DNA Electrochemical Behaviors, Recognition and Sensing by Combining with PCR Technique

Abstract
:Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
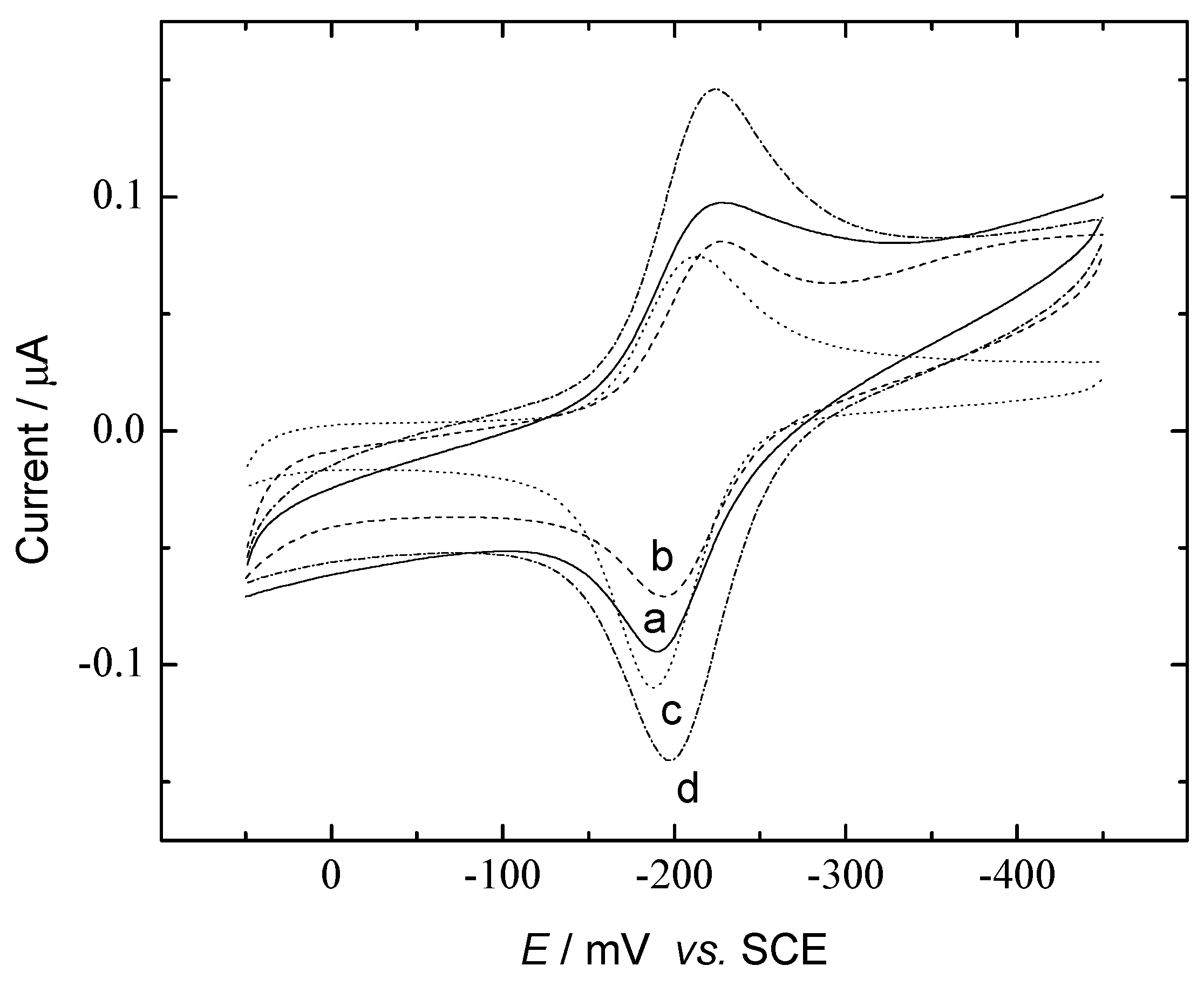{kind=link}
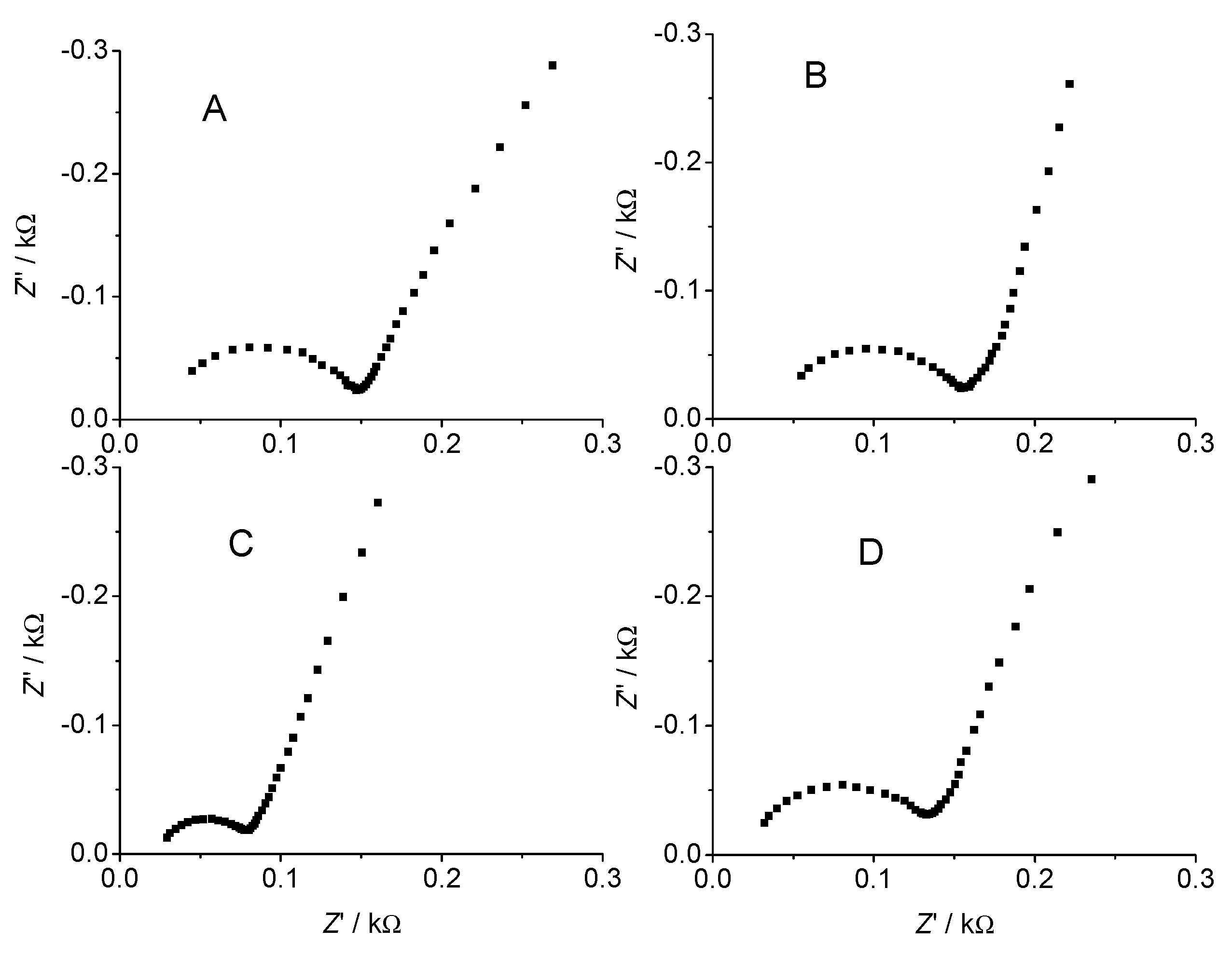{kind=link}
