
The ?-Lactamase Assay: Harnessing a FRET Biosensor to Analyse Viral Fusion Mechanisms


Abstract
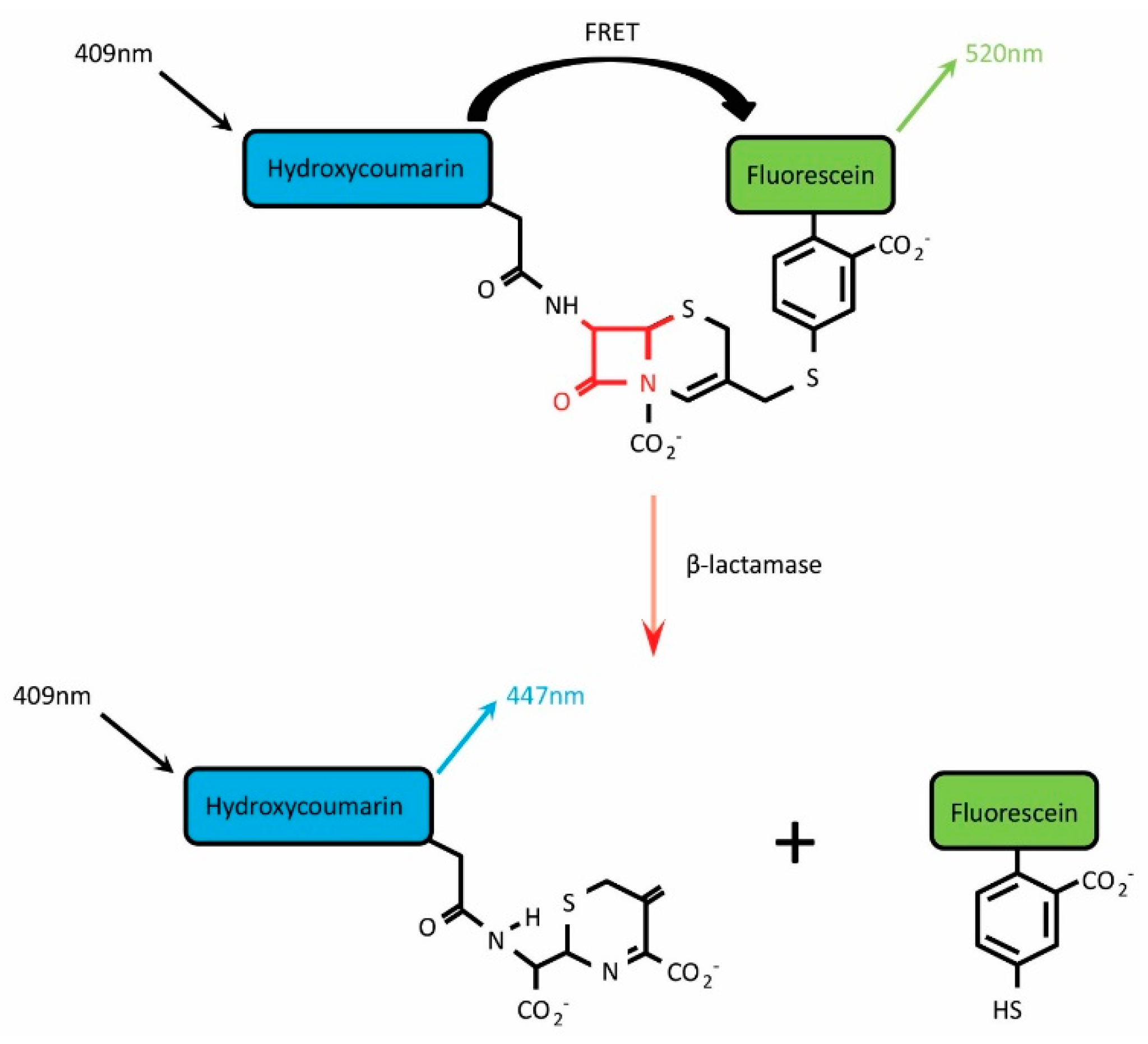
:1. Introduction
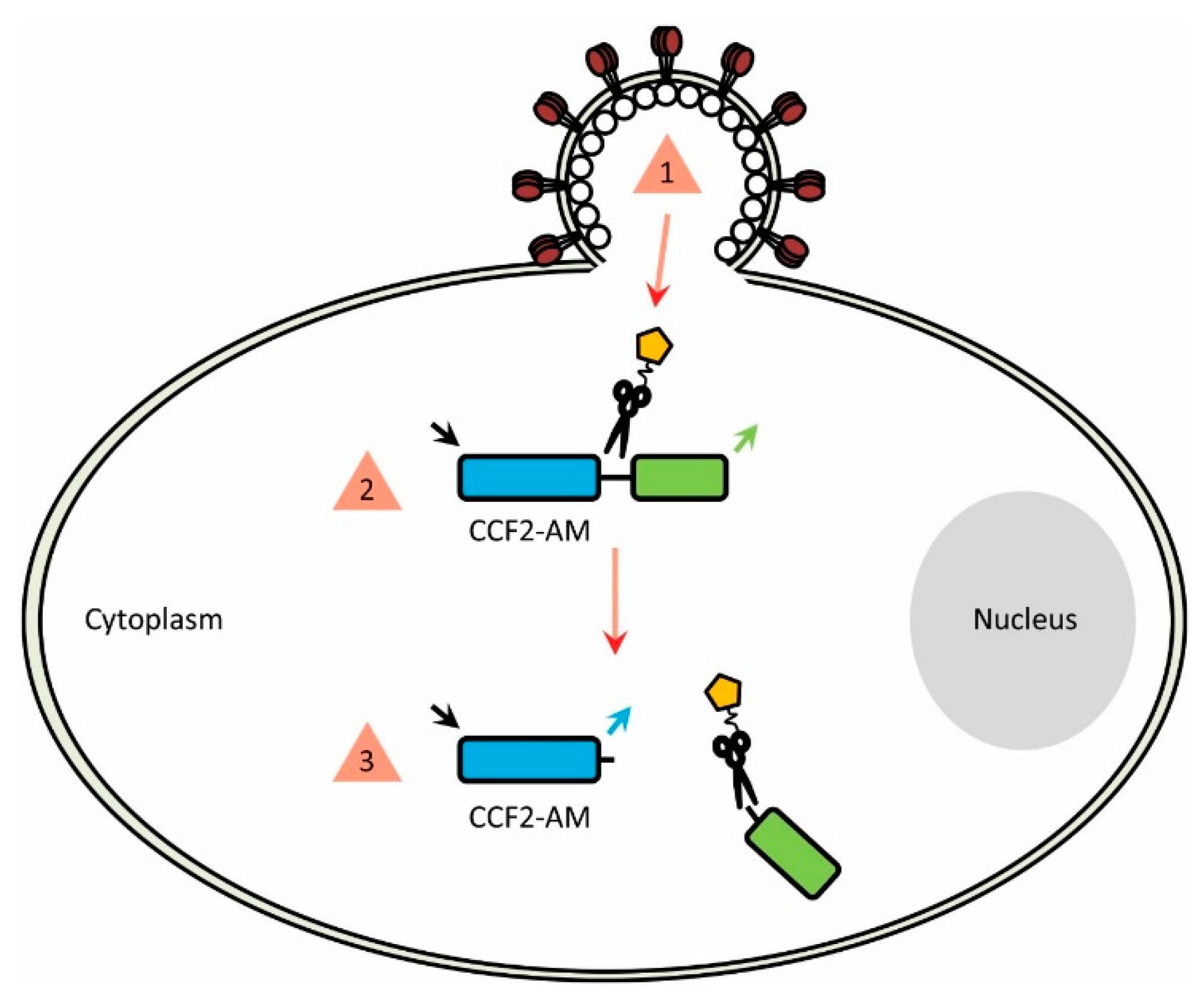
2. Producing BlaM-Compatible Viruses
