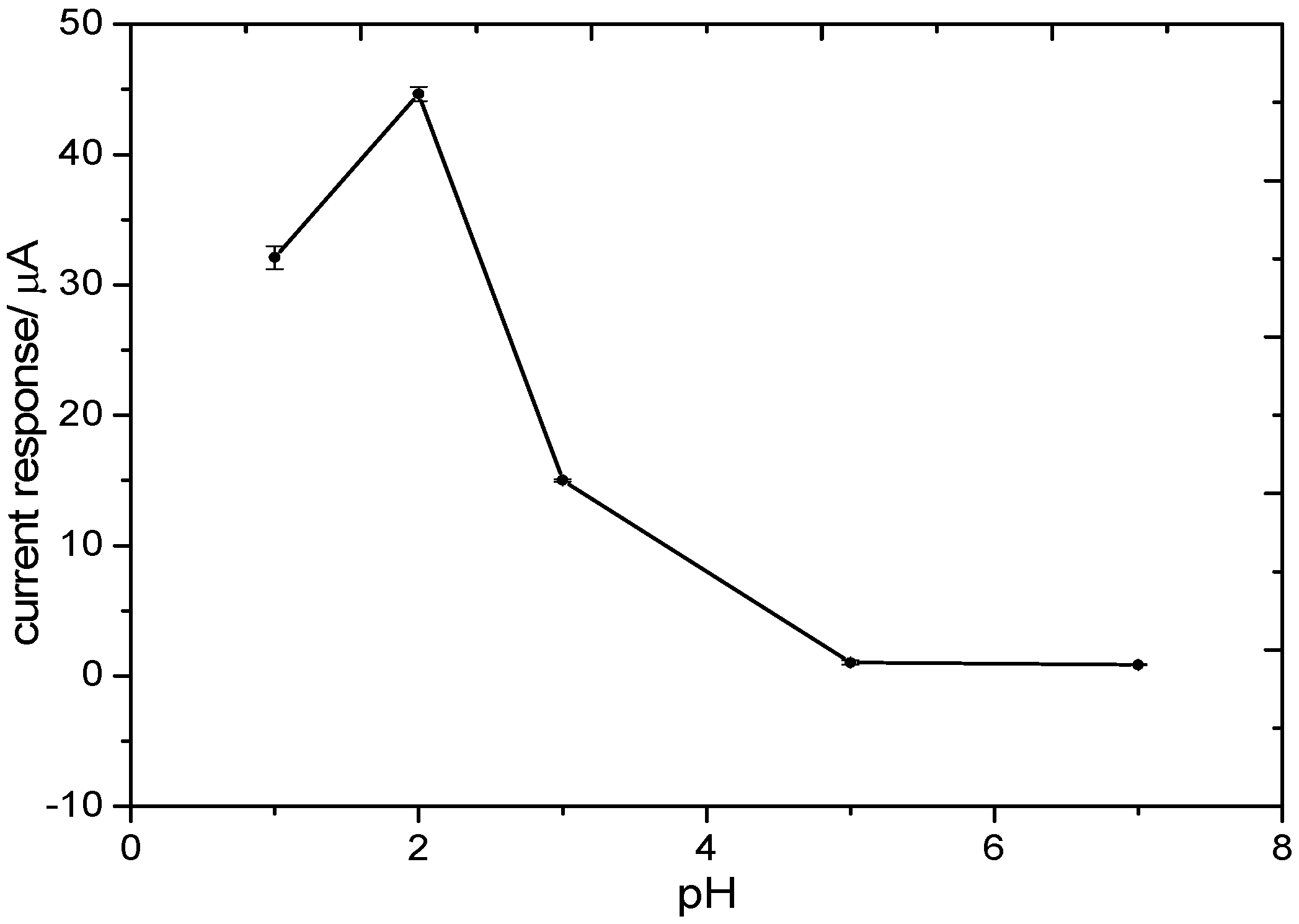
3.2.2. Optimization of the Electrolyte pH

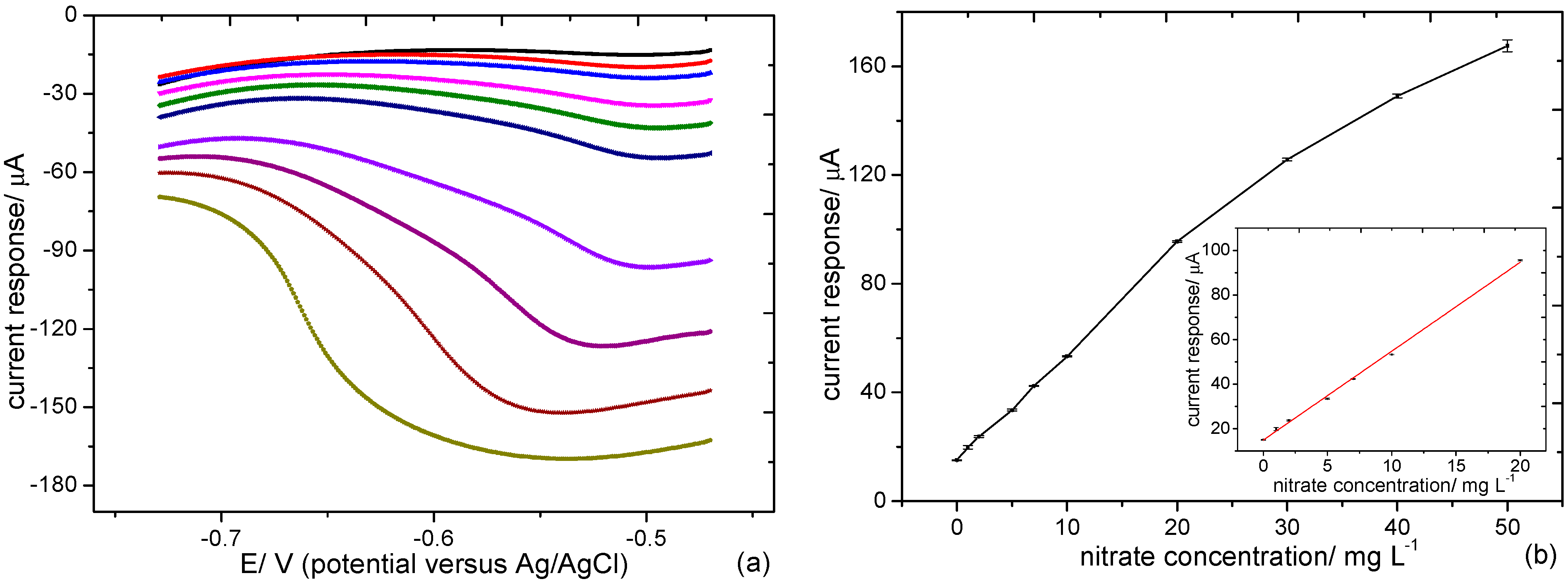
3.2.3. Nitrate Determination at the Pd-Sn Modified Electrode

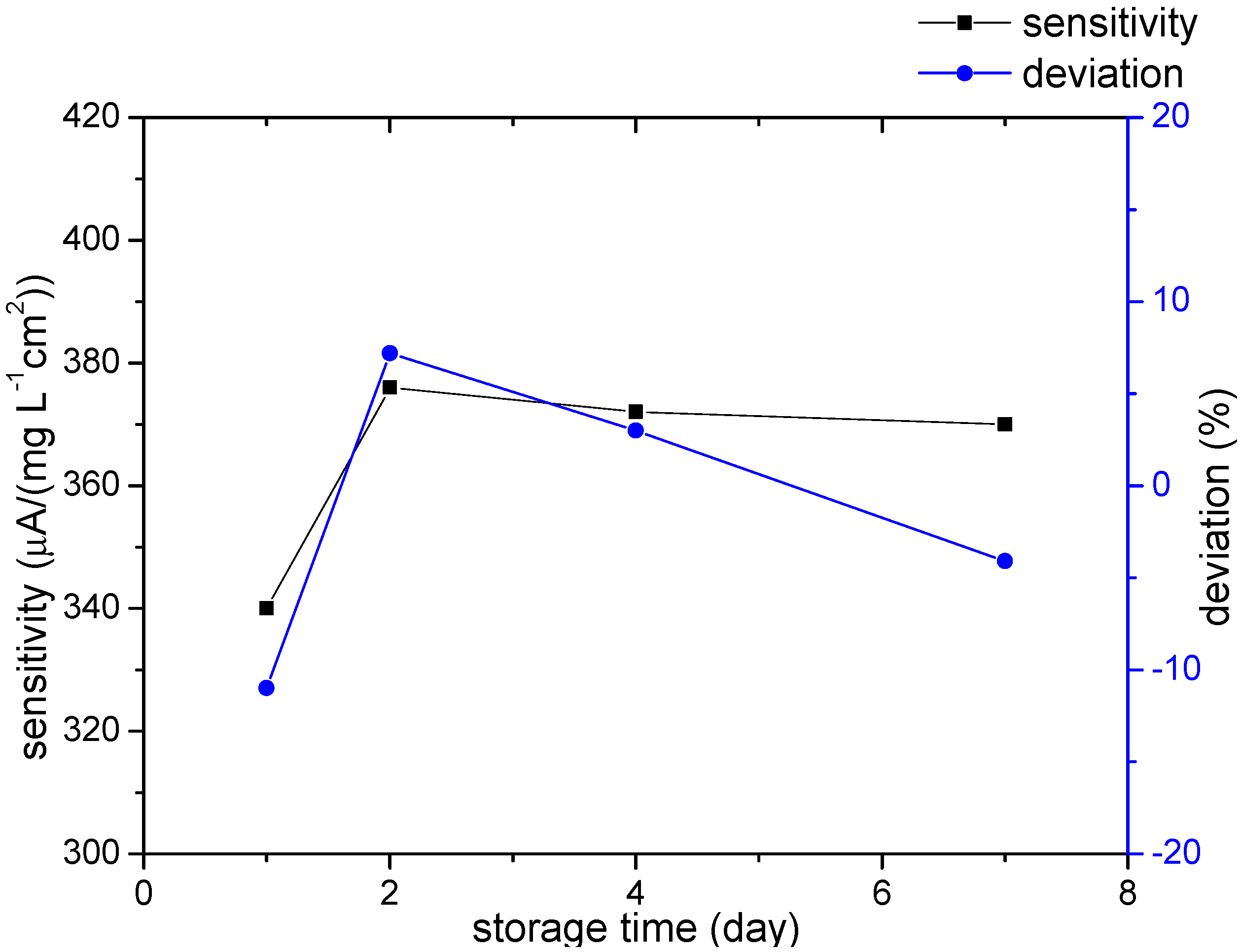
3.2.4. Repeatability and Lifetime


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Working Electrode | Supporting Electrolyte | pH | Sensitivity (μA/(mg·L−1·cm2)) | Lifetime | Literature |
|---|---|---|---|---|---|---|
| Palladium-tin | Platinum microband electrode array | 0.01 M HClO4 | 2 | 398 | More than 60 days | This paper |
| Palladium-tin | GC disk electrode (d = 3 mm) | 0.05 M H2SO4 | 1 | 167 | - | 2006, [24] |
| Copper | Platinum microband electrode array | 0.1 M Na2SO4 | 2 | 315 | - | 2015, [31] |
| Copper | Copper (d = 3 mm) | 0.1 M Na2SO4 | 2 | 147 | Renewable in situ | 2007, [36] |
| Palladium-copper | Epoxy-copper electrode (d = 3 mm) | 0.1 M PBS | 7 | 17 | - | 2013, [37] |
| AgNPs | GC disk electrode (d = 3 mm) | 0.1 M PBS | 6.7 | 52 | 1 day | 2013, [9] |
| PPy/Ag | Epoxy-glassy carbon rod | 0.1 M Na2SO4 | 7 | 1.6 | More than 60 days | 2011, [38] |
| Macroporous Ag | ITO (0.6 cm × 1.2 cm) | 1 M NaOH | 14 | 9.29 | - | 2010, [8] |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Birdja, Y.Y.; Yang, J.; Koper, M.T.M. Electrocatalytic Reduction of Nitrate on Tin-modified Palladium Electrodes. Electrochim. Acta. 2014, 140, 518–524. [Google Scholar] [CrossRef]
- Badea, G.E. Electrocatalytic reduction of nitrate on copper electrode in alkaline solution. Electrochim. Acta. 2009, 54, 996–1001. [Google Scholar] [CrossRef]
- Peel, J.W.; Reddy, K.J.; Sullivan, B.P.; Bowen, J.M. Electrocatalytic reduction of nitrate in water. Water Res. 2003, 37, 2512–2519. [Google Scholar] [CrossRef]
