Q3: A Compact Device for Quick, High Precision qPCR

 ,
,  , and
, and
Abstract
:1. Introduction
2. Q3: System Description
2.1. Non-Disposable Unit (Instrument)
2.2. Disposable Unit (LoC Cartridge)
2.3. Thermal Control
2.4. Optical Read-Out
2.5. Software
3. Materials and Methods
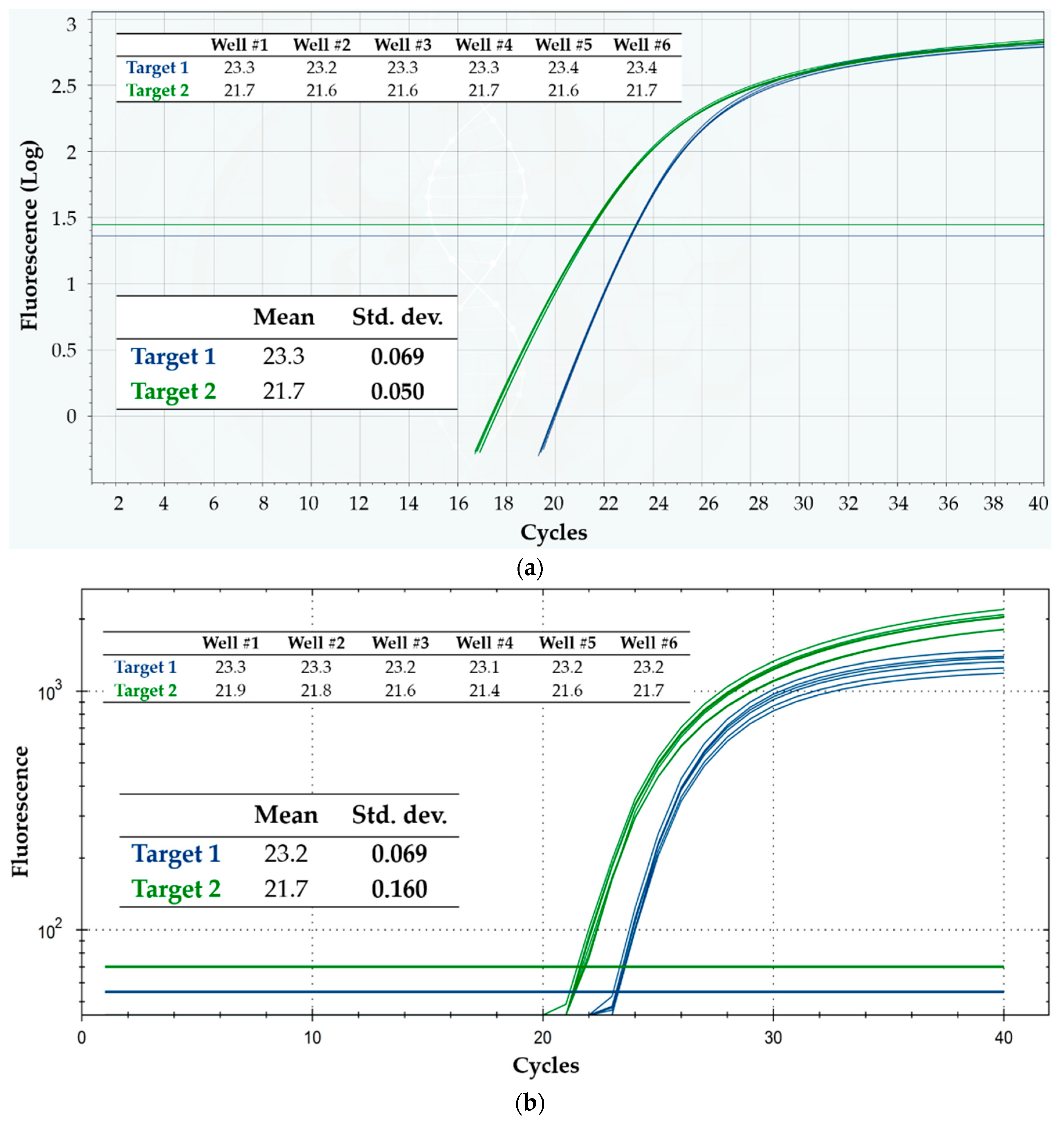
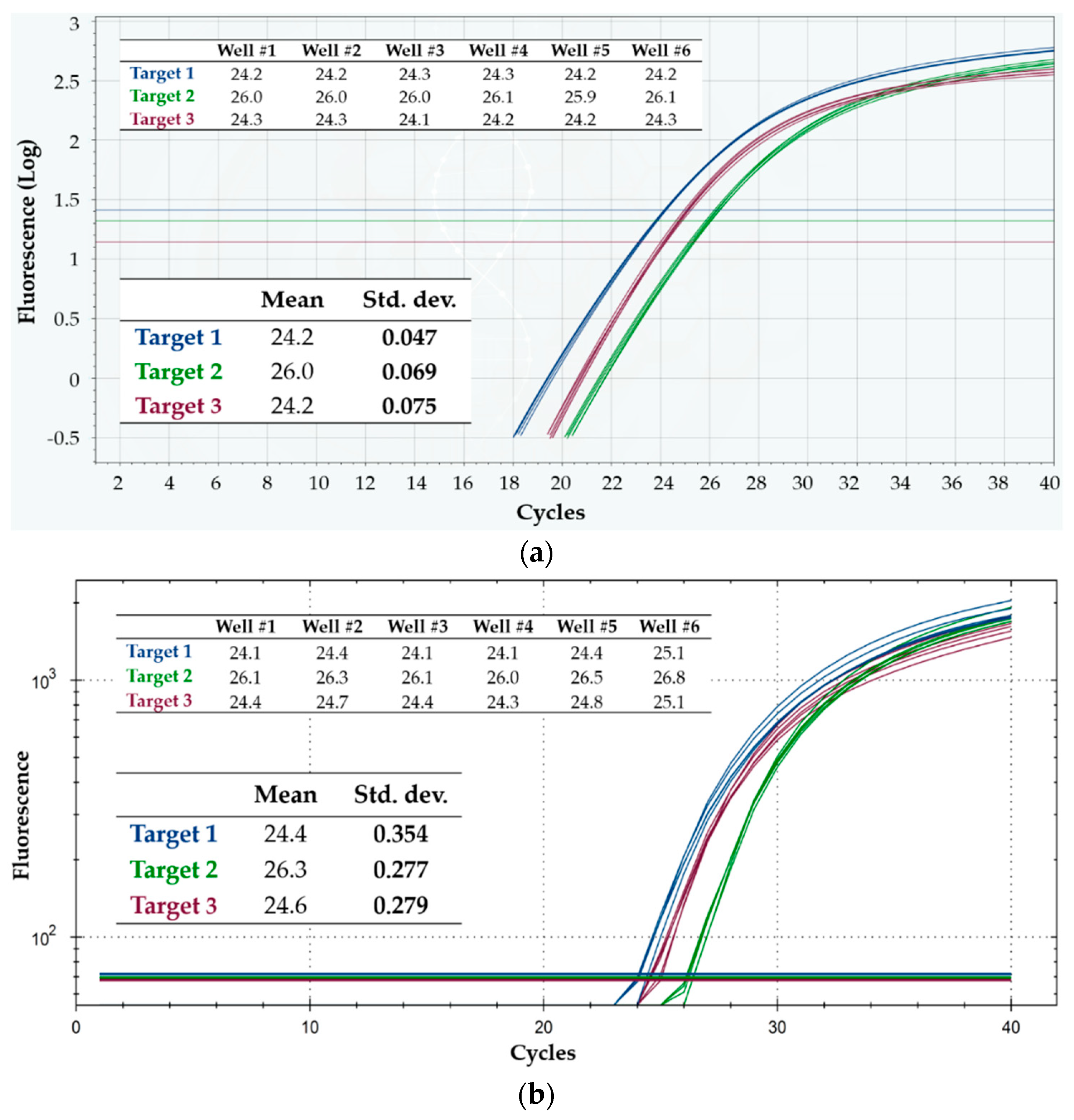
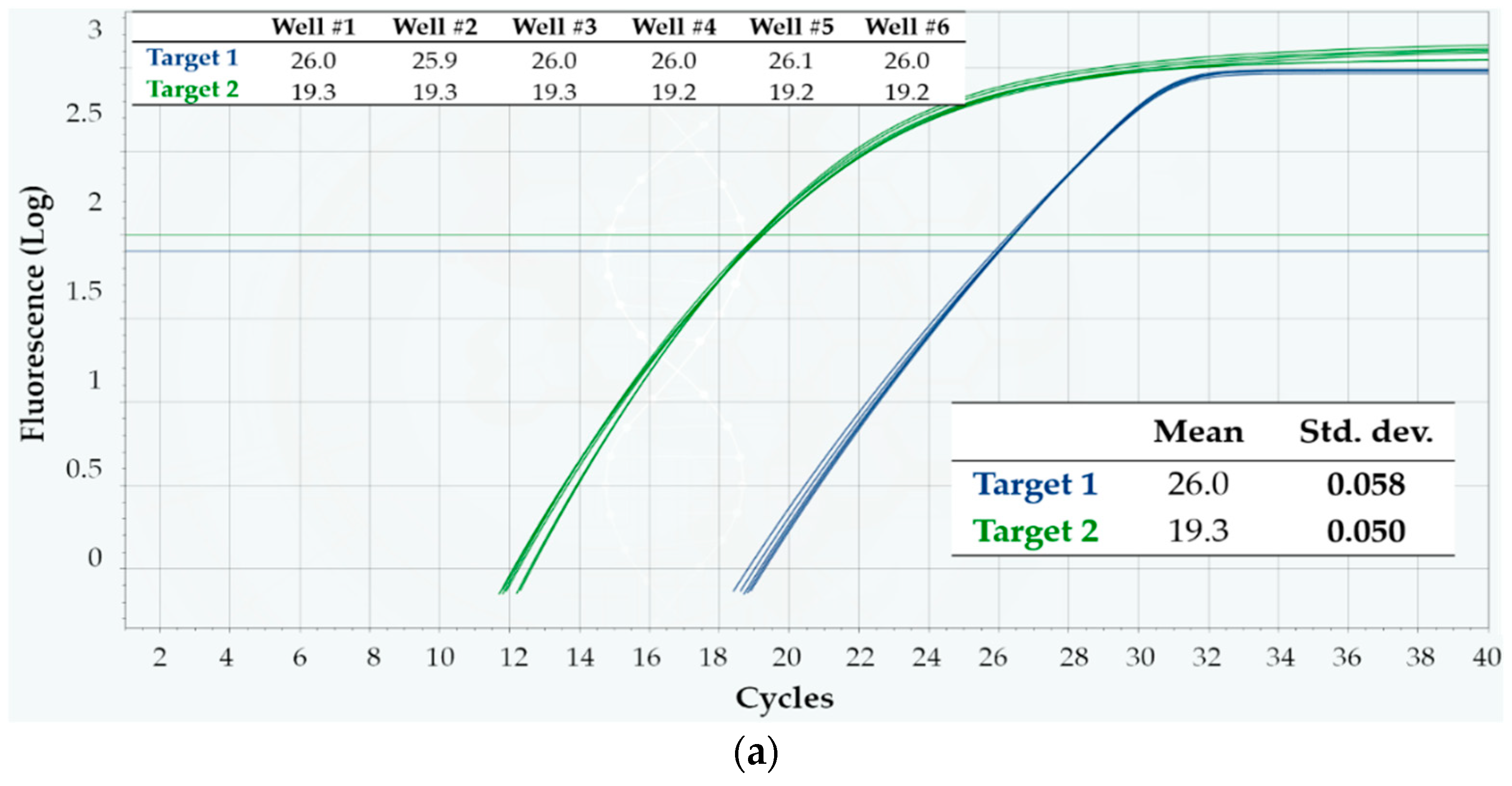
3.1. Intra-Run Repeatability and Inter-Run Reproducibility: Comparison with a Gold Standard
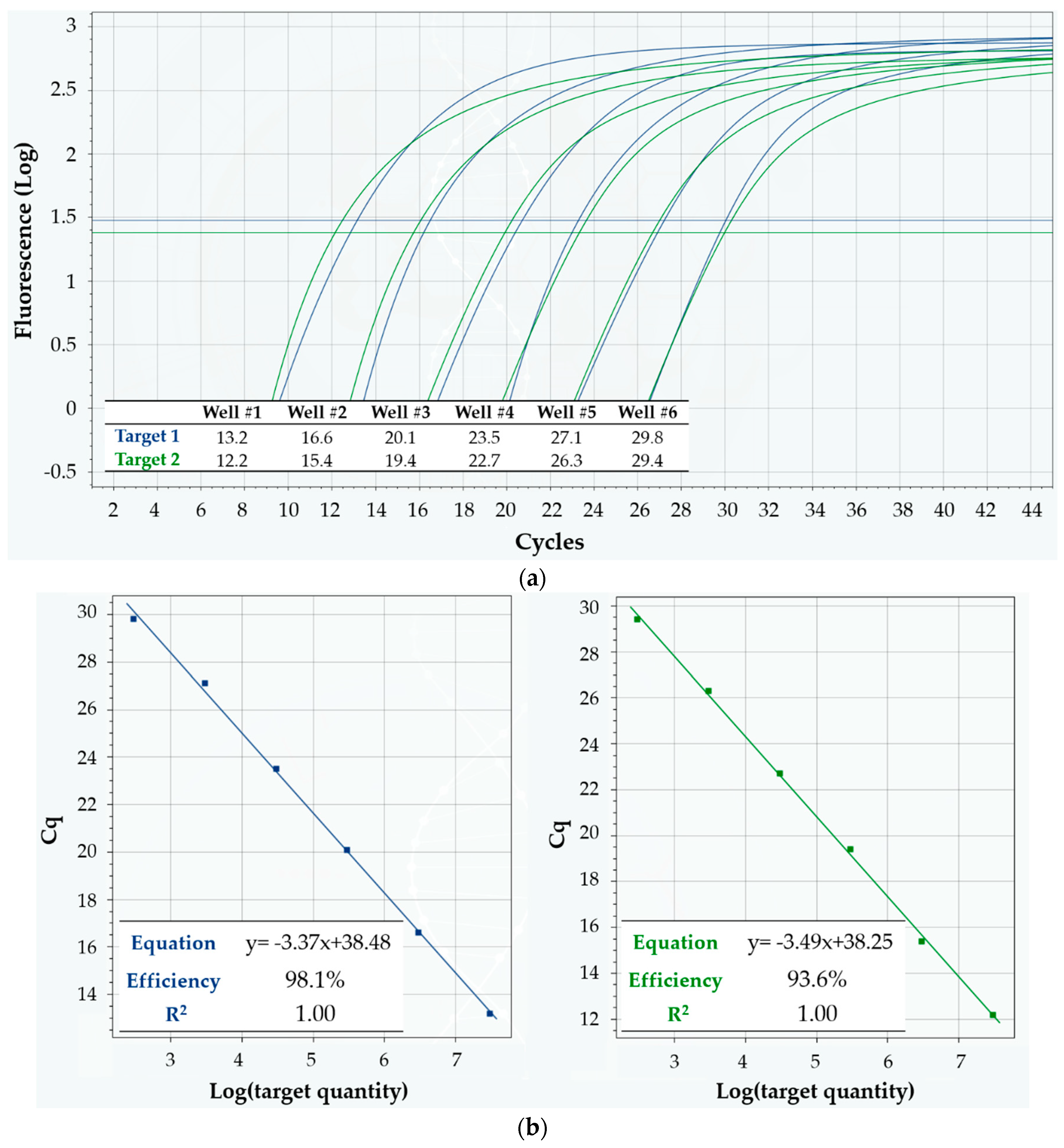
3.2. Linearity and Sensitivity
4. Results
4.1. Intra-Run Repeatability and Inter-Run Reproducibility: Comparison with a Gold Standard
4.2. Linearity and Sensitivity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Ehrlich, K. Specific Enzymatic Amplification of DNA in Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Ishmael, F.T.; Stellato, C. Principles and Applications of Polymerase Chain Reaction: Basic Science for the Practicing Physician. Ann. Allergy Asthma Immunol. 2008, 101, 437–443. [Google Scholar] [CrossRef]
- Valasek, M.A.; Repa, J.J. The Power of Real-Time PCR. Adv. Physiol. Educ. 2005, 29, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Vajpayee, M.; Mohan, T. Current Practices in Laboratory Monitoring of HIV Infection. Indian J. Med. Res. 2011, 134, 801–822. [Google Scholar] [CrossRef] [PubMed]
- Salihah, N.T.; Hossain, M.M.; Lubis, H.; Ahmed, M.U. Trends and Advances in Food Analysis by Real-Time Polymerase Chain Reaction. J. Food Sci. Technol. 2016, 53, 2196–2209. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.U.; de Mello, J.; Manz, A. Chemical Amplification: Continuous-Flow PCR on a Chip. Science 1998, 280, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Ahrberg, C.D.; Manz, A.; Chung, B.G. Polymerase Chain Reaction in Microfluidic Devices. Lab Chip 2016, 16, 3866–3884. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Chen, P.J.; Lee, G.B. The Evolution of Real-Time PCR Machines to Real-Time PCR Chips. Biosens. Bioelectron. 2010, 25, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Consolandi, C.; Severgnini, M.; Frosini, A.; Caramenti, G.; De Fazio, M.; Ferrara, F.; Zocco, A.; Fischetti, A.; Palmieri, M.; De Bellis, G. Polymerase Chain Reaction of 2-kb Cyanobacterial Gene and Human Anti-Alpha1-Chymotrypsin Gene from Genomic DNA on the In-Check Single-Use Microfabricated Silicon Chip. Anal. Biochem. 2006, 353, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [PubMed]
