

Electrochemical Impedance Spectroscopy for the Sensing of the Kinetic Parameters of Engineered Enzymes
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Impedance Measurements
2.2. Mutations of hEKL
2.3. SDS-PAGE and Testing of Activity on Trx-DCD1 Substrate
3. Results and Discussion
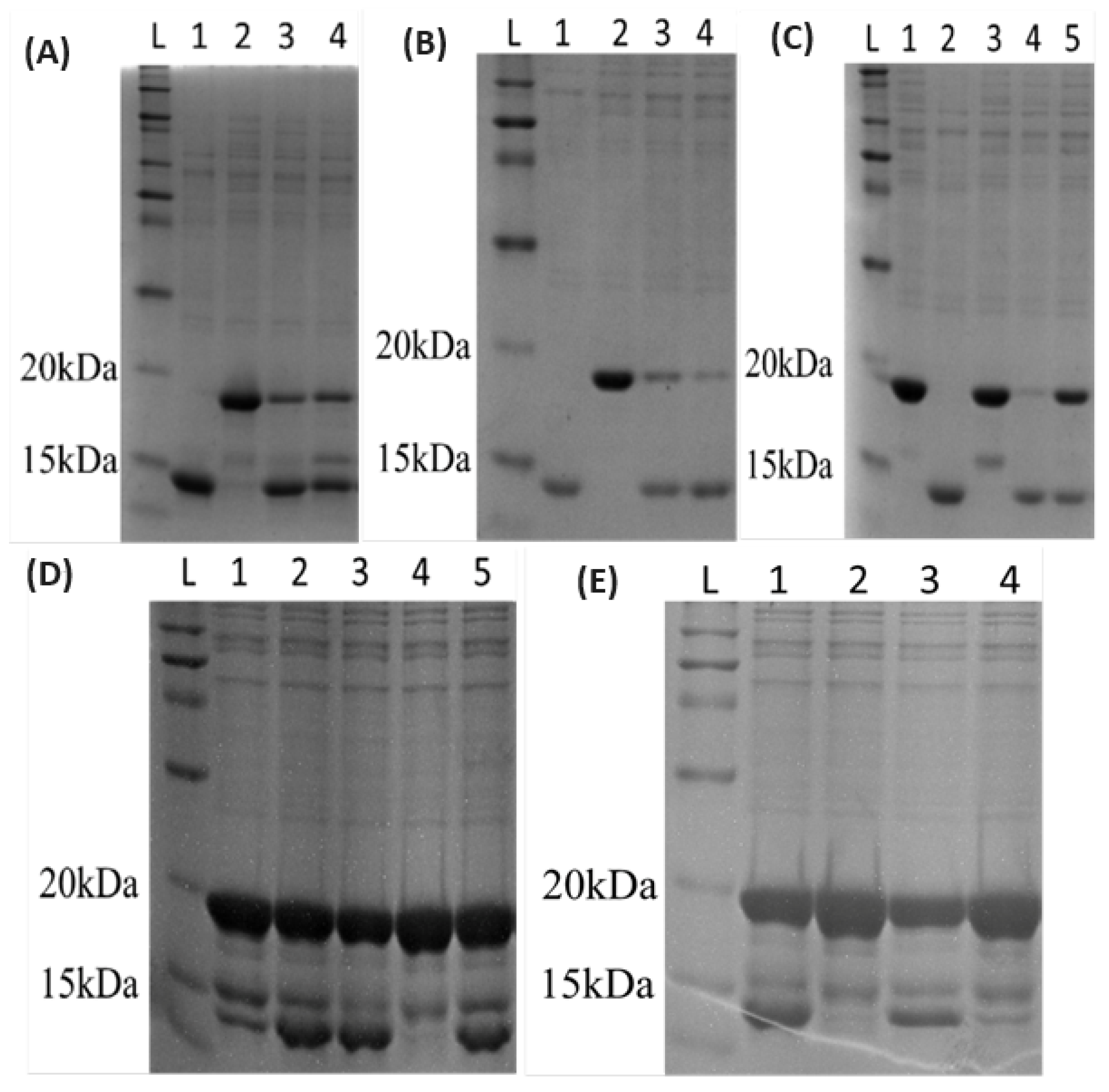
3.1. Preparation of Mutant Forms of the Enzyme
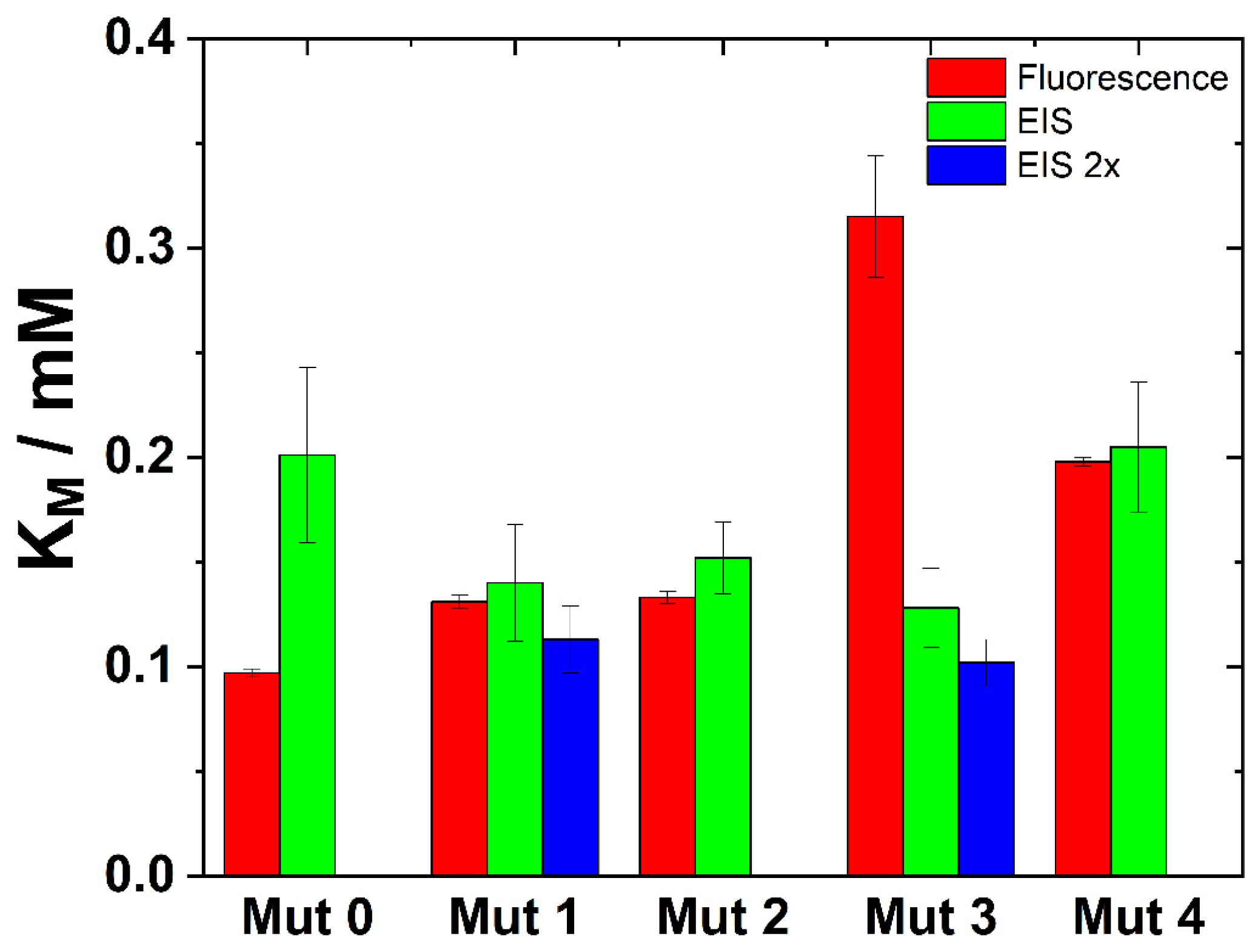
3.2. Characterization of Basic Kinetic Parameters of Mutant Enzymes
3.2.1. Fluorescence Measurements
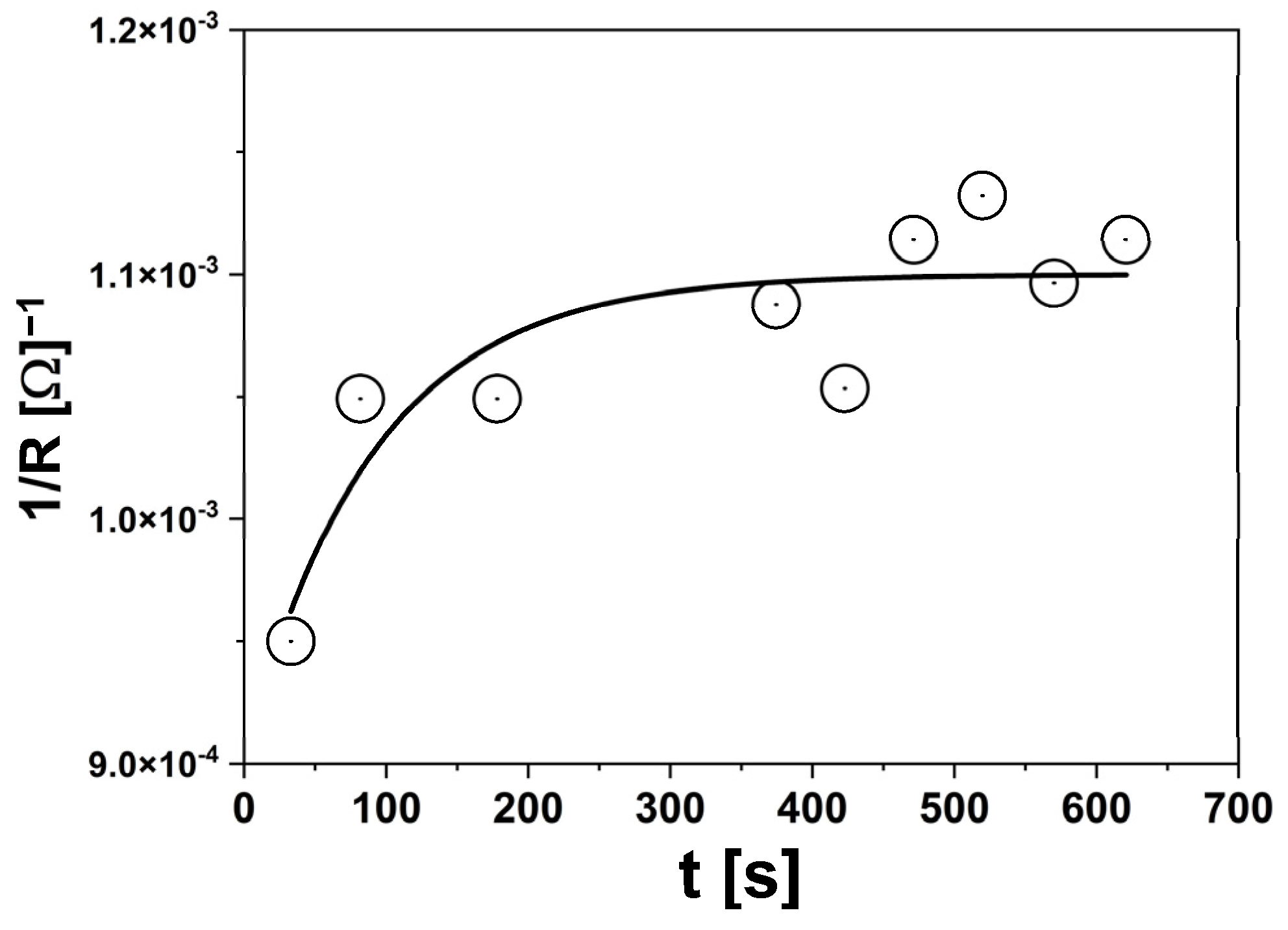
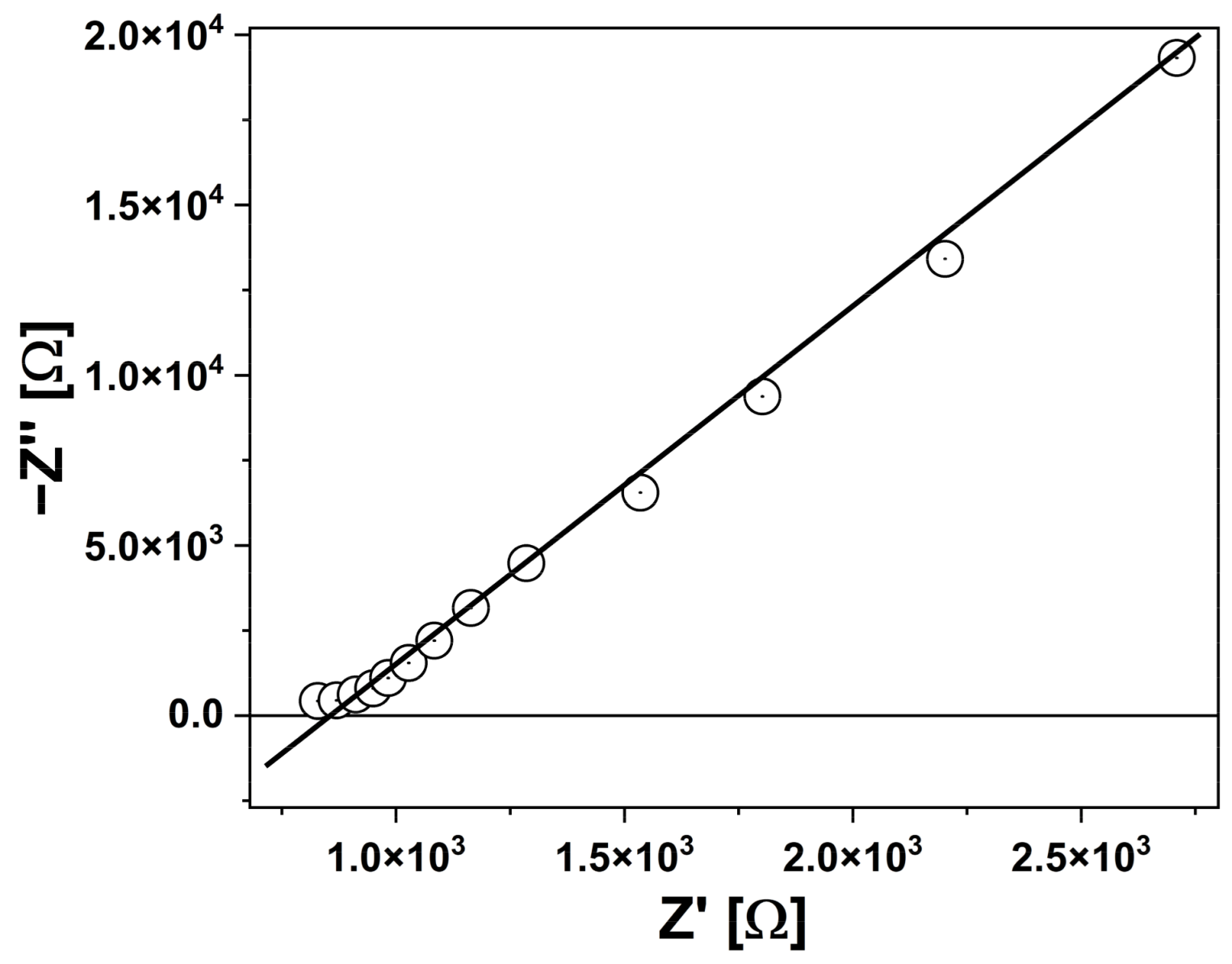
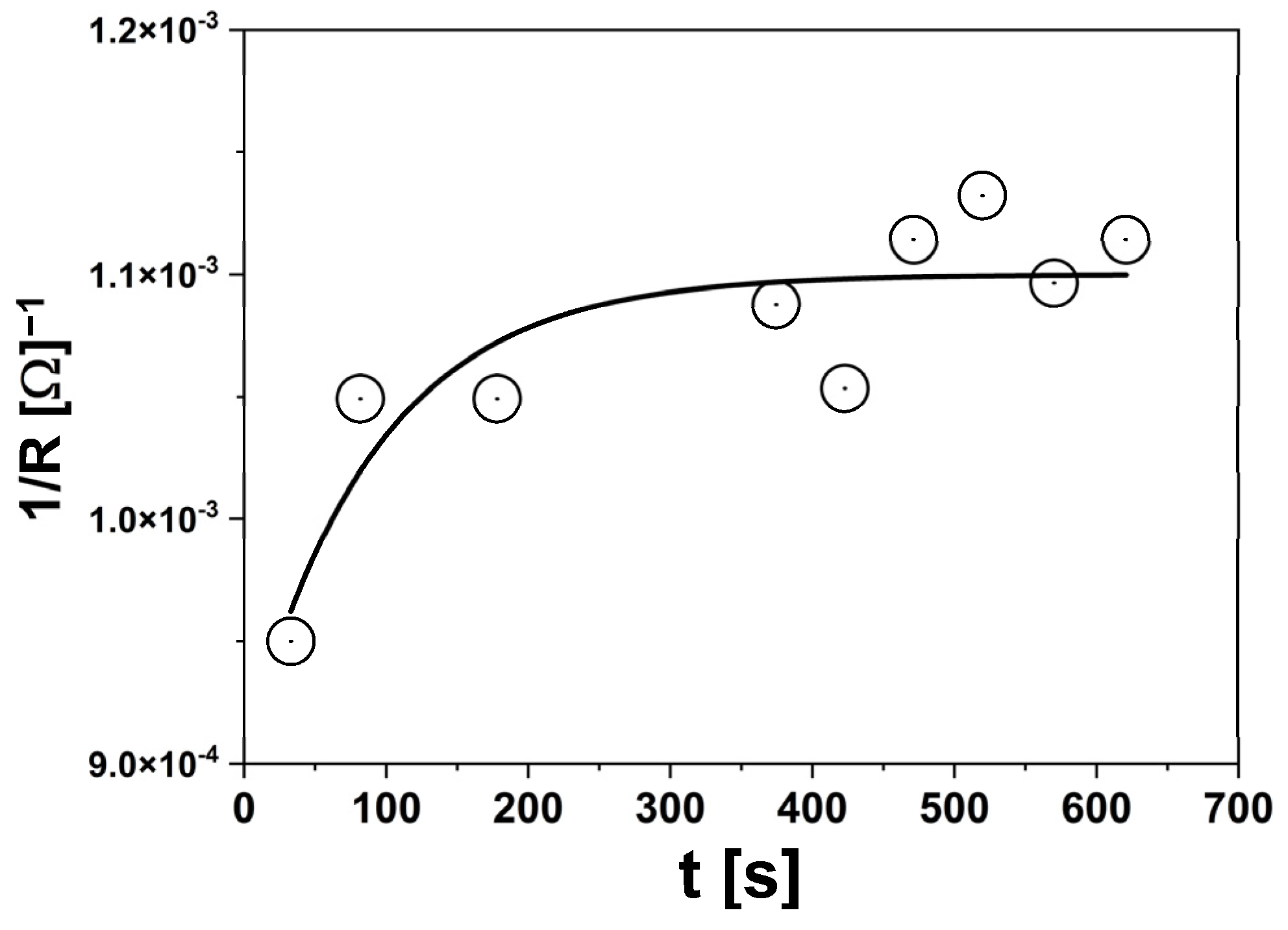
3.2.2. EIS Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, X.; Yin, H.; Peng, L.; Zhang, D.; Li, K.; Cui, F.; Xia, C.; Huang, H.; Li, Z. The Global Status and Trends of Enteropeptidase: A Bibliometric Study. Front. Med. 2022, 9, 779722. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.T.; Kruppa, M.; Johnson, D.A.; Van Haeften, J.; Chen, X.; Leahy, D.; Peake, J.; Harris, J.M. High Yield Expression in Pichia Pastoris of Human Neutrophil Elastase Fused to Cytochrome B5. Protein Expr. Purif. 