
Fast and Inexpensive Detection of Bacterial Viability and Drug Effectiveness through Metabolic Monitoring


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Working Principle
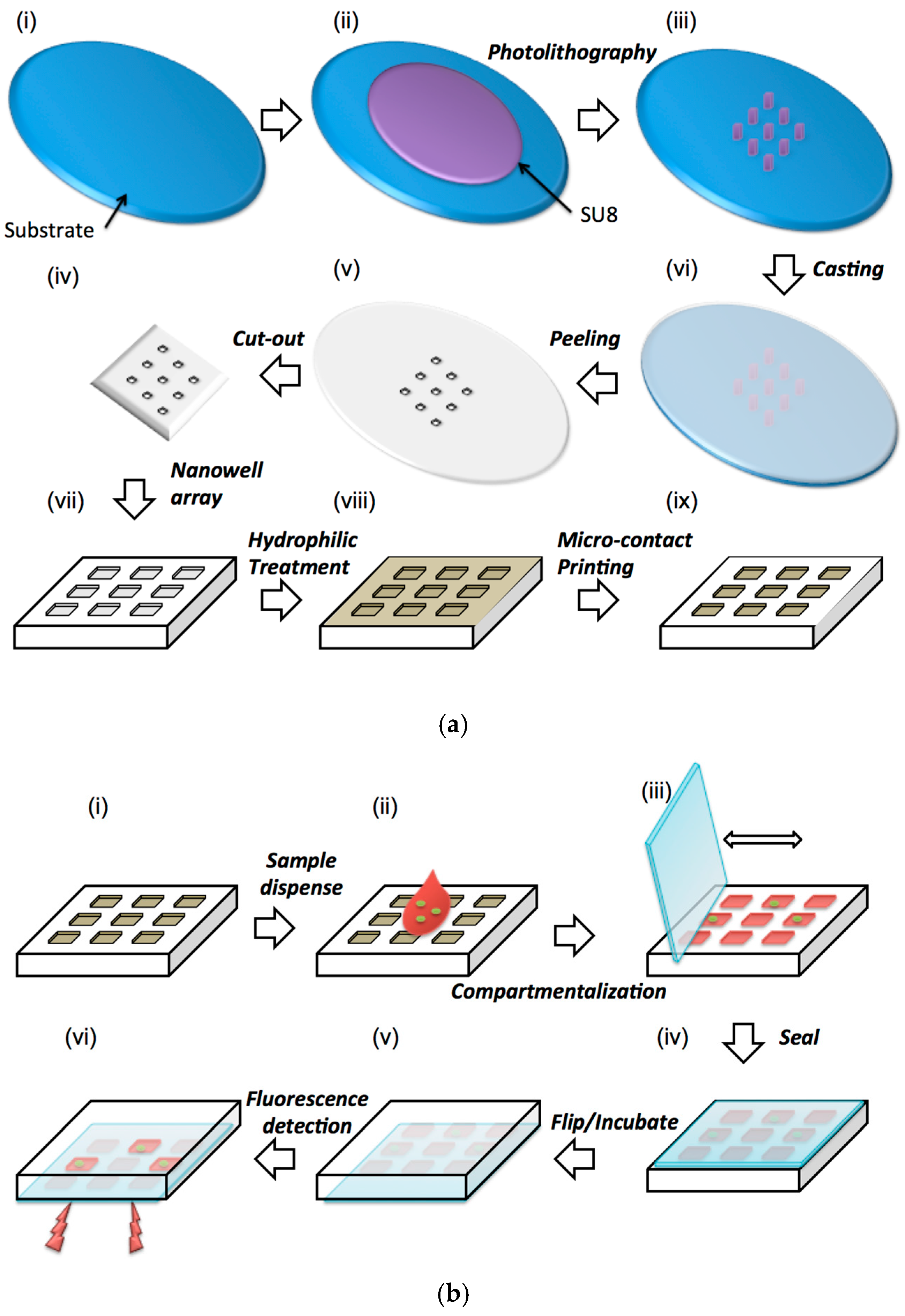
3. Materials and Methods
4. Results and Discussion
4.1. Sample Dispensing and Compartmentalization
4.2. Distribution of Bacteria
4.3. Oxygen Sensing in Nanowells
4.4. Growth of Bacteria in Nanowells
4.5. Measurement of Metabolic Activity
4.6. Effect of Nanowell Size on Change in Intensity
4.7. Drug Effectiveness
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. The Global Burden of Disease. Available online: http://www.who.int/topics/global_burden_of_disease/en/ (accessed on 27 October 2016).
- Infectious Diseases. The CSIS Global Health Policy Center 2015. Available online: https://www.csis.org/topics/global-health/infectious-disease (accessed on 27 October 2016).
