
Trends in Single-Molecule Total Internal Reflection Fluorescence Imaging and Their Biological Applications with Lab-on-a-Chip Technology

 ,
,
Abstract
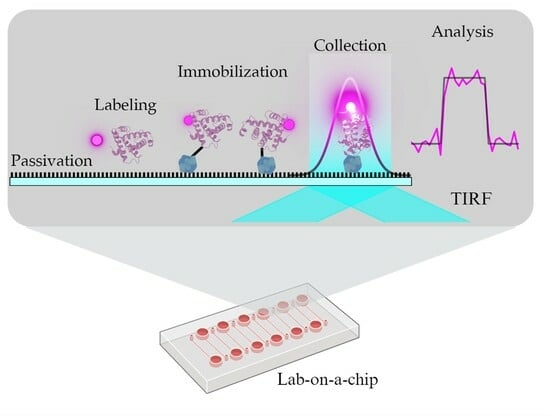
:
1. Introduction
2. Optical Systems for Single-Molecule Fluorescence Imaging
3. Sample Preparation
3.1. Surface Passivation
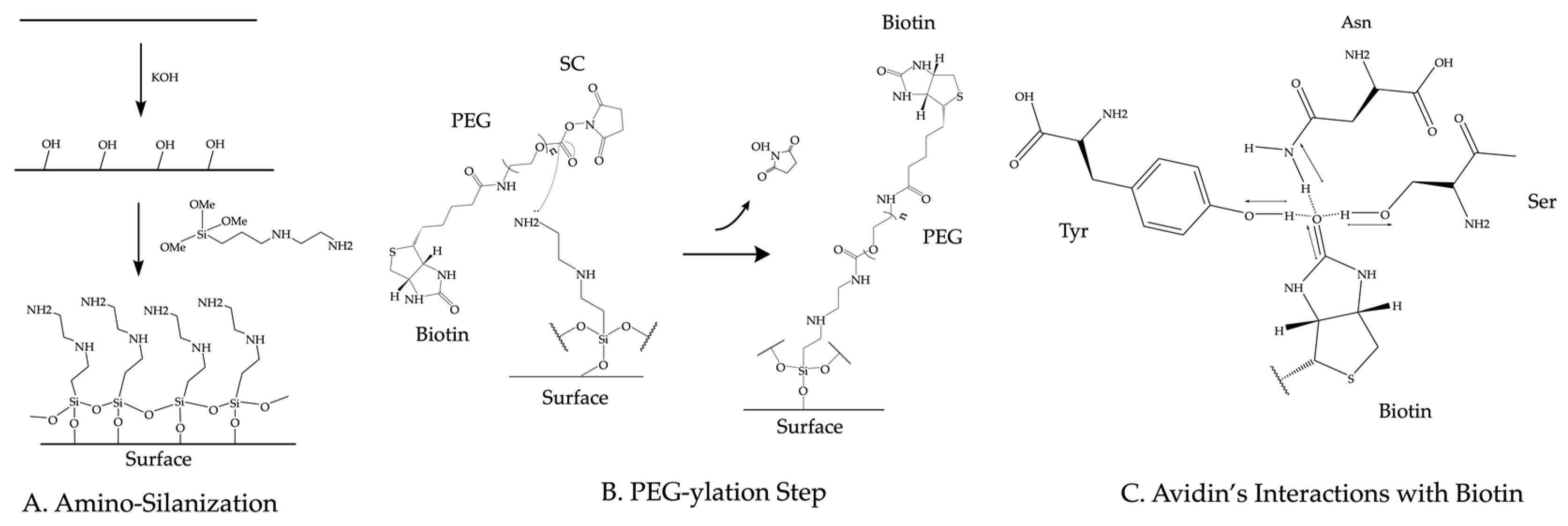
3.2. Surface Functionalization
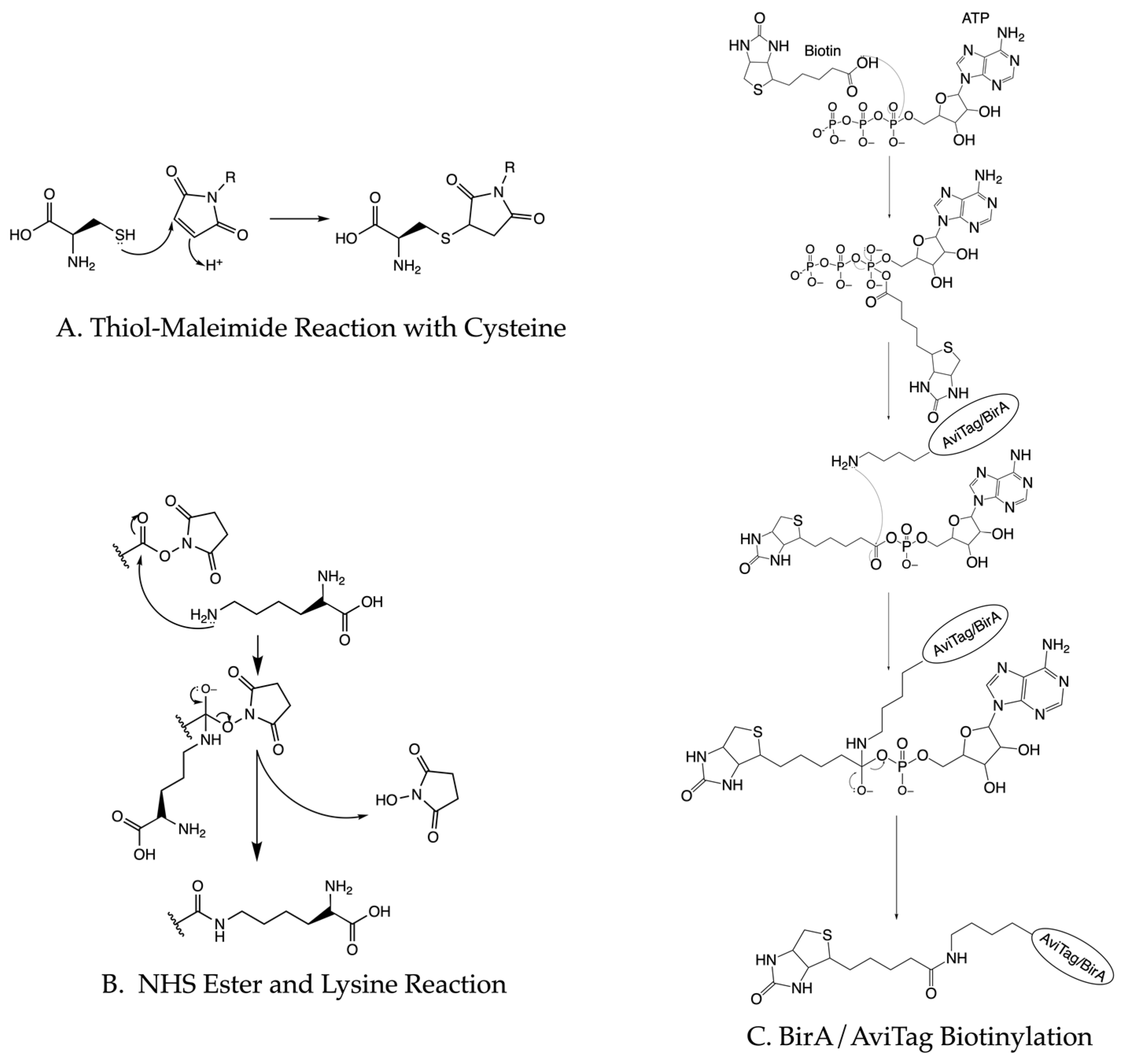
3.3. Protein Biotinylation
3.4. Protein Fluorescence Labeling
4. Analysis Methods
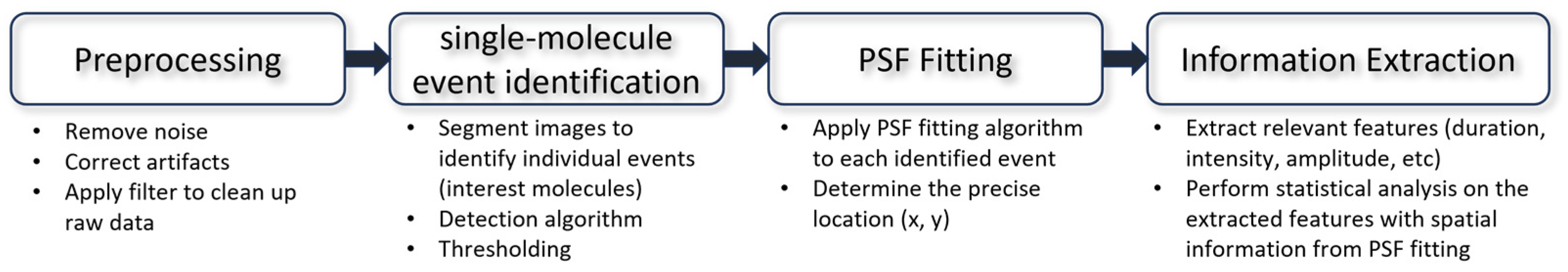
4.1. Point Spread Function Fitting
4.2. Extracting Information from Signal
4.3. Deep Learning
5. Biological Application
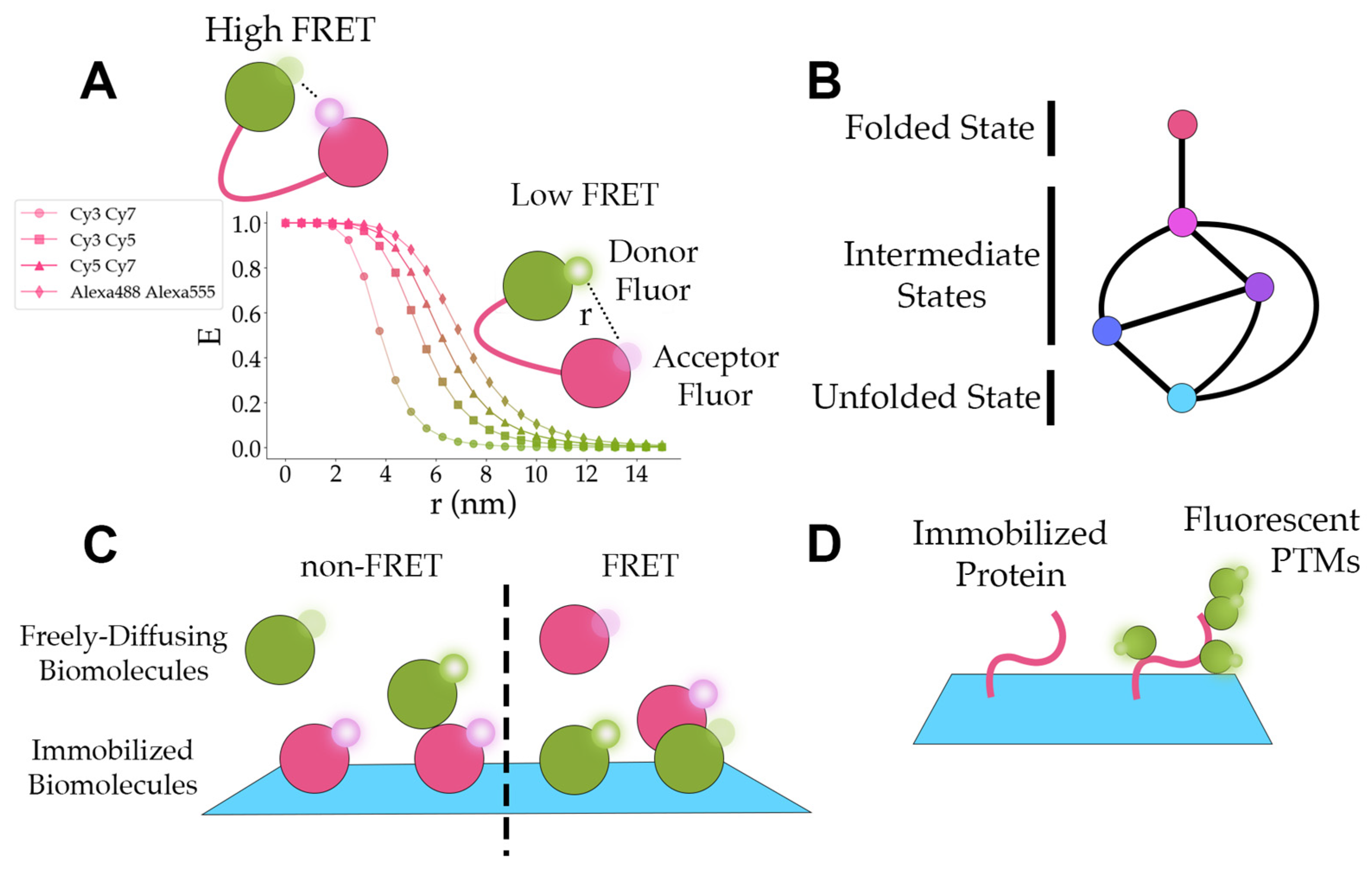
5.1. Conformation Dynamics
5.2. Protein Folding/Unfolding
5.3. Protein Interactions
5.4. Protein Post-Translational Modifications
6. Application of Lab-on-a-Chip Techniques for Single-Molecule Fluorescence Imaging
6.1. Enhance Signal-to-Noise Ratio
6.2. Increase Sample Concentration
6.3. On-Chip Single-Molecule Manipulation
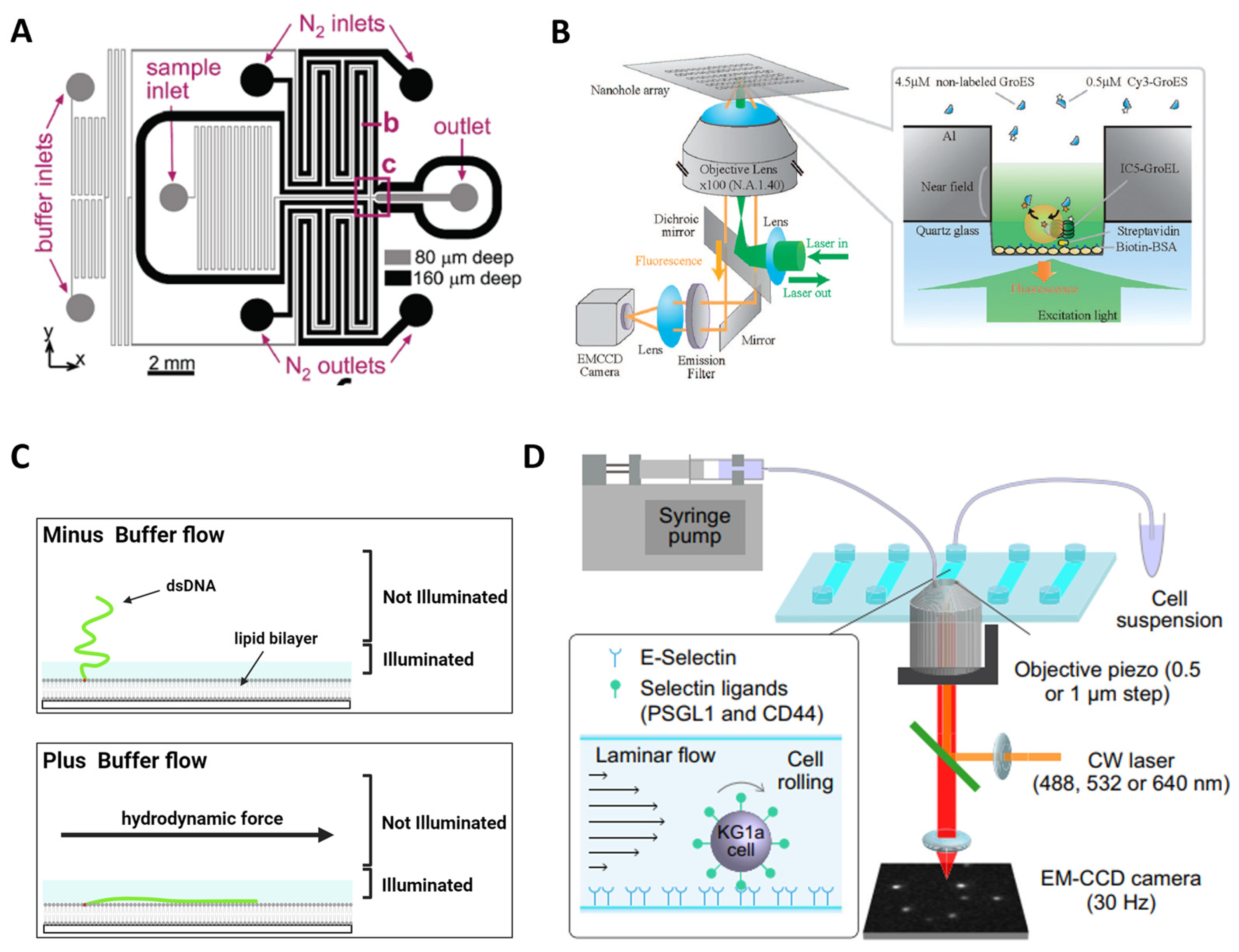
6.4. Microenvironment Control
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, K.; Kim, K.; Jung, J.; Heo, J.; Cho, S.; Lee, S.; Chang, G.; Jo, Y.; Park, H.; Park, Y.; et al. Quantitative phase imaging techniques for the study of cell pathophysiology: From principles to applications. Sensors 2013, 13, 4170–4191. [Google Scholar] [CrossRef] [PubMed]
- Royer, L.A.; Lemon, W.C.; Chhetri, R.K.; Wan, Y.; Coleman, M.; Myers, E.W.; Keller, P.J. Adaptive light-sheet microscopy for long-term, high-resolution imaging in living organisms. Nat. Biotechnol. 2016, 34, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Wassie, A.T.; Zhao, Y.; Boyden, E.S. Expansion microscopy: Principles and uses in biological research. Nat. Methods 2019, 16, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Fluorescence lifetime imaging–techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Zhou, J.; Zhu, W.S.; Manley, P.W.; Wang, Y.K.; Hood, T.; Wylie, A.; Xie, X.S. Imaging the intracellular distribution of tyrosine kinase inhibitors in living cells with quantitative hyperspectral stimulated Raman scattering. Nat. Chem. 2014, 6, 614–622. [Google Scholar] [CrossRef]
- Lu, F.K.; Basu, S.; Igras, V.; Hoang, M.P.; Ji, M.; Fu, D.; Holtom, G.R.; Neel, V.A.; Freudiger, C.W.; Fisher, D.E.; et al. Label-free DNA imaging in vivo with stimulated Raman scattering microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 11624–11629. [Google Scholar] [CrossRef]
- Roeffaers, M.B.; Zhang, X.; Freudiger, C.W.; Saar, B.G.; van Ruijven, M.; van Dalen, G.; Xiao, C.; Xie, X.S. Label-free imaging of biomolecules in food products using stimulated Raman microscopy. J. Biomed. Opt. 2010, 15, 066016. [Google Scholar] [CrossRef]
- Shi, L.; Zheng, C.; Shen, Y.; Chen, Z.; Silveira, E.S.; Zhang, L.; Wei, M.; Liu, C.; de Sena-Tomas, C.; Targoff, K.; et al. Optical imaging of metabolic dynamics in animals. Nat. Commun. 2018, 9, 2995. [Google Scholar] [CrossRef]
- Jalili, N.; Laxminarayana, K. A review of atomic force microscopy imaging systems: Application to molecular metrology and biological sciences. Mechatronics 2004, 14, 907–945. [Google Scholar] [CrossRef]
- Kherlopian, A.R.; Song, T.; Duan, Q.; Neimark, M.A.; Po, M.J.; Gohagan, J.K.; Laine, A.F. A review of imaging techniques for systems biology. BMC Syst. Biol. 2008, 2, 74. [Google Scholar] [CrossRef]