2023, 206, 106255. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.; Chang, W.W.; Hu, M.Y.; Stumpp, M. Characterization of Digestive Proteases in the Gut of a Basal Deuterostome. J. Exp. Biol. 2023, 226, jeb245789. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, Y.; Veile, R.A.; Donis-Keller, H.; Sadler, J.E. CDNA Sequence and Chromosomal Localization of Human Enterokinase, the Proteolytic Activator of Trypsinogen. Biochemistry 1995, 34, 4562–4568. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, Y.; Yuan, X.; Wu, Q.; McCourt, D.W.; Sadler, J.E. Enterokinase, the Initiator of Intestinal Digestion, Is a Mosaic Protease Composed of a Distinctive Assortment of Domains. Proc. Natl. Acad. Sci. USA 1994, 91, 7588–7592. [Google Scholar] [CrossRef]
- Gasparian, M.E.; Ostapchenko, V.G.; Schulga, A.A.; Dolgikh, D.A.; Kirpichnikov, M.P. Expression, Purification, and Characterization of Human Enteropeptidase Catalytic Subunit in Escherichia coli. Protein Expr. Purif. 2003, 31, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Bricteux-Grégoire, S.; Florkin, R.S.M. Phylogeny of Trypsinogen Activation Peptides. Comp. Biochem. Physiol. Part B Comp. Biochem. 