- World Health Organization. Tuberculosis. Available online: http://www.who.int/mediacentre/factsheets/fs104/en/ (accessed on 27 October 2016).
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Wolfson, L.J.; Watt, J.P.; Henkle, E.; Deloria-Knoll, M.; McCall, N.; Lee, E.; Mulholland, K.; Levine, O.S.; Cherian, T. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 2009, 374, 893–902. [Google Scholar] [CrossRef]
- Pachecker, H.H. The Immigrant’s Universe; Xlibris: Bloomington, IN, USA, 2010. [Google Scholar]
- Jay, M.T.; Garrett, V.; Mohle-Boetani, J.C.; Barros, M.; Farrar, J.A.; Rios, R.; Abbott, S.; Sowadsky, R.; Komatsu, K.; Mandrell, R.; et al. A multistate outbreak of Escherichia coli O157:H7 infection linked to consumption of beef tacos at a fast-food restaurant chain. Clin. Infect. Dis. 2004, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Messelhäusser, U.; Kämpf, P.; Hörmansdorfer, S.; Wagner, B.; Schalch, B.; Busch, U.; Höller, C.; Wallner, P.; Barth, G.; Rampp, A. Culture and molecular method for detection of Mycobacterium tuberculosis complex and Mycobacterium avium subsp. paratuberculosis in milk and dairy products. Appl. Environ. Microbiol. 2012, 78, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Toze, S. PCR and the detection of microbial pathogens in water and wastewater. Water Res. 1999, 33, 3545–3556. [Google Scholar] [CrossRef]
- Allegranzi, B.; Nejad, S.B.; Combescure, C.; Graafmans, W.; Attar, H.; Donaldson, L.; Pittet, D. Burden of endemic health-care associated infection in developing countries: Systematic review and meta-analysis. Lancet 2011, 377, 228–241. [Google Scholar] [CrossRef]
- Polin, R.A.; Denson, S.; Brady, M.T.; Papile, L.A.; Baley, J.E.; Carlo, W.A.; Cummings, J.J.; Kumar, P.; Tan, R.C.; Watterberg, K.L. Epidemiology and diagnosis of health care-associated infections in the NICU. Pediatrics 2012, 129, e1104–e1109. [Google Scholar] [CrossRef] [PubMed]
- Kochi, A. The global tuberculosis situation and the new control strategy of the World Health Organization. Bull. World Health Organ. 2001, 79, 71–75. [Google Scholar] [CrossRef]
- Chung, H.J.; Castro, C.M.; Im, H.; Lee, H.; Weissleder, R. A magneto-DNA nanoparticle system for rapid detection and phenotyping of bacteria. Nat. Nanotechnol. 2013, 8, 369–375. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cutting-Edge Science and the Future of Tuberculosis Control. Available online: http://www.who.int/bulletin/volumes/85/5/06-035386/en/ (accessed on 27 October 2016).
- Gracias, K.S.; McKillip, J.L. A review of conventional detection and enumeration methods for pathogenic bacteria in food. Can. J. Microbiol. 2004, 50, 883–890. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Diagnostics for Tuberculosis: Global Demand and Market Potential. Available online: http://www.who.int/tdr/publications/documents/tbdi.pdf?ua=1 (accessed on 27 October 2016).
- Lee, H.; Park, J.; Kim, J.; Jung, H.; Kawai, T. Well-oriented nanowell array metrics for integrated digital nanobiosensors. Appl. Phys. Lett. 2006, 89, 113901. [Google Scholar] [CrossRef]
- Liang, P.-S.; Park, T.S.; Yoon, J.-Y. Rapid and reagentless detection of microbial contamination within meat utilizing a smartphone-based biosensor. Sci. Rep. 2014, 4, 5953. [Google Scholar] [CrossRef] [PubMed]
- Kik, S.V.; Denkinger, C.M.; Casenghi, M.; Vadnais, C.; Pai, M. Tuberculosis diagnostics: Which target product profiles should be prioritised? Eur. Respir. J. 2014, 44, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Francois, P.; Tangomo, M.; Hibbs, J.; Bonetti, E.-J.; Boehme, C.C.; Notomi, T.; Perkins, M.D.; Schrenzel, J. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Mic. 2011, 62, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Aellen, S.; Que, Y.-A.; Guignard, B.; Haenni, M.; Moreillon, P. Detection of live and antibiotic-killed bacteria by quantitative real-time PCR of specific fragments of rRNA. Antimicrob. Agents Chemother. 2006, 50, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Sprayberry, K.A.; Robinson, N.E. Robinson’s Current Therapy in Equine Medicine; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Torres, A.J.; Hill, A.S.; Love, J.C. Nanowell-based immunoassays for measuring single-cell secretion: Characterization of transport and surface binding. Anal. Chem. 2014, 86, 11562–11569. [Google Scholar] [CrossRef] [PubMed]