- Moorcroft, M.J.; Davis, J.; Compton, R.G. Detection and determination of nitrate and nitrite: A review. Talanta 2011, 54, 785–803. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Bian, C.; Tong, J.; Xia, S. Electrodeposition of copper nano-clusters at a platinum microelectrode for trace nitrate determination. Procedia Eng. 2010, 5, 339–342. [Google Scholar] [CrossRef]
- Ward-Jones, S.; Banks, C.E.; Simm, A.O.; Jiang, L.; Compton, R.G. An in situ copper plated boron-doped diamond microelectrode array for the sensitive electrochemical detection of nitrate. Electroanalysis 2005, 17, 1806–1815. [Google Scholar] [CrossRef]
- Stortini, A.M.; Moretto, L.M.; Mardegan, A.; Ongaro, M.; Ugo, P. Arrays of copper nanowire electrodes: Preparation, characterization and application as nitrate sensor. Sens. Actuators B: Chem. 2015, 207, 186–192. [Google Scholar] [CrossRef]
- Tsai, M.C.; Zhuang, D.X.; Chen, P.Y. Electrodeposition of macroporous silver films from ionic liquids and assessment of these films in the electrocatalytic reduction of nitrate. Electrochim. Acta. 2010, 55, 1019–1027. [Google Scholar] [CrossRef]
- Guadagnini, L.; Tonelli, D. Carbon electrodes unmodified and decorated with silver nanoparticles for the determination of nitrite, nitrate and iodate. Sens. Actuators B: Chem. 2013, 188, 806–814. [Google Scholar] [CrossRef]
- Dhanya, S.; Saumya, V.; Rao, T.P. Synthesis of silver nanoclusters, characterization and application to trace level sensing of nitrate in aqueous media. Electrochim. Acta. 2013, 102, 299–305. [Google Scholar] [CrossRef]