- Marziliano, N.; Notarangelo, M.F.; Cereda, M.; Caporale, V.; Coppini, L.; Demola, M.A.; Guidorossi, A.; Crocamo, A.; Pigazzani, F.; Boffetti, F.; et al. Rapid and Portable, Lab-On-Chip, Point-Of-Care Genotyping for Evaluating Clopidogrel Metabolism. Clin. Chim. Acta 2015, 451, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, F.M.; Maglietta, G.; Bevilacqua, P.; Cereda, M.; Merlini, P.A.; Villani, G.Q.; Moruzzi, P.; Patrizi, G.; Tagliazucchi, G.M.; Crocamo, A.; et al. Pharmacogenomic Approach to Selecting Antiplatelet Therapy in Acute Coronary Syndromes: PHARMCLO trial. J. Am. Coll. Cardiol. 2018, 71, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Biava, M.; Colavita, F.; Marzorati, A.; Russo, D.; Pirola, D.; Cocci, A.; Petrocelli, A.; Delli Guanti, M.; Cataldi, G.; Kamara, T.A.; et al. Evaluation of a Rapid and Sensitive RT-qPCR Assay for the Detection of Ebola Virus. J. Virol. Methods 2018, 252, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Dube, S.; Mir, A.; Qin, J.; Sun, G.; Ramakrishnan, R.; Jones, R.C.; Livak, K.J. Taking qPCR to a Higher Level: Analysis of CNV Reveals the Power of High Throughput qPCR to Enhance Quantitative Resolution. Methods 2010, 50, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TaqMan® Fast Advanced Master Mix on the Bio-Rad CFX96™ Real-Time PCR System. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/Specification-Sheets/cms_087006.pdf (accessed on 27 July 2018).
- TaqMan® Fast Advanced Master MixUSER GUIDE. Available online: https://tools.thermofisher.com/content/sfs/manuals/cms_084554.pdf (accessed on 27 July 2018).