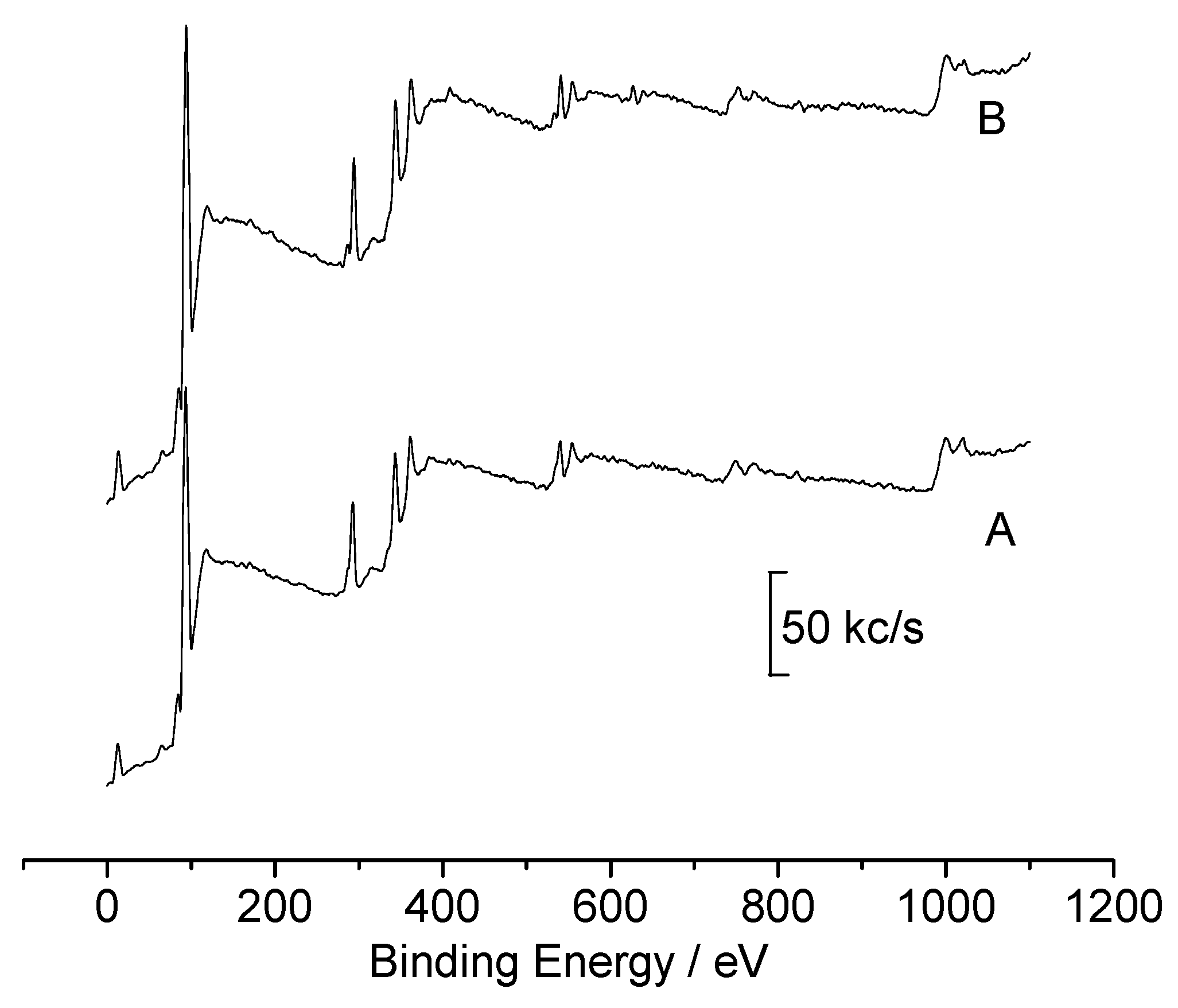{kind=link}
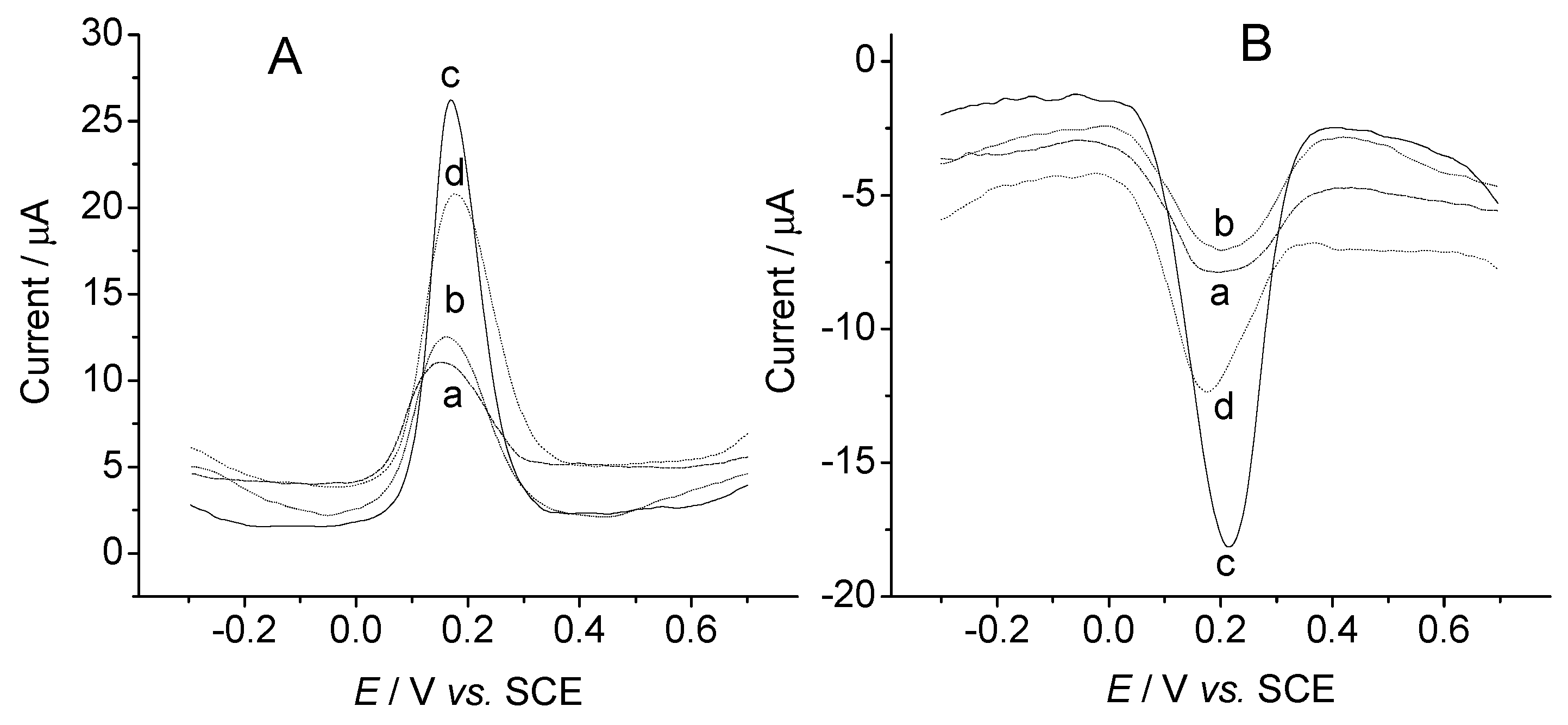{kind=link}
{kind=link}
{kind=link}
| DNA Sensors | Cited References | ||
|---|---|---|---|
| Electrode materials | Mercury | Dropping mercury | 11-13 |
| Hanging mercury drop | 14-24 | ||