1972, 42, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, M.E.; Bychkov, M.L.; Dolgikh, D.A.; Kirpichnikov, M.P. Strategy for Improvement of Enteropeptidase Efficiency in Tag Removal Processes. Protein Expr. Purif. 2011, 79, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Sakin, E.; Yanardağ, İ.H.; Ramazanoğlu, E.; Yalçın, H. Enzyme Activities and Heavy Metal Interactions in Calcareous Soils under Different Land Uses. Int. J. Phytoremediation 2024, 26, 273–286. [Google Scholar] [CrossRef]
- Shaposhnikov, L.A.; Chikurova, N.Y.; Chernobrovkina, A.V.; Tishkov, V.I.; Pometun, A.A. Development of an Approach to Determining Enzymatic Activity of Ribonucleoside Hydrolase c Using Hydrophilic Interaction Liquid Chromatography. J. Chromatogr. A 2024, 1715, 464561. [Google Scholar] [CrossRef]
- Kuziemska, B.; Wysokiński, A.; Trębicka, J. The Effect of Different Copper Doses and Organic Fertilisation on Soil’s Enzymatic Activity. Plant Soil Environ. 2020, 66, 93–98. [Google Scholar] [CrossRef]
- Tang, J.; Zhang, L.; Zhang, J.; Ren, L.; Zhou, Y.; Zheng, Y.; Luo, L.; Yang, Y.; Huang, H.; Chen, A. Physicochemical Features, Metal Availability and Enzyme Activity in Heavy Metal-Polluted Soil Remediated by Biochar and Compost. Sci. Total Environ. 2020, 701, 134751. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, A.G.; Likhareva, V.V.; Vaskovsky, B.V.; Garanin, S.K.; Onoprienko, L.V.; Prudchenko, I.A.; Chikin, L.D.; Rumsh, L.D. Study of Secondary Specificity of Enteropeptidase in Comparison with Trypsin. Biochemistry 2004, 69, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, A.G.; Likhareva, V.V.; Rumsh, L.D. A Rate-Limiting Step of Enteropeptidase Hydrolysis. Russ. J. Bioorg. Chem. 2008, 34, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, A.G.; Likhareva, V.V.; Teich, N.; Rumsh, L.D. The Ways of Realization of High Specificity and Efficiency of Enteropeptidase. Protein Pept. Lett. 2007, 14, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yuan, X.; Zheng, X.; Sadler, J.E. Bovine Proenteropeptidase Is Activated by Trypsin, and the Specificity of Enteropeptidase Depends on the Heavy Chain. J. Biol. Chem. 1997, 272, 31293–31300. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, A.G.; Rumsh, L.D. Enteropeptidase: Structure, Function, and Application in Biotechnology. Appl. Biochem. Biotechnol. 2000, 88, 159–174. [Google Scholar] [CrossRef]
- Gasparian, M.E.; Ostapchenko, V.G.; Dolgikh, D.A.; Kirpichnikov, M.P. Biochemical Characterization of Human Enteropeptidase Light Chain. Biochemistry 2006, 71, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Jiríčková, K.; Gál, M.; Krahulec, J.; Híveš, J. Electrochemical Determination of Basic Biochemical Properties of Enzyme Enterokinase. Monatshefte Chem. Chem. Mon. 2015, 146, 755–759. [Google Scholar] [CrossRef]
- Dai, T.; Qiu, M.; Li, H.; Yang, M.; Gao, J.; Li, X.; Lv, J.; Yang, M.; Yuan, Z.; Luo, J. A Simple and Sensitive Electrochemical Sensor for the Detection of Peptidase Activity. Anal. Bioanal. Chem. 2023, 415, 2209–2215. [Google Scholar] [CrossRef]
- Sajedeh Jafarypour, S.; Rahdar, M.; Farnoosh, G. Implementation of the Electrochemical Impedance Method for Measuring Troponin Enzyme in the Laboratory on a Chip. J. Appl. Biotechnol. Rep. 2023, 10, 934–942. [Google Scholar]