3. Choosing a BlaM-Based Viral Entry Assay
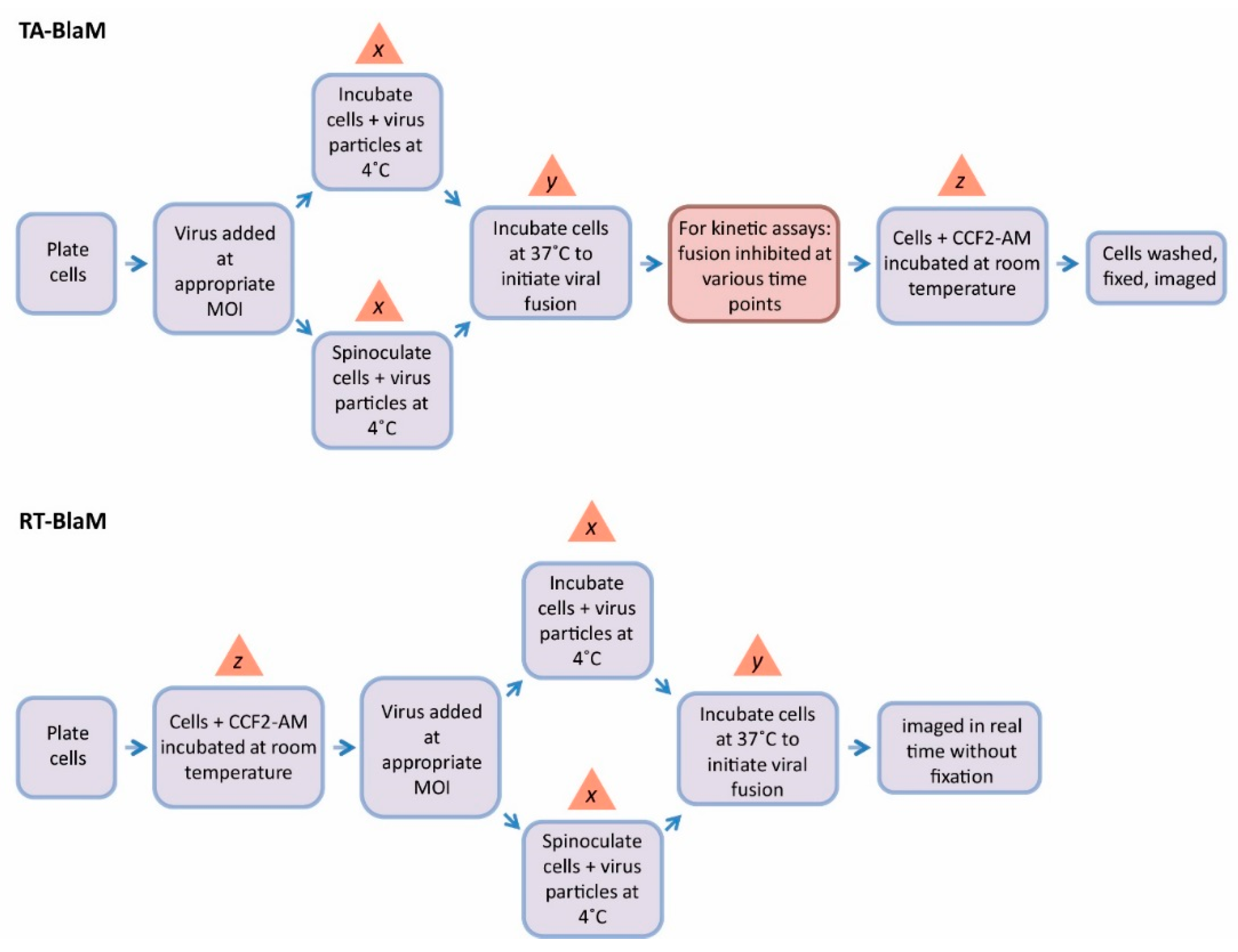
- The ratio of infectious virus particles to cells is referred to as the multiplicity of infection (MOI).
- At 4 °C, virus particles are able to bind to target cells but are not able to enter them [41]. A 4 °C incubation step is therefore often used to prime virions on the surface of the cell, meaning any unbound particles can be washed away.
- Viral fusion is initiated by a temperature shift from 4 °C to 37 °C [42].