- Shashkova, S.; Leake, M.C. Single-molecule fluorescence microscopy review: Shedding new light on old problems. Biosci. Rep. 2017, 37, BSR20170031. [Google Scholar] [CrossRef]
- Cho, Y.; Zhao, C.L.; Lu, H. Trends in high-throughput and functional neuroimaging in Caenorhabditis elegans. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1376. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lee, B.-h.; King, R.W.; Finley, D.; Kirschner, M.W. Substrate degradation by the proteasome: A single-molecule kinetic analysis. Science 2015, 348, 1250834. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, W.; Kirschner, M.W. Specificity of the anaphase-promoting complex: A single-molecule study. Science 2015, 348, 1248737. [Google Scholar] [CrossRef]
- Rief, M.; Žoldák, G. Single-molecule mechanical studies of chaperones and their clients. Biophys. Rev. 2022, 3, 041301. [Google Scholar] [CrossRef]
- Georgescu, R.E.; Yao, N.Y.; O’Donnell, M. Single-molecule analysis of the Escherichia coli replisome and use of clamps to bypass replication barriers. FEBS Lett. 2010, 584, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Visnapuu, M.-L.; Greene, E.C. Single-molecule imaging of DNA curtains reveals intrinsic energy landscapes for nucleosome deposition. Nat. Struct. Mol. Biol. 2009, 16, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.; Wang, F.; Redding, S.; Plys, A.J.; Fazio, T.; Wind, S.; Alani, E.E.; Greene, E.C. Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proc. Natl. Acad. Sci. USA 2012, 109, E3074–E3083. [Google Scholar] [CrossRef]
- Hon, J.; Lu, Y. Single-molecule methods for measuring ubiquitination and protein stability. Methods Enzymol. 2019, 619, 225–247. [Google Scholar]
- van Ginkel, J.; Filius, M.; Szczepaniak, M.; Tulinski, P.; Meyer, A.S.; Joo, C. Single-molecule peptide fingerprinting. Proc. Natl. Acad. Sci. USA 2018, 115, 3338–3343. [Google Scholar] [CrossRef]
- Gust, A.; Zander, A.; Gietl, A.; Holzmeister, P.; Schulz, S.; Lalkens, B.; Tinnefeld, P.; Grohmann, D. A starting point for fluorescence-based single-molecule measurements in biomolecular research. Molecules 2014, 19, 15824–15865. [Google Scholar] [CrossRef] [PubMed]
- Moerner, W.; Fromm, D.P. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev. Sci. Instrum. 2003, 74, 3597–3619. [Google Scholar] [CrossRef]
- Axelrod, D. Total internal reflection fluorescence microscopy. Methods Cell Biol. 1989, 30, 245–270. [Google Scholar]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Traffic 2001, 2, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Schneckenburger, H. Total internal reflection fluorescence microscopy: Technical innovations and novel applications. Curr. Opin. Biotechnol. 2005, 16, 13–18. [Google Scholar] [CrossRef]
- Endesfelder, U.; Heilemann, M. Art and artifacts in single-molecule localization microscopy: Beyond attractive images. Nat. Methods 2014, 11, 235–238. [Google Scholar] [CrossRef]
- Mortensen, K.I.; Churchman, L.S.; Spudich, J.A.; Flyvbjerg, H. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 2010, 7, 377–381. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Tschanz, A.; Krupnik, L.; Ries, J. Quantitative data analysis in single-molecule localization microscopy. Trends Cell Biol. 2020, 30, 837–851. [Google Scholar] [CrossRef]
- Sage, D.; Kirshner, H.; Pengo, T.; Stuurman, N.; Min, J.; Manley, S.; Unser, M. Quantitative evaluation of software packages for single-molecule localization microscopy. Nat. Methods 2015, 12, 717–724. [Google Scholar] [CrossRef]