- Gamboa, J.C.M.; Peña, R.C.; Paixão, T.R.L.C.; Bertotti, M. A renewable copper electrode as an amperometric flow detector for nitrate determination in mineral water and soft drink samples. Talanta 2009, 80, 581–585. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Song, X.; Yang, H. Study on Sensor of Nitrate Based on Copper Plated Electrode. Chin. J. Sens. Actuat. 2012, 25, 1216–1220. [Google Scholar]
- Kaminskaya, O.V.; Zakharova, E.A.; Slepchenko, G.B. Simultaneous Voltammetric Determination of Nitrites and Nitrates in Waters. J. Anal. Chem. 2004, 59, 1091–1096. [Google Scholar] [CrossRef]
- Davis, J.; Moorcroft, M.J.; Wilkins, S.J.; Compton, R.G.; Cardosi, M.F. Electrochemical detection of nitrate and nitrite at a copper modified electrode. Analyst 2000, 125, 737–742. [Google Scholar] [CrossRef]
- Trawczyński, J.; Gheek, P.; Okal, J.; Zawadzki, M.; Gomez, M.J.I. Reduction of nitrate on active carbon supported Pd-Cu catalysts. Appl. Catal. A: Gen. 2011, 409, 39–47. [Google Scholar] [CrossRef]
- De Vooys, A.C.A.; Van Santen, R.A.; van Veen, J.A.R. Electrocatalytic reduction of NO3− on palladium/copper electrodes. J. Mol. Catal. A: Chem. 2000, 154, 203–215. [Google Scholar] [CrossRef]
- Da Cunha, M.; De Souza, J.P.I.; Nart, F.C. Reaction pathways for reduction of nitrate ions on platinum, rhodium, and platinum-rhodium alloy electrodes. Langmuir 2000, 16, 771–777. [Google Scholar] [CrossRef]
- Hasnat, M.A.; Ahamad, N.; Uddin, S.M.N.; Mohamed, N. Silver modified platinum surface/H+ conducting Nafion membrane for cathodic reduction of nitrate ions. Appl. Surf. Sci. 2012, 258, 3309–3314. [Google Scholar] [CrossRef]