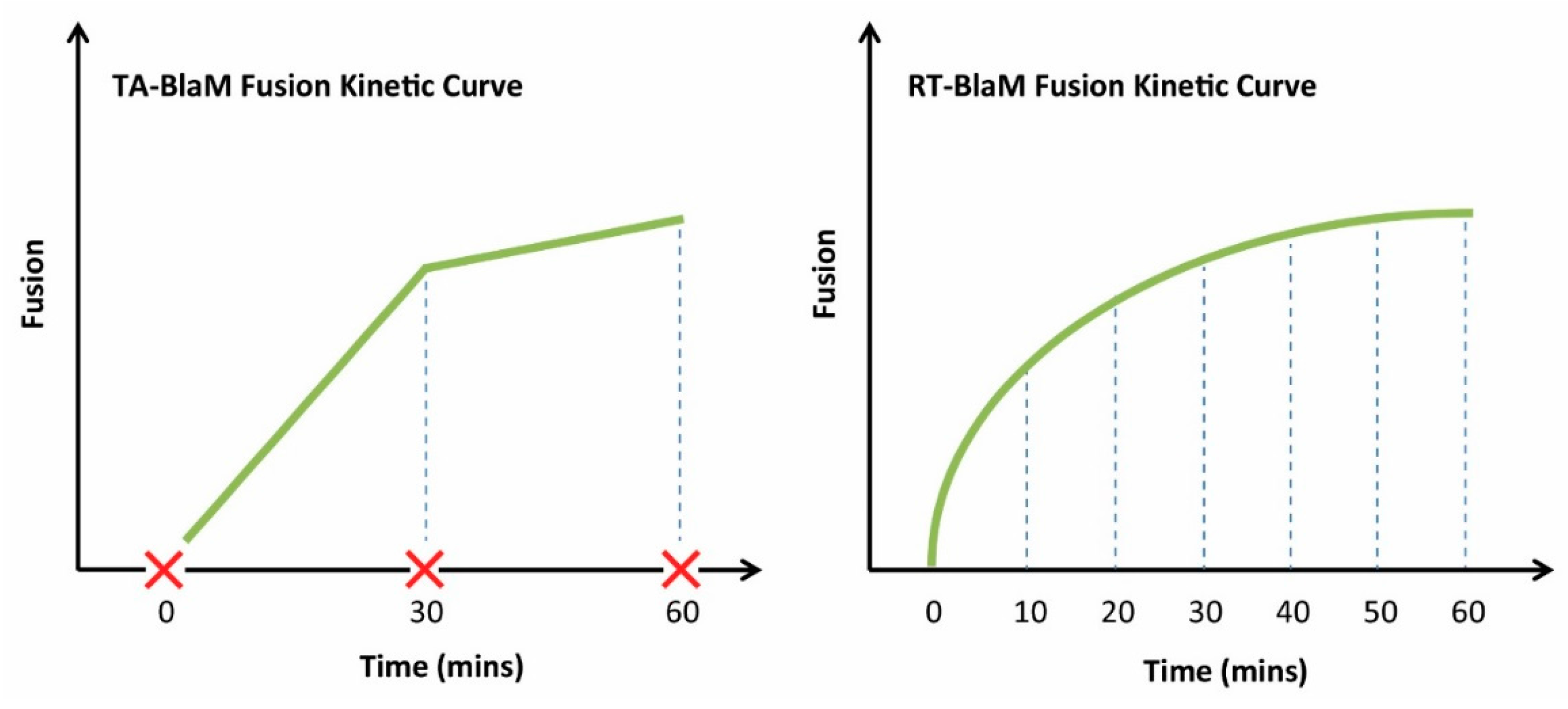
4. BlaM Assay Intricacies and Discrepancies
5. BlaM and Other Methods of Viral Fusion Analysis
6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ASLV | Avian sarcoma and leukosis virus |
| FRET | Förster resonance energy transfer |
| EBOV | Ebola virus |
| EBV | Epstein-Barr virus |
| GFP | Green fluorescent protein |
| HeV | Hendra virus |
| HIV-1 | Human immunodeficiency virus-1 |
| HTS | High-throughput screening |
| IAV | Influenza A virus |
| IFITM | Interferon-induced transmembrane protein |
| LASV | Lassa virus |
| MARV | Marburg virus |
| MOI | Multiplicity of infection |
| NiV | Nipah virus |
| QD | Quantum dot |
| RT-BlaM | Real-time BlaM assay |
| TA-BlaM | Time-of-addition BlaM assay |
| VLP | Virus-like particle |
| Vpr | Viral protein R |
| VSV | Vesicular stomatitis virus |
References
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. Rev. Infect. Dis. 1988, 10, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Zlokarnik, G.; Negulescu, P.A.; Knapp, T.E.; Mere, L.; Burres, N.; Feng, L.; Whitney, M.; Roemer, K.; Tsien, R.Y. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science 1998, 279, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Krausslich, H.G. The molecular architecture of hiv. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary t lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Tessmer, U.; Schubert, U.; Krausslich, H.G. Human immunodeficiency virus type 1 vpr protein is incorporated into the virion in significantly smaller amounts than gag and is phosphorylated in infected cells. J. Virol. 2000, 74, 9727–9731. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Tungaturthi, P.; Cartas, M.; Tomkowicz, B.; Rizvi, T.A.; Khan, S.A.; Kalyanaraman, V.S.; Srinivasan, A. Virion-associated hiv-1 vpr: Variable amount in virus particles derived from cells upon virus infection or proviral DNA transfection. Virology 2001, 283, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.A.; Dehni, G.; Sodroski, J.G.; Haseltine, W.A. Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J. Virol. 1990, 64, 3097–3099. [Google Scholar] [PubMed]
- Selig, L.; Pages, J.C.; Tanchou, V.; Preveral, S.; Berlioz-Torrent, C.; Liu, L.X.; Erdtmann, L.; Darlix, J.; Benarous, R.; Benichou, S. Interaction with the p6 domain of the gag precursor mediates incorporation into virions of vpr and vpx proteins from primate lentiviruses. J. Virol. 1999, 73, 592–600. [Google Scholar] [PubMed]
- Jenkins, Y.; Pornillos, O.; Rich, R.L.; Myszka, D.G.; Sundquist, W.I.; Malim, M.H. Biochemical analyses of the interactions between human immunodeficiency virus type 1 vpr and p6(gag). J. Virol. 2001, 75, 10537–10542. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Mammano, F.; Cohen, E.A.; Gottlinger, H.G. The p6gag domain of human immunodeficiency virus type 1 is sufficient for the incorporation of vpr into heterologous viral particles. J. Virol. 1995, 69, 2759–2764. [Google Scholar] [PubMed]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. Ifitm3 restricts influenza a virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [PubMed]