- Khater, I.M.; Nabi, I.R.; Hamarneh, G. A review of super-resolution single-molecule localization microscopy cluster analysis and quantification methods. Patterns 2020, 1, 100038. [Google Scholar] [CrossRef]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffié, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-molecule localization microscopy. Nat. Rev. Methods Primers 2021, 1, 39. [Google Scholar] [CrossRef]
- Nicovich, P.R.; Owen, D.M.; Gaus, K. Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat. Protoc. 2017, 12, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Imran, A.; Zhu, Q.; Sulaman, M.; Bukhtiar, A.; Xu, M. Electric-Dipole Gated Two Terminal Phototransistor for Charge-Coupled Device. Adv. Opt. Mater. 2023, 2300910. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Zhang, Z.; Hu, K.; Fang, X. Nanostructured Photodetectors: From Ultraviolet to Terahertz. Adv. Mater. 2016, 28, 403–433. [Google Scholar] [CrossRef] [PubMed]
- Lanza, M.; Smets, Q.; Huyghebaert, C.; Li, L.-J. Yield, variability, reliability, and stability of two-dimensional materials based solid-state electronic devices. Nat. Commun. 2020, 11, 5689. [Google Scholar] [CrossRef]
- Ha, T.; Tinnefeld, P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu. Rev. Phys. Chem. 2012, 63, 595–617. [Google Scholar] [CrossRef]
- Ha, T. Probing Nature’s Nanomachines One Molecule at a Time. Biophys. J. 2016, 110, 1004–1007. [Google Scholar] [CrossRef]
- Wilson, H.; Wang, Q. ABEL-FRET: Tether-free single-molecule FRET with hydrodynamic profiling. Nat. Methods 2021, 18, 816–820. [Google Scholar] [CrossRef]
- Ye, Y.; Blaser, G.; Horrocks, M.H.; Ruedas-Rama, M.J.; Ibrahim, S.; Zhukov, A.A.; Orte, A.; Klenerman, D.; Jackson, S.E.; Komander, D. Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature 2012, 492, 266–270. [Google Scholar] [CrossRef]
- Chandradoss, S.D.; Haagsma, A.C.; Lee, Y.K.; Hwang, J.-H.; Nam, J.-M.; Joo, C. Surface Passivation for Single-molecule Protein Studies. J. Vis. Exp. 2014, 86, 50549. [Google Scholar]
- Hua, B.; Han, K.Y.; Zhou, R.; Kim, H.; Shi, X.; Abeysirigunawardena, S.C.; Jain, A.; Singh, D.; Aggarwal, V.; Woodson, S.A.; et al. An improved surface passivation method for single-molecule studies. Nat. Methods 2014, 11, 1233–1236. [Google Scholar] [CrossRef]
- Paul, T.; Ha, T.; Myong, S. Regeneration of PEG slide for multiple rounds of single-molecule measurements. Biophys. J. 2021, 120, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Louis, J.M.; Chung, H.S. Diverse Folding Pathways of HIV-1 Protease Monomer on a Rugged Energy Landscape. Biophys. J. 2019, 117, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; McHale, K.; Louis, J.M.; Eaton, W.A. Single-Molecule Fluorescence Experiments Determine Protein Folding Transition Path Times. Science 2012, 335, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Pirchi, M.; Ziv, G.; Riven, I.; Cohen, S.S.; Zohar, N.; Barak, Y.; Haran, G. Single-molecule fluorescence spectroscopy maps the folding landscape of a large protein. Nat. Commun. 2011, 2, 493. [Google Scholar] [CrossRef]
- Granéli, A.; Yeykal, C.C.; Prasad, T.K.; Greene, E.C. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir 2006, 22, 292–299. [Google Scholar] [CrossRef]
- Green, N.M. Avidin. 1. The use of (14-C)biotin for kinetic studies and for assay. Biochem. J. 1963, 89, 585–591. [Google Scholar] [CrossRef]