- Lou, Y.; Shao, Y.; Li, P.; Li, Z.; Niu, Z. Electrocatalytic reduction of in a neutral solution on an electrodeposited film of amorphous Pd33Ni60P7 alloy. J. Electroanal. Chem. 2008, 624, 33–38. [Google Scholar] [CrossRef]
- Hossain, M.M.; Nakata, K.; Kawaguchi, T.; Shimazu, K. Reduction of nitrate on electrochemically pre-reduced tin-modified palladium electrodes. J. Electroanal. Chem. 2013, 707, 59–65. [Google Scholar] [CrossRef]
- Tada, K.; Kawaguchi, T.; Shimazu, K. High electrocatalytic performance of Pd/Sn/Au electrodes for nitrate reduction. J. Electroanal. Chem. 2004, 572, 93–99. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, J.; Wu, R.; Lei, P. The electrocatalytic reduction of nitrate in water on Pd/Sn-modified activated carbon fiber electrode. Water Res. 2006, 40, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, K.; Goto, R.; Piao, S.; Kayama, R.; Nakata, K.; Yoshinaga, Y. Reduction of nitrate ions on tin-modified palladium thin film electrodes. J. Electroanal. Chem. 2007, 601, 161–168. [Google Scholar] [CrossRef]
- Piao, S.; Kayama, Y.; Nakano, Y.; Nakata, K.; Yoshinaga, Y.; Shimazu, K. Nitrate reduction on tin-modified rhodium, ruthenium, and iridium electrodes. J. Electroanal. Chem. 2009, 629, 110–116. [Google Scholar] [CrossRef]
- Katsounaros, I.; Dortsiou, M.; Polatides, C.; Preston, S.; Kypraios, T.; Kyriacou, T. Reaction pathways in the electrochemical reduction of nitrate on tin. Electrochim. Acta. 2012, 71, 270–276. [Google Scholar] [CrossRef]
- Casella, I.G.; Contursi, M. An electrochemical and XPS study of the electrodeposited binary Pd–Sn catalyst: The electroreduction of nitrate ions in acid medium. J. Electroanal. Chem. 2006, 588, 147–154. [Google Scholar] [CrossRef]