- Landowski, M.; Dabundo, J.; Liu, Q.; Nicola, A.V.; Aguilar, H.C. Nipah virion entry kinetics, composition, and conformational changes determined by enzymatic virus-like particles and new flow virometry tools. J. Virol. 2014, 88, 14197–14206. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.C.; Wang, Y.; Freiberg, A.N.; Aguilar, H.C.; Holbrook, M.R.; Lee, B. A catalytically and genetically optimized beta-lactamase-matrix based assay for sensitive, specific, and higher throughput analysis of native henipavirus entry characteristics. J. Virol. 2009, 6, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.K.; Latinovic, O.; Martin, E.; Novitskiy, G.; Marin, M.; Miyauchi, K.; Naughton, J.; Young, J.A.; Melikyan, G.B. Imaging single retrovirus entry through alternative receptor isoforms and intermediates of virus-endosome fusion. PLoS Pathog. 2011, 7, e1001260. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Marin, M.; Kondo, N.; Melikyan, G.B. Synchronized retrovirus fusion in cells expressing alternative receptor isoforms releases the viral core into distinct sub-cellular compartments. PLoS Pathog. 2012, 8, e1002694. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.J.; Narayan, S.; Dornadula, G.; Miller, M.D.; Young, J.A. Low ph is required for avian sarcoma and leukosis virus env-dependent viral penetration into the cytosol and not for viral uncoating. J. Virol. 2004, 78, 10433–10441. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; Neidleman, J.; Yonemoto, W.; Fenard, D.; Greene, W.C. HIV-1 virion fusion assay: Uncoating not required and no effect of nef on fusion. Virology 2004, 328, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Tobiume, M.; Lineberger, J.E.; Lundquist, C.A.; Miller, M.D.; Aiken, C. Nef does not affect the efficiency of human immunodeficiency virus type 1 fusion with target cells. J. Virol. 2003, 77, 10645–10650. [Google Scholar] [CrossRef] [PubMed]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Krausslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Demirkhanyan, L.H.; Marin, M.; Padilla-Parra, S.; Zhan, C.; Miyauchi, K.; Jean-Baptiste, M.; Novitskiy, G.; Lu, W.; Melikyan, G.B. Multifaceted mechanisms of hiv-1 entry inhibition by human alpha-defensin. J. Biol. Chem. 2012, 287, 28821–28838. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.; Du, Y.; Giroud, C.; Kim, J.H.; Qui, M.; Fu, H.; Melikyan, G.B. High-throughput hiv-cell fusion assay for discovery of virus entry inhibitors. Assay. Drug Dev. Technol. 2015, 13, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Cavrois, M.; Greene, W.C. Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: Involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J. Virol. 2005, 79, 918–926. [Google Scholar] [CrossRef] [PubMed]
- de la Vega, M.; Marin, M.; Kondo, N.; Miyauchi, K.; Kim, Y.; Epand, R.F.; Epand, R.M.; Melikyan, G.B. Inhibition of hiv-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology 2011, 8, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Sims, J.S.; John, S.P.; Chin, C.R.; Pertel, T.; Chen, L.M.; Gaiha, G.D.; Ryan, B.J.; Donis, R.O.; Elledge, S.J. Ifitm3 inhibits influenza a virus infection by preventing cytosolic entry. PLoS Pathog. 2011, 7, e1002337. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Manicassamy, B.; Garcia-Sastre, A. An enzymatic virus-like particle assay for sensitive detection of virus entry. J. Virol. Methods 2010, 163, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Dale, B.M.; McNerney, G.P.; Thompson, D.L.; Hubner, W.; de Los Reyes, K.; Chuang, F.Y.; Huser, T.; Chen, B.K. Cell-to-cell transfer of hiv-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 2011, 10, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. Ifitm proteins incorporated into hiv-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.; Soros, V.B.; Greene, W.C. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human cd4 t lymphocytes. J. Virol. 