| Mercury film | 25,26 | ||
| Mercury amalgam | 27-29 | ||
| Carbon-based | Carbon paste | 29-42 | |
| Graphite | 23,43-52 | ||
| Carbon fiber | 53-55 | ||
| Glassy carbon | 56-71 | ||
| Screen-printed | 72-75 | ||
| Pencil | 76 | ||
| Gold | 77-96 | ||
| Platinum | 97,98 | ||
| Silver | 99-101 | ||
| Indium tin oxide | 102,103 | ||
| C nanotube/nano-Au modified | 57,61,62 | ||
| Others | 104-106 | ||
| Probes Immobilization methods | Covalently attachment | 43,44,62,66,89,104 | |
| Adsorption | 21,32,39,46-48,52,57,65,67,68,80,86, 87,103,107 | ||
| Sol-gel embedding | 71 | ||
| Self-assembled monolayer | 77,78,82,84,85,96,108 | ||
| Indicators | Metal complexes | 14,19,21,22,28,29,31,34,35,39,42,45, 48,51,54,57,59-61,63,69,71,73,77,80, 82-86,88,90,92,97,102,107-110,113 | |
| Dyes and fluoresceins | 44,47,78,79,81,106,111 | ||
| Biomolecules (cytochrome c, enzyme) | 33,70,89,91,93,94,104,114, | ||
| Pharmic molecules | 11,12,17,18,20,30,40,43,49,52,58,62, 87,105 | ||
| Others | 37,66,107 | ||
| Usage | Sensors for DNA hybridization | ||
| Sensors for DNA damage | 15,16,21,32,51,73,115 | ||