- Roca, J.; Santiago-Frangos, A.; Woodson, S.A. Diversity of bacterial small RNAs drives competitive strategies for a mutual chaperone. Nat. Commun. 2022, 13, 2449. [Google Scholar] [CrossRef]
- Branigan, E.; Carlos Penedo, J.; Hay, R.T. Ubiquitin transfer by a RING E3 ligase occurs from a closed E2~ubiquitin conformation. Nat. Commun. 2020, 11, 2846. [Google Scholar] [CrossRef]
- Wang, Y.; Mallon, J.; Wang, H.; Singh, D.; Hyun Jo, M.; Hua, B.; Bailey, S.; Ha, T. Real-time observation of Cas9 postcatalytic domain motions. Proc. Natl. Acad. Sci. USA 2021, 118, 1. [Google Scholar] [CrossRef] [PubMed]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Cull, M.G.; Schatz, P.J. Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol. 2000, 326, 430–440. [Google Scholar] [PubMed]
- Jonsson, E.; Htet, Z.M.; Bard, J.A.M.; Dong, K.C.; Martin, A. Ubiquitin modulates 26S proteasome conformational dynamics and promotes substrate degradation. Sci. Adv. 2022, 8, eadd9520. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ade, C.; Nurik, C.E.; Carrejo, N.C.; Dutta, C.; Jayaraman, V.; Landes, C.F. Phosphorylation Induces Conformational Rigidity at the C-Terminal Domain of AMPA Receptors. J. Phys. Chem. B 2019, 123, 130–137. [Google Scholar] [CrossRef]
- Ciftci, D.; Huysmans, G.H.M.; Wang, X.; He, C.; Terry, D.; Zhou, Z.; Fitzgerald, G.; Blanchard, S.C.; Boudker, O. Single-molecule transport kinetics of a glutamate transporter homolog shows static disorder. Sci. Adv. 2020, 6, eaaz1949. [Google Scholar] [CrossRef]
- Asher, W.B.; Terry, D.S.; Gregorio, G.G.A.; Kahsai, A.W.; Borgia, A.; Xie, B.; Modak, A.; Zhu, Y.; Jang, W.; Govindaraju, A.; et al. GPCR-mediated β-arrestin activation deconvoluted with single-molecule precision. Cell 2022, 185, 1661–1675.e16. [Google Scholar] [CrossRef]
- Tran, T.; Mitra, J.; Ha, T.; Kavran, J.M. Increasing kinase domain proximity promotes MST2 autophosphorylation during Hippo signaling. J. Biol. Chem. 2020, 295, 16166–16179. [Google Scholar] [CrossRef]
- Poyton, M.F.; Feng, X.A.; Ranjan, A.; Lei, Q.; Wang, F.; Zarb, J.S.; Louder, R.K.; Park, G.; Jo, M.H.; Ye, J.; et al. Coordinated DNA and histone dynamics drive accurate histone H2A.Z exchange. Sci. Adv. 2022, 8, eabj5509. [Google Scholar] [CrossRef]
- Park, S.R.; Hauver, J.; Zhang, Y.; Revyakin, A.; Coleman, R.A.; Tjian, R.; Chu, S.; Pertsinidis, A. A Single-Molecule Surface-Based Platform to Detect the Assembly and Function of the Human RNA Polymerase II Transcription Machinery. Structrue 2020, 28, 1337–1343.e4. [Google Scholar] [CrossRef]
- Song, E.; Uhm, H.; Munasingha, P.R.; Hwang, S.; Seo, Y.-S.; Kang, J.Y.; Kang, C.; Hohng, S. Rho-dependent transcription termination proceeds via three routes. Nat. Commun. 2022, 13, 1663. [Google Scholar] [CrossRef] [PubMed]
- Liauw, B.W.-H.; Afsari, H.S.; Vafabakhsh, R. Conformational rearrangement during activation of a metabotropic glutamate receptor. Nat. Chem. Biol. 2021, 17, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Hon, J.; Zhou, M.; Lu, Y. An empirical energy landscape reveals mechanism of proteasome in polypeptide translocation. eLife 2022, 11, e71911. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fard, A.P.; Davies, A. Characterization of instrument drift using a spherical artifact. Precis. Eng. 2014, 38, 443–447. [Google Scholar] [CrossRef]