2004, 78, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. Hiv enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Putcharoen, O.; Lee, S.H.; Henrich, T.J.; Hu, Z.; Vanichanan, J.; Coakley, E.; Greaves, W.; Gulick, R.M.; Kuritzkes, D.R.; Tsibris, A.M. Hiv-1 clinical isolates resistant to ccr5 antagonists exhibit delayed entry kinetics that are corrected in the presence of drug. J. Virol. 2012, 86, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kozlov, M.M.; Melikyan, G.B. Early steps of hiv-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 2009, 5, e1000585. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Le Duff, Y.; Wang, Y.; Pan, Q.; Ding, S.; Zheng, Y.M.; Liu, S.L.; Liang, C. Primate lentiviruses are differentially inhibited by interferon-induced transmembrane proteins. Virology 2015, 474, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, K.; Mueller, S.; Sticht, H.; Jung, S.; Zou, P.; Jiang, S.; Gross, A.; Eichler, J.; Fleckenstein, B.; Reil, H. HIV-1 fusion is blocked through binding of GB virus C E2-D peptides to the HIV-1 gp41 disulfide loop. PLoS ONE 2013, 8, e54452. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Padilla-Parra, S. Imaging real-time HIV-1 virion fusion with FRET-based biosensors. Sci. Rep. [CrossRef] [PubMed]
- Gheysen, D.; Jacobs, E.; de Foresta, F.; Thiriart, C.; Francotte, M.; Thines, D.; De Wilde, M. Assembly and release of HIV-1 precursor Pr55 gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 1989, 59, 103–112. [Google Scholar] [CrossRef]
- Shioda, T.; Shibuta, H. Production of human immunodeficiency virus (HIV)-like particles from cells infected with recombinant vaccinia viruses carrying the gag gene of HIV. Virology 1990, 175, 139–148. [Google Scholar] [PubMed]
- Koedel, Y.; Eissmann, K.; Wend, H.; Fleckenstein, B.; Reil, H. Peptides derived from a distinct region of gb virus c glycoprotein e2 mediate strain-specific HIV-1 entry inhibition. J. Virol. 2011, 85, 7037–7047. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Leser, G.P.; Morita, E.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007, 81, 7111–7123. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, J.; Sun, W.; Martinez-Romero, C.; Tawa, G.; Shinn, P.; Chen, C.Z.; Schimmer, A.; Sanderson, P.; McKew, J.C.; Zheng, W.; et al. Identification of 53 compounds that block ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 2014, 3, e84. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, S.R.; Markosyan, R.M.; Eadon, M.T.; Moore, J.P.; Melikyan, G.B.; Cohen, F.S. Ternary complex formation of human immunodeficiency virus type 1 env, cd4, and chemokine receptor captured as an intermediate of membrane fusion. J. Virol. 2005, 79, 11161–11169. [Google Scholar] [CrossRef] [PubMed]
- Herold, N.; Anders-Osswein, M.; Glass, B.; Eckhardt, M.; Muller, B.; Krausslich, H.G. HIV-1 entry in supt1-r5, cem-ss, and primary cd4+ t cells occurs at the plasma membrane and does not require endocytosis. J. Virol. 2014, 88, 13956–13970. [Google Scholar] [CrossRef] [PubMed]
- O'Doherty, U.; Swiggard, W.J.; Malim, M.H. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 2000, 74, 10074–10080. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.B.; Misra, V.; Mosmann, T.R. Cytomegalovirus infectivity: Analysis of the phenomenon of centrifugal enhancement of infectivity. Virology 1976, 72, 235–243. [Google Scholar] [CrossRef]