- Zhao, H.; Wan, H.; Cai, W.; Ha, D.; Wang, P. Microelectrode array sensor for detection of heavy metals in water pollution. J. Zhejiang Univ. (Eng. Sci.) 2013, 47, 984–989. [Google Scholar]
- Huang, X.J.; O'Mahony, A.M.; Compton, R.G. Microelectrode arrays for electrochemistry: Approaches to fabrication. Small 2009, 5, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, Y.; Li, D.; Long, Y. Recent developments and applications of screen-printed electrodes in environmental assays-A review. Anal. Chim. Acta. 2000, 734, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Sun, J.; Bian, C.; Tong, J.; Xia, S. 3D Dendritic Nanostructure of Silver-Array: Preparation, Growth Mechanism and Application in Nitrate Sensor. Electroanalysis 2013, 25, 546–556. [Google Scholar] [CrossRef]
- Jian, K.; Yang, L.; Bian, C.; Tong, J.; Sun, J.; Xia, S. Microelectrode arrays modified with copper for nitrate determination. High Power Laser Part. Beam. 2015, 27, 024122-1–024122-5. [Google Scholar] [CrossRef]
- Aristizábal, A.; Contrerasa, S.; Barrabés, N.; Llorca, J.; Tichit, D.; Medina, F. Catalytic reduction of nitrates in water on Pt promoted Cu hydrotalcite-derived catalysts: Effect of the Pt–Cu alloy formation. Appl. Catal. B: Environ. 2011, 110, 58–70. [Google Scholar] [CrossRef]
- Low, C.T.J.; Walsh, F.C. Electrodeposition of tin, copper and tin–copper alloys from a methanesulfonic acid electrolyte containing a perfluorinated cationic surfactant. Surf. Coat. Technol. 2008, 202, 1339–1349. [Google Scholar] [CrossRef]
- Rosca, V.; Duca, M.; de Groot, M.T.; Koper, M.T.M. Nitrogen cycle electrocatalysis. Chem. Rev. 2009, 109, 2209–2244. [Google Scholar] [CrossRef] [PubMed]