- Wang, Y.; Schnitzbauer, J.; Hu, Z.; Li, X.; Cheng, Y.; Huang, Z.-L.; Huang, B. Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Opt. Express 2014, 22, 15982–15991. [Google Scholar] [CrossRef]
- Davenport, W.B.; Root, W.L. Random Signals and Noise; McGraw-Hill: New York, NY, USA, 1958. [Google Scholar]
- Kou, S.C.; Xie, X.S. Generalized Langevin equation with fractional Gaussian noise: Subdiffusion within a single protein molecule. Phys. Rev. Lett. 2004, 93, 180603. [Google Scholar] [CrossRef]
- Jakeman, E. On the statistics of K-distributed noise. J. Phys. A Math. General. 1980, 13, 31. [Google Scholar] [CrossRef]
- Chan, R.; Yang, H.; Zeng, T. A two-stage image segmentation method for blurry images with poisson or multiplicative gamma noise. SIAM J. Imaging Sci. 2014, 7, 98–127. [Google Scholar] [CrossRef]
- Kassam, S.A. Signal Detection in Non—Gaussian Noise; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Shnerb, N.; Bettelheim, E.; Louzoun, Y.; Agam, O.; Solomon, S. Adaptation of autocatalytic fluctuations to diffusive noise. Phys. Rev. E 2001, 63, 021103. [Google Scholar] [CrossRef]
- Flegg, M.B.; Rüdiger, S.; Erban, R. Diffusive spatio-temporal noise in a first-passage time model for intracellular calcium release. J. Chem. Phys. 2013, 138, 154103. [Google Scholar] [CrossRef]
- Colomb, W.; Sarkar, S.K. Extracting physics of life at the molecular level: A review of single-molecule data analyses. Phys. Life Rev. 2015, 13, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Rieckh, G.; Tkačik, G. Noise and information transmission in promoters with multiple internal states. Biophys. J. 2014, 106, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; He, K.; Cheng, M.; Yu, J.; Fang, X. Analysis of the Steps in Single-Molecule Photobleaching Traces by Using the Hidden Markov Model and Maximum-Likelihood Clustering. Chem.—An Asian J. 2014, 9, 2303–2308. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Johnson-Buck, A.; Walter, N. Hidden Markov modeling in single-molecule biophysics. In Encyclopedia of Biophysics; Springer: Berlin/Heidelberg, Germany, 2013; pp. 971–975. [Google Scholar]
- Tao, L.; Ozarkar, S.; Beck, J.M.; Bhandawat, V. Statistical structure of locomotion and its modulation by odors. Elife 2019, 8, e41235. [Google Scholar] [CrossRef]
- Okamoto, K.; Sako, Y. Variational Bayes analysis of a photon-based hidden Markov model for single-molecule FRET trajectories. Biophys. J. 2012, 103, 1315–1324. [Google Scholar] [CrossRef]
- Sungkaworn, T.; Jobin, M.-L.; Burnecki, K.; Weron, A.; Lohse, M.J.; Calebiro, D. Single-molecule imaging reveals receptor–G protein interactions at cell surface hot spots. Nature 2017, 550, 543–547. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, Y.; Cui, Y.; Yuan, J.; Fang, X. Deep learning in single-molecule imaging and analysis recent advances and prospects. Chem. Sci. 2022, 13, 11964–11980. [Google Scholar] [CrossRef]
- Wang, Q.; He, H.; Zhang, Q.; Feng, Z.; Li, J.; Chen, X.; Liu, L.; Wang, X.; Ge, B.; Yu, D. Deep-learning-assisted single-molecule tracking on a live cell membrane. Anal. Chem. 2021, 93, 8810–8816. [Google Scholar] [CrossRef]
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.-W.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G.; et al. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908. [Google Scholar] [CrossRef]