- Ye, L.; Wang, X.; Wang, S.; Luo, G.; Wang, Y.; Liang, H.; Ho, W. Centrifugal enhancement of hepatitis c virus infection of human hepatocytes. J. Virol. Methods 2008, 148, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, C.M.; Rawat, S.S.; Puri, A.; Wang, J.M.; Ruscetti, F.W.; Blumenthal, R. Ceramide, a target for antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2004, 101, 15452–15457. [Google Scholar] [CrossRef] [PubMed]
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced cd4-cxcr4 interaction, f-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, M.; Lopez-Huertas, M.R.; Garcia-Perez, J.; Climent, N.; Descours, B.; Ambrosioni, J.; Mateos, E.; Rodriguez-Mora, S.; Rus-Bercial, L.; Benkirane, M.; et al. Dasatinib inhibits hiv-1 replication through the interference of samhd1 phosphorylation in cd4+ t cells. Biochem. Pharmacol. 2016, 106, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Yao, H.; Ren, H.; Subbian, S.; Cirillo, S.L.; Sacchettini, J.C.; Rao, J.; Cirillo, J.D. Imaging tuberculosis with endogenous beta-lactamase reporter enzyme fluorescence in live mice. Proc. Natl. Acad. Sci. USA 2010, 107, 12239–12244. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xing, B.; Rao, J. A self-assembled quantum dot probe for detecting beta-lactamase activity. Biochem. Biophys. Res. Commun. 2006, 344, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, B.; Zhuang, X. Virus trafficking-learning from single-virus tracking. Nat. Rev. Microbiol. 2007, 5, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Ewers, H.; Smith, A.E.; Sbalzarini, I.F.; Lilie, H.; Koumoutsakos, P.; Helenius, A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 15110–15115. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Marin, M.; Gahlaut, N.; Suter, R.; Kondo, N.; Melikyan, G.B. Fusion of mature hiv-1 particles leads to complete release of a gag-gfp-based content marker and raises the intraviral pH. PLoS ONE 2013, 8, e71002. [Google Scholar]
- Xie, D. Fluorescent dye labeled influenza virus mainly infects innate immune cells and activated lymphocytes and can be used in cell-mediated immune response assay. J. Immunol. Methods 2009, 343, 42–48. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sun, E.; Bujny, M.V.; Kim, D.; Davidson, M.W.; Zhuang, X. Dual function of cd81 in influenza virus uncoating and budding. PLoS Pathog. 2013, 9, e1003701. [Google Scholar] [CrossRef] [PubMed]
- van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Bennett, M.C.; Bercot, T.; Singh, I.R. Functional analysis of hepatitis c virus envelope proteins, using a cell-cell fusion assay. J. Virol. 2006, 80, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Hofmann, J.; Uberla, K.; Liebert, U.G. Rubella virus pseudotypes and a cell-cell fusion assay as tools for functional analysis of the rubella virus e2 and e1 envelope glycoproteins. J. Gen. Virol. 2006, 87, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Huerta, L.; Lamoyi, E.; Baez-Saldana, A.; Larralde, C. Human immunodeficiency virus envelope-dependent cell-cell fusion: A quantitative fluorescence cytometric assay. Cytometry 2002, 47, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Markosyan, R.M.; Miao, C.; Zheng, Y.M.; Melikyan, G.B.; Liu, S.L.; Cohen, F.S. Induction of cell-cell fusion by ebola virus glycoprotein: Low pH is not a trigger. PLoS Pathog. 2016, 12, e1005373. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Takada, A.; Kawaoka, Y.; Alizon, M. Detection of cell-cell fusion mediated by ebola virus glycoproteins. J. Virol. 2006, 80, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- McShane, M.P.; Longnecker, R. Cell-surface expression of a mutated epstein-barr virus glycoprotein b allows fusion independent of other viral proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 17474–17479. [Google Scholar] [CrossRef] [PubMed]