- De Groot, M.T.; Koper, M.T.M. The influence of nitrate concentration and acidity on the electrocatalytic reduction of nitrate on platinum. J. Electroanal. Chem. 2004, 562, 81–94. [Google Scholar] [CrossRef]
- Paixao, T.R.L.C.; Cardoso, J.L.; Bertotti, M. Determination of nitrate in mineral water and sausage samples by using a renewable in situ copper modified electrode. Talanta 2007, 71, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Gutés, A.; Carraro, C.; Maboudian, R. Nitrate amperometric sensor in neutral pH based on Pd nanoparticles on epoxy-copper electrodes. Electrochim. Acta. 2013, 103, 38–43. [Google Scholar] [CrossRef]
- Atmeh, M.; Alcock-Earley, B.E. A conducting polymer/Ag nanoparticle composite as a nitrate sensor. J. Appl. Electrochem. 2011, 41, 1341–1347. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Bian, C.; Kuang, J.; Wang, J.; Tong, J.; Xia, S. A Palladium-Tin Modified Microband Electrode Array for Nitrate Determination. Sensors 2015, 15, 23249-23261. https://doi.org/10.3390/s150923249
Fu Y, Bian C, Kuang J, Wang J, Tong J, Xia S. A Palladium-Tin Modified Microband Electrode Array for Nitrate Determination. Sensors. 2015; 15(9):23249-23261. https://doi.org/10.3390/s150923249
Chicago/Turabian StyleFu, Yexiang, Chao Bian, Jian Kuang, Jinfen Wang, Jianhua Tong, and Shanhong Xia. 2015. "A Palladium-Tin Modified Microband Electrode Array for Nitrate Determination" Sensors 15, no. 9: 23249-23261. https://doi.org/10.3390/s150923249
APA StyleFu, Y., Bian, C., Kuang, J., Wang, J., Tong, J., & Xia, S. (2015). A Palladium-Tin Modified Microband Electrode Array for Nitrate Determination. Sensors, 15(9), 23249-23261. https://doi.org/10.3390/s150923249