- Chen, C.; Stevens, B.; Kaur, J.; Cabral, D.; Liu, H.; Wang, Y.; Zhang, H.; Rosenblum, G.; Smilansky, Z.; Goldman, Y.E.; et al. Single-Molecule Fluorescence Measurements of Ribosomal Translocation Dynamics. Mol. Cell 2011, 42, 367–377. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Hohng, S. Single-Molecule Three-Color FRET with Both Negligible Spectral Overlap and Long Observation Time. PLoS ONE 2010, 5, e12270. [Google Scholar] [CrossRef] [PubMed]
- Bibeau, J.P.; Gray, S.; De La Cruz, E.M. Clusters of a Few Bound Cofilins Sever Actin Filaments. J. Mol. Biol. 2021, 433, 166833. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Bhattacharya, A.; Ghelfi, M.D.; Li, H.; Fritsch, C.; Chenoweth, D.M.; Goldman, Y.E.; Cooperman, B.S. Ataluren binds to multiple protein synthesis apparatus sites and competitively inhibits release factor-dependent termination. Nat. Commun. 2022, 13, 2413. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, A.A.; Bow, H.; Hou, H.W.; Tan, S.J.; Han, J.; Lim, C.T. Microfluidics for cell separation. Med. Biol. Eng. Comput. 2010, 48, 999–1014. [Google Scholar] [CrossRef]
- Kuntaegowdanahalli, S.S.; Bhagat, A.A.; Kumar, G.; Papautsky, I. Inertial microfluidics for continuous particle separation in spiral microchannels. Lab. Chip 2009, 9, 2973–2980. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, L.; Wei, W.; Shi, Q. Microchip-based single-cell functional proteomics for biomedical applications. Lab. Chip 2017, 17, 1250–1263. [Google Scholar] [CrossRef]
- Shields, C.W.; Reyes, C.D.; Lopez, G.P. Microfluidic cell sorting: A review of the advances in the separation of cells from debulking to rare cell isolation. Lab. Chip 2015, 15, 1230–1249. [Google Scholar] [CrossRef]
- Xi, H.D.; Zheng, H.; Guo, W.; Ganan-Calvo, A.M.; Ai, Y.; Tsao, C.W.; Zhou, J.; Li, W.; Huang, Y.; Nguyen, N.T.; et al. Active droplet sorting in microfluidics: A review. Lab. Chip 2017, 17, 751–771. [Google Scholar] [CrossRef]
- Yin, H.; Marshall, D. Microfluidics for single cell analysis. Curr. Opin. Biotechnol. 2012, 23, 110–119. [Google Scholar] [CrossRef]
- Young, E.W.; Beebe, D.J. Fundamentals of microfluidic cell culture in controlled microenvironments. Chem. Soc. Rev. 2010, 39, 1036–1048. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, D.; Yue, H.; French, J.B.; Rufo, J.; Benkovic, S.J.; Huang, T.J. Lab-on-a-chip technologies for single-molecule studies. Lab. Chip 2013, 13, 2183–2198. [Google Scholar] [CrossRef] [PubMed]
- Rasnik, I.; McKinney, S.A.; Ha, T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nat. Methods 2006, 3, 891–893. [Google Scholar] [CrossRef] [PubMed]
- Vasdekis, A.E.; Laporte, G.P. Enhancing single molecule imaging in optofluidics and microfluidics. Int. J. Mol. Sci. 2011, 12, 5135–5156. [Google Scholar] [CrossRef]
- Campos, L.A.; Liu, J.; Wang, X.; Ramanathan, R.; English, D.S.; Munoz, V. A photoprotection strategy for microsecond-resolution single-molecule fluorescence spectroscopy. Nat. Methods 2011, 8, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Lemke, E.A.; Gambin, Y.; Vandelinder, V.; Brustad, E.M.; Liu, H.-W.; Schultz, P.G.; Groisman, A.; Deniz, A.A. Microfluidic device for single-molecule experiments with enhanced photostability. J. Am. Chem. Soc. 2009, 131, 13610–13612. [Google Scholar] [CrossRef] [PubMed]
- Gambin, Y.; VanDelinder, V.; Ferreon, A.C.; Lemke, E.A.; Groisman, A.; Deniz, A.A. Visualizing a one-way protein encounter complex by ultrafast single-molecule mixing. Nat. Methods 2011, 8, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Levene, M.J.; Korlach, J.; Turner, S.W.; Foquet, M.; Craighead, H.G.; Webb, W.W. Zero-mode waveguides for single-molecule analysis at high concentrations. Science 2003, 299, 682–686. [Google Scholar] [CrossRef]