- Didonè, L.; Shin, Y.; Silvestri, A.; Prato, M.; Park, S.; Bianco, A. Electrochemical Impedance Spectroscopy, Another Arrow in the Arsenal to Study the Biodegradability of Two-Dimensional Materials. Nanoscale 2024, 16, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Pilz, F.H.; Kielb, P. Cyclic Voltammetry, Square Wave Voltammetry or Electrochemical Impedance Spectroscopy? Interrogating Electrochemical Approaches for the Determination of Electron Transfer Rates of Immobilized Redox Proteins. BBA Adv. 2023, 4, 100095. [Google Scholar] [CrossRef] [PubMed]
- Torrigino, F.; Herkendell, K. Investigating Enzyme-Based Electrode Processes Using Electrochemical Impedance Spectroscopy and Distribution of Relaxation Time Analysis. In Proceedings of the Electrochemical Society Meeting Abstracts 244, Gothenburg, Sweden, 8–12 October 2023; The Electrochemical Society, Inc.: Pennington, NJ, USA, 2023; p. 2491. [Google Scholar]
- Smith, B.J. SDS Polyacrylamide Gel Electrophoresis of Proteins. Proteins 1984, 1, 41–55. [Google Scholar]
- Pepeliaev, S.; Krahulec, J.; Černý, Z.; Jílková, J.; Tlustá, M.; Dostálová, J. High Level Expression of Human Enteropeptidase Light Chain in Pichia Pastoris. J. Biotechnol. 2011, 156, 67–75. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Change | Position | Amino Acid | Specific Activity (U∙mg−1) |
|---|---|---|---|---|
| Mut0 | K99R | 99 | Lysin ⟶ arginine | 7729 |
| Mut1 | N101S | 101 | Asparagine ⟶ serine | 3615 |
| Mut2 | N95E | 95 | Asparagine ⟶ glutamic acid | 6068 |
| Mut3 | N95D | 95 | Asparagine ⟶ aspartic acid | 4453 |
| Mut4 | D100N | 100 | Aspartic acid ⟶ asparagine | 4804 |
| Enterokinase | Kinetic Parameters | ||
|---|---|---|---|
| Km [∙mM] | kcat [∙s−1] | kcat/Km [∙mM−1∙s−1] | |
| hEKL | 0.116 ± 0.004 | 131.75 ± 20.660 | 1129.354 |
| Mut0 | 0.097 ± 0.002 | 14.116 ± 0.716 | 144.535 |
| Mut1 | 0.131 ± 0.003 | 54.050 ± 10.765 | 410.506 |
| Mut2 | 0.133 ± 0.003 | 66.603 ± 8.208 | 498.275 |
| Mut3 | 0.315 ± 0.029 | 15.098 ± 1.098 | 47.880 |
| Mut4 | 0.198 ± 0.002 | 39.170 ± 2.269 | 197.828 |
| Enterokinase | Kinetic Parameters | ||
|---|---|---|---|
| Km [∙mM] | kcat [∙s−1] | kcat/Km [∙mM−1∙s−1] | |
| Mut0 | 0.206 ± 0.042 | 15.5 ± 2.0 | 75.2 |
| Mut1 | 0.140 ± 0.028 | 75.9 ± 13.2 | 542.1 |
| Mut2 | 0.152 ± 0.017 | 64.2 ± 11.1 | 422.3 |