- Broeders, S.; Huber, I.; Grohmann, L.; Berben, G.; Taverniers, I.; Mazzara, M.; Roosens, N.; Morisset, D. Guidelines for Validation of Qualitative Real-Time PCR Methods. Trends Food Sci. Technol. 2014, 37, 115–126. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intra-Run | Inter-Run | ||||
|---|---|---|---|---|---|
| Mean | Std. Dev. | Mean Std. Dev. | Mean | Std. Dev. | |
| Q3, Target 1 | 23.2 | 0.090 | 0.074 | 23.2 | 0.094 |
| 23.3 | 0.069 | ||||
| 23.2 | 0.069 | ||||
| 23.2 | 0.069 | ||||
| CFX96, Target 1 | 23.2 | 0.069 | 0.065 | 23.4 | 0.163 |
| 23.6 | 0.047 | ||||
| 23.4 | 0.075 | ||||
| 23.4 | 0.069 | ||||
| Q3, Target 2 | 21.6 | 0.047 | 0.060 | 21.7 | 0.087 |
| 21.7 | 0.050 | ||||
| 21.8 | 0.069 | ||||
| 21.7 | 0.075 | ||||
| CFX96, Target 2 | 21.7 | 0.160 | 0.144 | 22.2 | 0.346 |
| 22.5 | 0.134 | ||||
| 22.1 | 0.149 | ||||
| 22.3 | 0.134 | ||||
| Intra-Run | Inter-Run | ||||
|---|---|---|---|---|---|
| Mean | Std. Dev. | Mean Std. Dev. | Mean | Std. Dev. | |
| Q3, Target 1 | 24.1 | 0.075 | 0.069 | 24.1 | 0.143 |
| 24.2 | 0.047 | ||||
| 24.0 | 0.080 | ||||
| 23.9 | 0.075 | ||||
| CFX96, Target 1 | 24.4 | 0.354 | 0.322 | 24.5 | 0.369 |
| 24.3 | 0.315 | ||||
| 24.7 | 0.309 | ||||
| 24.5 | 0.309 | ||||
| Q3, Target 2 | 25.9 | 0.047 | 0.073 | 26.0 | 0.096 |
| 26.0 | 0.069 | ||||
| 26.0 | 0.075 | ||||
| 26.1 | 0.100 | ||||
| CFX96, Target 2 | 26.3 | 0.277 | 0.260 | 26.5 | 0.300 |
| 26.4 | 0.267 | ||||
| 26.7 | 0.219 | ||||
| 26.6 | 0.275 | ||||
| Q3, Target 3 | 24.2 | 0.111 | 0.109 | 24.2 | 0.127 |
| 24.2 | 0.075 | ||||
| 24.1 | 0.115 | ||||
| 24.1 | 0.137 | ||||
| CFX96, Target 3 | 24.6 | 0.279 | 0.315 | 24.7 | 0.324 |
| 24.8 | 0.350 | ||||
| 24.5 | 0.345 | ||||
| 24.6 | 0.292 | ||||
| Intra-Run | Inter-Run | ||||
|---|---|---|---|---|---|
| Mean | Std. Dev. | Mean Std. Dev. | Mean | Std. Dev. | |
| Q3, Target 1 | 26.0 | 0.058 | 0.063 | 26.0 | 0.076 |
| 26.0 | 0.075 | ||||
| 26.1 | 0.058 | ||||
| CFX96, Target 1 | 26.6 | 0.273 | 0.158 | 26.5 | 0.207 |
| 26.5 | 0.126 | ||||
| 26.4 | 0.075 | ||||
| Q3, Target 2 | 19.3 | 0.050 | 0.069 | 19.2 | 0.073 |
| 19.2 | 0.090 | ||||
| 19.2 | 0.069 | ||||
| CFX96, Target 2 | 19.4 | 0.223 | 0.192 | 19.3 | 0.217 |
| 19.3 | 0.149 | ||||
| 19.1 | 0.205 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cereda, M.; Cocci, A.; Cucchi, D.; Raia, L.; Pirola, D.; Bruno, L.; Ferrari, P.; Pavanati, V.; Calisti, G.; Ferrara, F.; et al. Q3: A Compact Device for Quick, High Precision qPCR. Sensors 2018, 18, 2583. https://doi.org/10.3390/s18082583
Cereda M, Cocci A, Cucchi D, Raia L, Pirola D, Bruno L, Ferrari P, Pavanati V, Calisti G, Ferrara F, et al. Q3: A Compact Device for Quick, High Precision qPCR. Sensors. 2018; 18(8):2583. https://doi.org/10.3390/s18082583
Chicago/Turabian StyleCereda, Marco, Alessandro Cocci, Davide Cucchi, Lillo Raia, Danilo Pirola, Lorenzo Bruno, Pietro Ferrari, Valentina Pavanati, Giorgia Calisti, Francesco Ferrara, and et al. 2018. "Q3: A Compact Device for Quick, High Precision qPCR" Sensors 18, no. 8: 2583. https://doi.org/10.3390/s18082583