- Zhu, P.; Craighead, H.G. Zero-mode waveguides for single-molecule analysis. Annu. Rev. Biophys. 2012, 41, 269–293. [Google Scholar] [CrossRef]
- Miyake, T.; Tanii, T.; Sonobe, H.; Akahori, R.; Shimamoto, N.; Ueno, T.; Funatsu, T.; Ohdomari, I. Real-time imaging of single-molecule fluorescence with a zero-mode waveguide for the analysis of protein− protein interaction. Anal. Chem. 2008, 80, 6018–6022. [Google Scholar] [CrossRef]
- Uemura, S.; Aitken, C.E.; Korlach, J.; Flusberg, B.A.; Turner, S.W.; Puglisi, J.D. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature 2010, 464, 1012–1017. [Google Scholar] [CrossRef]
- Fazio, T.; Visnapuu, M.-L.; Wind, S.; Greene, E.C. DNA curtains and nanoscale curtain rods: High-throughput tools for single molecule imaging. Langmuir 2008, 24, 10524–10531. [Google Scholar] [CrossRef] [PubMed]
- Al Alwan, B.; AbuZineh, K.; Nozue, S.; Rakhmatulina, A.; Aldehaiman, M.; Al-Amoodi, A.S.; Serag, M.F.; Aleisa, F.A.; Merzaban, J.S.; Habuchi, S. Single-molecule imaging and microfluidic platform reveal molecular mechanisms of leukemic cell rolling. Commun. Biol. 2021, 4, 868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jiang, K.; Liu, F.; Doyle, P.S.; van Kan, J.A.; van der Maarel, J.R. A nanofluidic device for single molecule studies with in situ control of environmental solution conditions. Lab. A Chip 2013, 13, 2821–2826. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Aristov, A.; Lelek, M.; Hao, X.; Zimmer, C. Deep learning massively accelerates super-resolution localization microscopy. Nat. Biotechnol. 2018, 36, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Small, A.; Stahlheber, S. Fluorophore localization algorithms for super-resolution microscopy. Nat. Methods 2014, 11, 267–279. [Google Scholar] [CrossRef]
- Möckl, L.; Moerner, W. Super-resolution microscopy with single molecules in biology and beyond–essentials, current trends, and future challenges. J. Am. Chem. Soc. 2020, 142, 17828–17844. [Google Scholar] [CrossRef]
- Sydor, A.M.; Czymmek, K.J.; Puchner, E.M.; Mennella, V. Super-resolution microscopy: From single molecules to supramolecular assemblies. Trends Cell Biol. 2015, 25, 730–748. [Google Scholar] [CrossRef]
- Shivanandan, A.; Deschout, H.; Scarselli, M.; Radenovic, A. Challenges in quantitative single molecule localization microscopy. FEBS Lett. 2014, 588, 3595–3602. [Google Scholar] [CrossRef]
- Funatsu, T.; Harada, Y.; Tokunaga, M.; Saito, K.; Yanagida, T. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature 1995, 374, 555–559. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Biomolecule(s) | Fluorophore Labeling | Fluorophores | Biotin Conjugation | Surface | Analysis Software | Results | Camera | Ref |
|---|---|---|---|---|---|---|---|---|
| Conformational Dynamics | ||||||||
| Streptococcus pyogenes CRISPR Cas9 | Cysteine–maleimide | Cy3 LD750 | Biotinylated DNA | PEG | Custom | Cas9′s HNH domain exhibits dynamics coupled with non-target strand cleavage | EMCCD | Wang [51] |
| Saccharomyces cerevisiae 26S Proteasome | Cysteine–maleimide Unnatural amino acid “click” chemistry | Cy3 LD555 LD655 | AviTag fusion reacted with BirA in vitro | PEG | SPARTAN | Ubiquitin chain binding to the 26S proteasome reduces the rate of conformational transitions | EMCCD | Jonsson [54] |