- Papkovsky, D.B.; Dmitriev, R.I. Biological detection by optical oxygen sensing. Chem. Soc. Rev. 2013, 42, 8700–8732. [Google Scholar] [CrossRef] [PubMed]
- Arain, S. Microrespirometry with Sensor-Equipped Microtiterplates. Ph.D. Thesis, University of Regensberg, Regensburg, Germany, February 2006. [Google Scholar]
- Chen, C.H.; Lu, Y.; Sin, M.L.Y.; Mach, K.E.; Zhang, D.D.; Gau, V.; Liao, J.C.; Wong, P.K. Antimicrobial susceptibility testing using high surface- to-volume ratio microchannels. Anal. Chem. 2010, 82, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Somoskövi, Á.; Ködmön, C.; Lantos, Á.; Bártfai, Z.; Tamási, L.; Füzy, J.; Magyar, P. Comparison of recoveries of mycobacterium tuberculosis using the automated BACTEC MGIT 960 system, the BACTEC 460 TB System, and Löwenstein-Jensen medium. J. Clin. Microbiol. 2000, 38, 2395–2397. [Google Scholar] [PubMed]
- George, W.L.; Sutter, V.L.; Citron, D.; Finegold, S.M. Selective and differential medium for isolation of Clostridium difficile. J. Clin. Microbiol. 1979, 9, 214–219. [Google Scholar] [PubMed]
- Rosenfeld, L.; Lin, T.; Derda, R.; Tang, S.K.Y. Review and analysis of performance metrics of droplet microfluidics systems. Microfluid. Nanofluid. 2014, 16, 921–939. [Google Scholar] [CrossRef]
- Rettig, J.R.; Folch, A. large-scale single-cell trapping and imaging using microwell arrays. Anal. Chem. 2005, 77, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Rigante, S.; Pisano, A.P.; Kuypers, F.A. Large-scale arrays of picolitre chambers for single-cell analysis of large cell populations. Lab Chip 2010, 10, 2952–2958. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Y.H.; Ma, X.; Qin, L. Rapid identification and drug susceptibility screening of ESAT-6 secreting Mycobacteria by a NanoELIwell assay. Sci. Rep. 2012, 2, 635. [Google Scholar] [CrossRef] [PubMed]
- Balsam, J.; Ossandon, M.; Bruck, H.; Rasooly, A. Low-cost technologies for medical diagnostics in low-resource settings. Expert. Opin. Med. Diagn. 2013, 7, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.R.; Davis, G.L.; Oden, Z.M.; Razavi, M.R.; Fateh, A.; Ghazanfari, M.; Abdolrahimi, F.; Poorazar, S.; Sakhaie, F.; Olsen, R.J.; et al. Portable, battery-operated, low-cost, bright field and fluorescence microscope. PLoS ONE 2010, 5, e11890. [Google Scholar] [CrossRef] [PubMed]
- Methyl Cellulose: Product Information Sheet. Available online: http://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/2/m0512pis.pdf (accessed on 27 October 2016).
- Lopez-Vidriero, M.T.; Charman, J.; Keal, E.; De Silva, D.J.; Reid, L. Sputum viscosity: Correlation with chemical and clinical features in chronic bronchitis. Thorax 1973, 28, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.C.; Walt, D.R. Application of High-Density Optical Microwell Arrays in a Live-Cell Biosensing System. Anal. Biochem. 2000, 278, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Mitarai, S.; Aguiman, L.; Matsumoto, H.; Fujiki, A. Preparation of mycobacteria-containing artificial sputum for TB panel testing and microscopy of sputum smears. Int. J. Tuberc. Lung Dis. 2006, 10, 899–905. [Google Scholar] [PubMed]
- Keer, J.T.; Birch, L. Molecular methods for the assessment of bacterial viability. J. Microbiolog. Methods 2003, 53, 175–183. [Google Scholar] [CrossRef]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayyash, S.; Wu, W.-I.; Selvaganapathy, P.R. Fast and Inexpensive Detection of Bacterial Viability and Drug Effectiveness through Metabolic Monitoring. Sensors 2016, 16, 1879. https://doi.org/10.3390/s16111879
Ayyash S, Wu W-I, Selvaganapathy PR. Fast and Inexpensive Detection of Bacterial Viability and Drug Effectiveness through Metabolic Monitoring. Sensors. 2016; 16(11):1879. https://doi.org/10.3390/s16111879
Chicago/Turabian StyleAyyash, Sondos, Wen-I Wu, and Ponnambalam Ravi Selvaganapathy. 2016. "Fast and Inexpensive Detection of Bacterial Viability and Drug Effectiveness through Metabolic Monitoring" Sensors 16, no. 11: 1879. https://doi.org/10.3390/s16111879