- McShane, M.P.; Longnecker, R. Analysis of fusion using a virus-free cell fusion assay. Methods Mol. Biol. 2005, 292, 187–196. [Google Scholar] [PubMed]
- Guo, J.; Wang, W.; Yu, D.; Wu, Y. Spinoculation triggers dynamic actin and cofilin activity that facilitates HIV-1 infection of transformed and resting cd4 t cells. J. Virol. 2011, 85, 9824–9833. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Literature Example |
|---|---|
| A549 | [11] |
| CD4+ T cells * | [4,5,6,7] |
| CEMss | [6,7,8] |
| CHO | [2,11,12,13] |
| COS-7 | [2] |
| CV-1 | [2,11,14,15] |
| HEK 293T | [2,16] |
| HeLa | [2,4,17,18,19,20,21,22] |
| HMVEC* | [13] |
| Jurkat | [2,4,23] |
| MDCK | [11,24,25] |
| MT-4 | [18,26] |
| PBMC * | [4,16,22,23,24] |
| PM-1 | [23] |
| SupT1 T cells | [4,17,27,28,29] |
| TZM-bl | [15,20,21,23,30,31,32,33,34,35] |
| U87 CD4 + CCR5 + | [23,31] |
| Main Feature | Literature Example | Advantages | Disadvantages | |
|---|---|---|---|---|
| TA-BlaM | Virus fusion precedes CCF2-AM loading | [4,11,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,32,33,34,35,38,47,48] | Best suited for fusion endpoint assays. Kinetic assays still possible Extensively validated Compatible with flow cytometry, microscopy and plate readers | One sample per time point required for kinetic analyses: greater reagent usage Kinetic measurements require the use of fusion inhibitors |
| RT-BlaM | CCF2-AM loading precedes virus fusion | [12,31] | Best suited to fusion kinetic assays Kinetic analyses can be conducted on a single sample Use of fusion inhibitors is optional Easier to acquire more measurements: more refined curves can be generated | Data collection platform should ideally support live cell analyses (37 °C, 5% CO2) Probenecid is likely required to limit CCF2-AM loss during 37 °C virus fusion step Data must be acquired in real time: No room for error |
| Advantages | Disadvantages | |
|---|---|---|
| BlaM Assay | Data acquirement not limited to microscopy—plate readers and flow cytometers work efficiently Flexible protocols can be applied | Does not provide single virus precision Fusion detection may be slightly delayed |
| SVT | Fusion measured with single virus precision Instantaneous observation of fusion event | Extremely Time consuming and cumbersome Requires high-end microscopy equipment and SVT expertise Multiple events required to draw firm conclusions |
| Cell-cell/Virus-free Assay | Virus production and characterisation not required Relatively straightforward protocol compared to BlaM and SVT | Cells unlikely to accurately mimic virus particles—caution with data interpretation required |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, D.M.; Padilla-Parra, S. The ?-Lactamase Assay: Harnessing a FRET Biosensor to Analyse Viral Fusion Mechanisms. Sensors 2016, 16, 950. https://doi.org/10.3390/s16070950
Jones DM, Padilla-Parra S. The ?-Lactamase Assay: Harnessing a FRET Biosensor to Analyse Viral Fusion Mechanisms. Sensors. 2016; 16(7):950. https://doi.org/10.3390/s16070950
Chicago/Turabian StyleJones, Daniel M., and Sergi Padilla-Parra. 2016. "The ?-Lactamase Assay: Harnessing a FRET Biosensor to Analyse Viral Fusion Mechanisms" Sensors 16, no. 7: 950. https://doi.org/10.3390/s16070950
APA StyleJones, D. M., & Padilla-Parra, S. (2016). The ?-Lactamase Assay: Harnessing a FRET Biosensor to Analyse Viral Fusion Mechanisms. Sensors, 16(7), 950. https://doi.org/10.3390/s16070950