| Mus musculus metabotropic glutamate receptor 2 and 3 | Unnatural amino acid “click” chemistry | Cy3 Cy5 | Commercial biotinylated antiFLAG antibody | PEG | smCamera software | Metabotropic glutamate receptor 2 displays four sequential conformational states | EMCCD | Liauw [63] |
| Protein Folding | ||||||||
| Escherichia coli adenylate kinase | Cysteine–maleimide | Alexa 488 Atto 590 | Biotinylated phosphoethanolamine | Lipids | Custom MATLAB | folding of adenylate kinase involves at least 6 states with sequential and non-sequential transitions | SPAD | Pirchi [46] |
| Protein Interactions | ||||||||
| Saccharomyces cerevisiae cofilin on actin | Cysteine–maleimide | Alexa 488 Alexa 647 | Biotinylated actin | Tween 20 | TrackMate MATLAB ImageJ | Cofilin clusters grow 2 times faster towards actin’s pointed end versus barbed end | EMCCD | Bibeau [87] |
| Bovine β-arrestin1 | Cysteine–maleimide | LD555p LD655 | Strep-tag fusion | PEG | SPARTAN | β-arrestin1 tail displacement by phosphorylated C-terminal receptor requires GPCR agonist | sCMOS | Asher [58] |
| S. cerevisiae Histone and SWR1 | Cysteine–maleimide | Cy3 Cy5 Cy7 | Biotinylated DNA | PEG | Custom MATLAB | H2A remains in complex with SWR1–nucleosome complex for tens of seconds after H2A.Z displacement | EMCCD | Poyton [60] |
| Release factor complex (RFC) | Lysine hydroxysuccinimide (NHS) ester | Cy3 Atto 647 | mRNA biotinylated at 3′ end | PEG | ImageJ Python | Ataluren, a translation readthrough-inducing drug, acts as a competitive inhibitor | EMCCD | Huang [88] |
| sRNA ChiX sRNA DsrA Escherichia coli Hfq sRNA chaperone | 5′ sRNA-free primary amine-NHS ester | Cy3 Cy5 | AviTag fusion biotinylated by endogenous BirA | Tween 20 | Imscroll in MATLAB | Sometimes two sRNAs can stably bind to Hfq. Most replacement occurs when a strongly competitive sRNA, ChiX, replaces a moderately competitive sRNA, DsrA. | EMCCD | Roca [49] |
| Protein post-translational | ||||||||
| Human Ubc13 E2 ubiquitin ligase | Cysteine–maleimide | Cy3B Alexa 647 | AviTag fusion reacted with BirA in vitro | PEG | Interactive data language (IDL) | Ubiquitin transfer proceeds from high FRET signal corresponding to the closed conformation of ubc13 | EMCCD | Branigan [50] |
| Human anaphase-promoting complex E3 ubiquitin ligase | Cysteine–maleimide | Alexa 488 DyLight 550 Alexa 647 | Intein-mediated protein ligation (IPL) of biotin-containing peptide to the C terminus | PEG | Custom MATLAB | Anaphase-promoting complex displays biphasic activity | EMCCD | Lu [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colson, L.; Kwon, Y.; Nam, S.; Bhandari, A.; Maya, N.M.; Lu, Y.; Cho, Y. Trends in Single-Molecule Total Internal Reflection Fluorescence Imaging and Their Biological Applications with Lab-on-a-Chip Technology. Sensors 2023, 23, 7691. https://doi.org/10.3390/s23187691
Colson L, Kwon Y, Nam S, Bhandari A, Maya NM, Lu Y, Cho Y. Trends in Single-Molecule Total Internal Reflection Fluorescence Imaging and Their Biological Applications with Lab-on-a-Chip Technology. Sensors. 2023; 23(18):7691. https://doi.org/10.3390/s23187691
Chicago/Turabian StyleColson, Louis, Youngeun Kwon, Soobin Nam, Avinashi Bhandari, Nolberto Martinez Maya, Ying Lu, and Yongmin Cho. 2023. "Trends in Single-Molecule Total Internal Reflection Fluorescence Imaging and Their Biological Applications with Lab-on-a-Chip Technology" Sensors 23, no. 18: 7691. https://doi.org/10.3390/s23187691