| Sensors for screening toxicants | 112 | ||
Materials and Methods
Electrode Materials
Probe and Its Immobilization
Electroactive Indicators and Labels

Detection of DNA Hybridization or Damage
Discussion
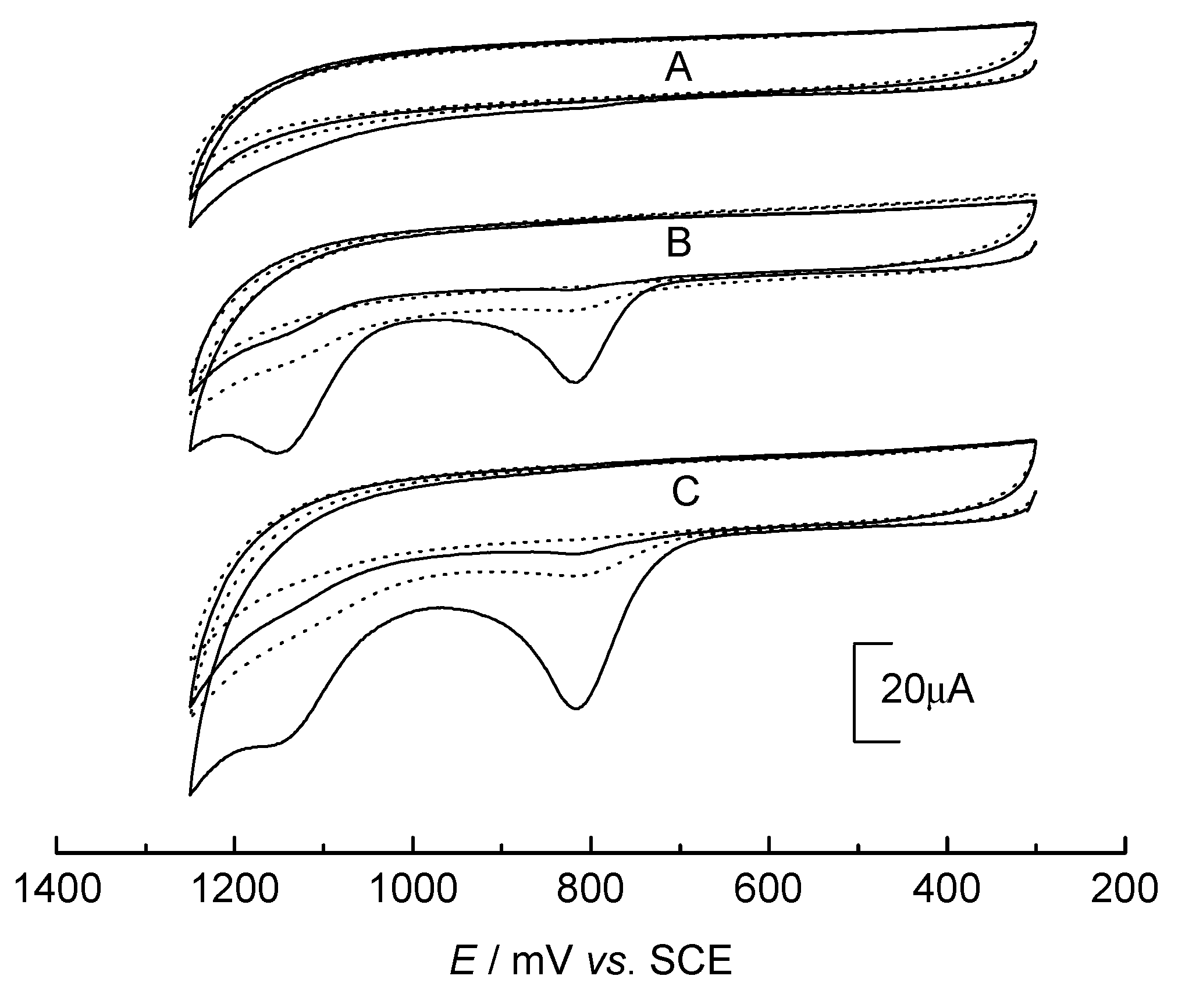