| Mut3 | 0.128 ± 0.019 | 21.9 ± 4.7 | 171.1 |
| Mut4 | 0.205 ± 0.031 | 35.0 ± 12.4 | 170.7 |
| Enterokinase | Kinetic Parameters | ||
|---|---|---|---|
| Km [∙mM] | kcat [∙s−1] | kcat/Km [∙mM−1∙s−1] | |
| Mut1 | 0.113 ± 0.016 | 66.6 ± 4.3 | 589.4 |
| Mut3 | 0.102 ± 0.011 | 31.1 ± 5.4 | 304.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dusíková, A.; Baranová, T.; Krahulec, J.; Dakošová, O.; Híveš, J.; Naumowicz, M.; Gál, M. Electrochemical Impedance Spectroscopy for the Sensing of the Kinetic Parameters of Engineered Enzymes. Sensors 2024, 24, 2643. https://doi.org/10.3390/s24082643
Dusíková A, Baranová T, Krahulec J, Dakošová O, Híveš J, Naumowicz M, Gál M. Electrochemical Impedance Spectroscopy for the Sensing of the Kinetic Parameters of Engineered Enzymes. Sensors. 2024; 24(8):2643. https://doi.org/10.3390/s24082643
Chicago/Turabian StyleDusíková, Adriána, Timea Baranová, Ján Krahulec, Olívia Dakošová, Ján Híveš, Monika Naumowicz, and Miroslav Gál. 2024. "Electrochemical Impedance Spectroscopy for the Sensing of the Kinetic Parameters of Engineered Enzymes" Sensors 24, no. 8: 2643. https://doi.org/10.3390/s24082643