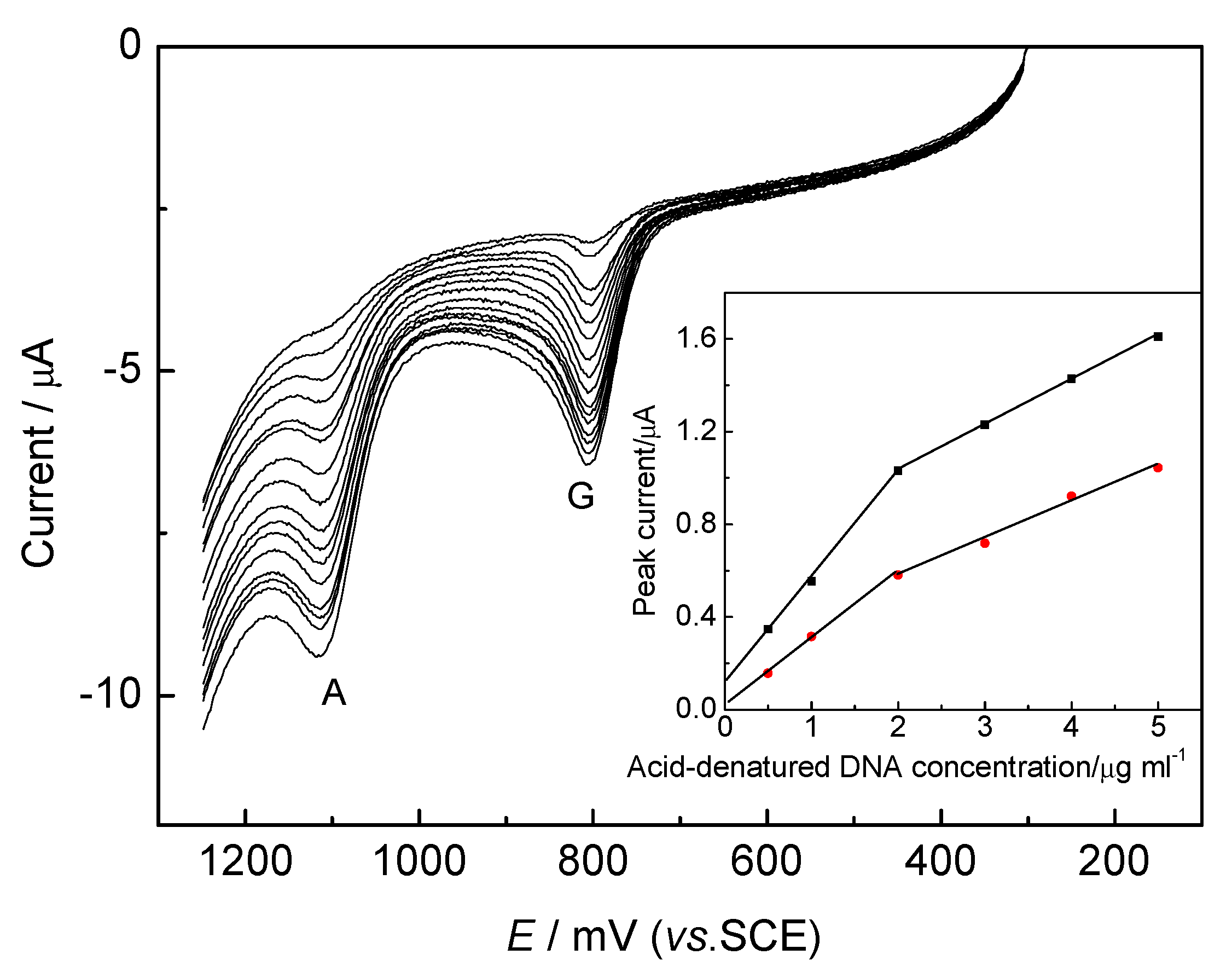
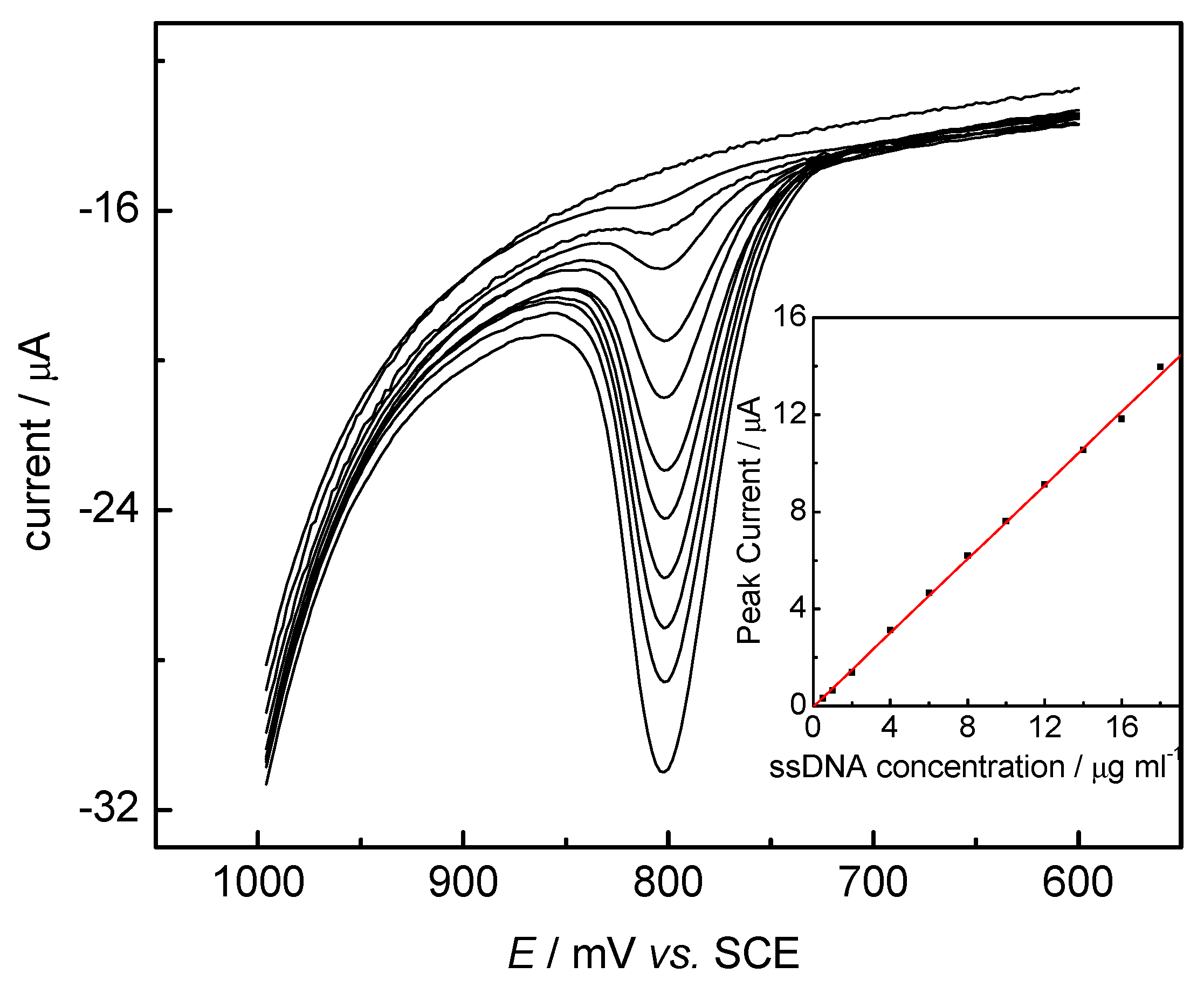
DNA Electrochemical Behaviors and Concentration Determination



DNA Sensors for Sequence Recognition

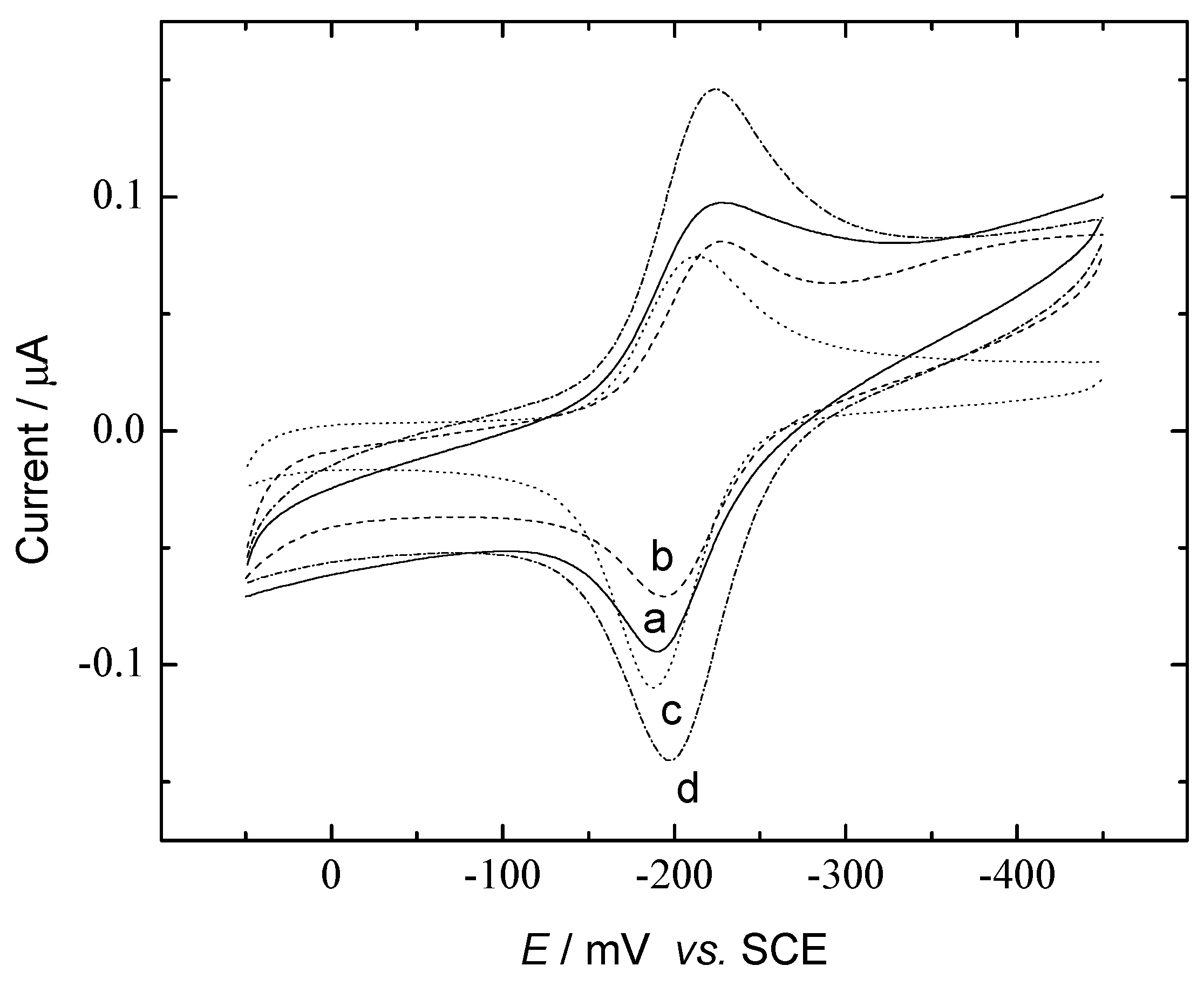
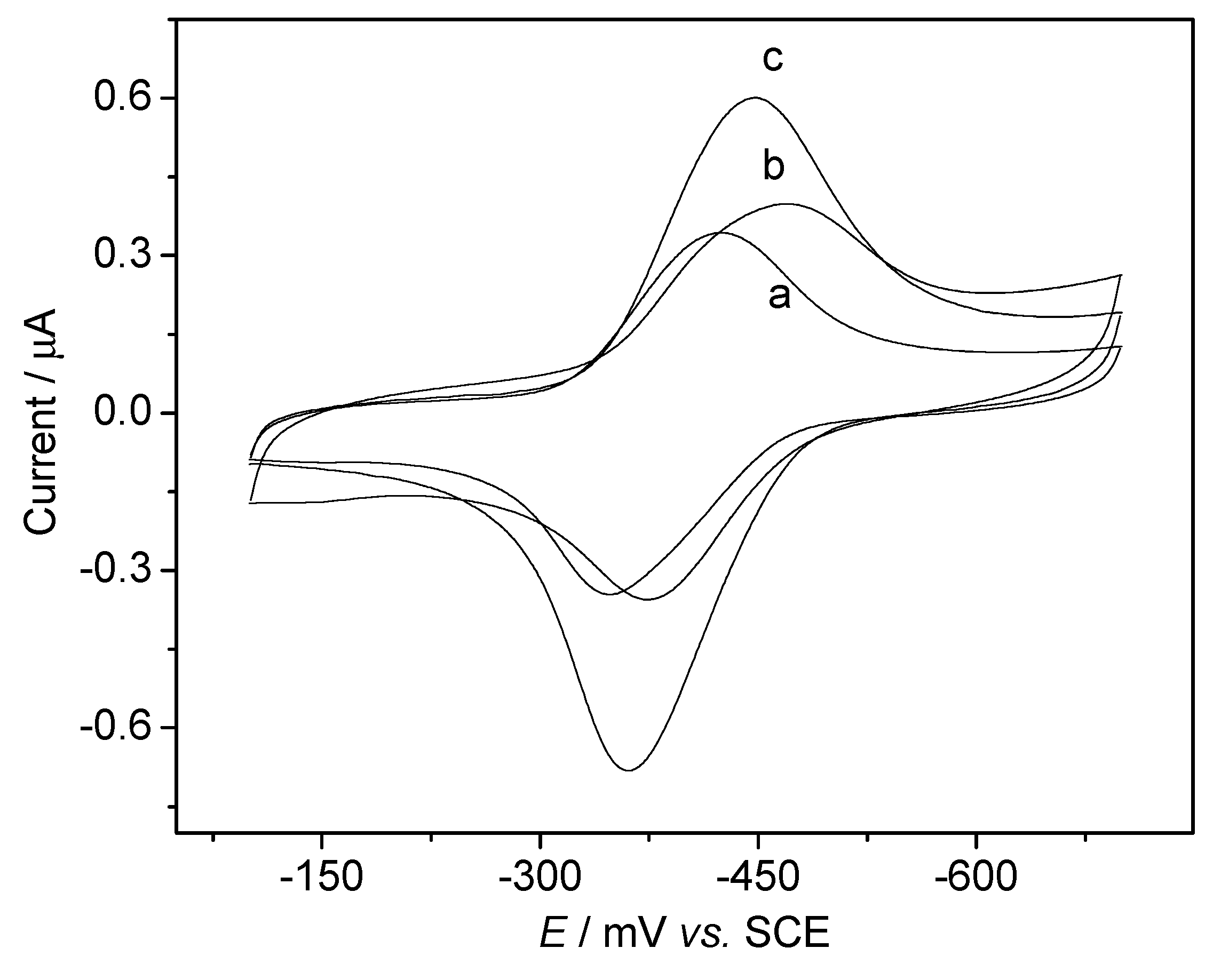
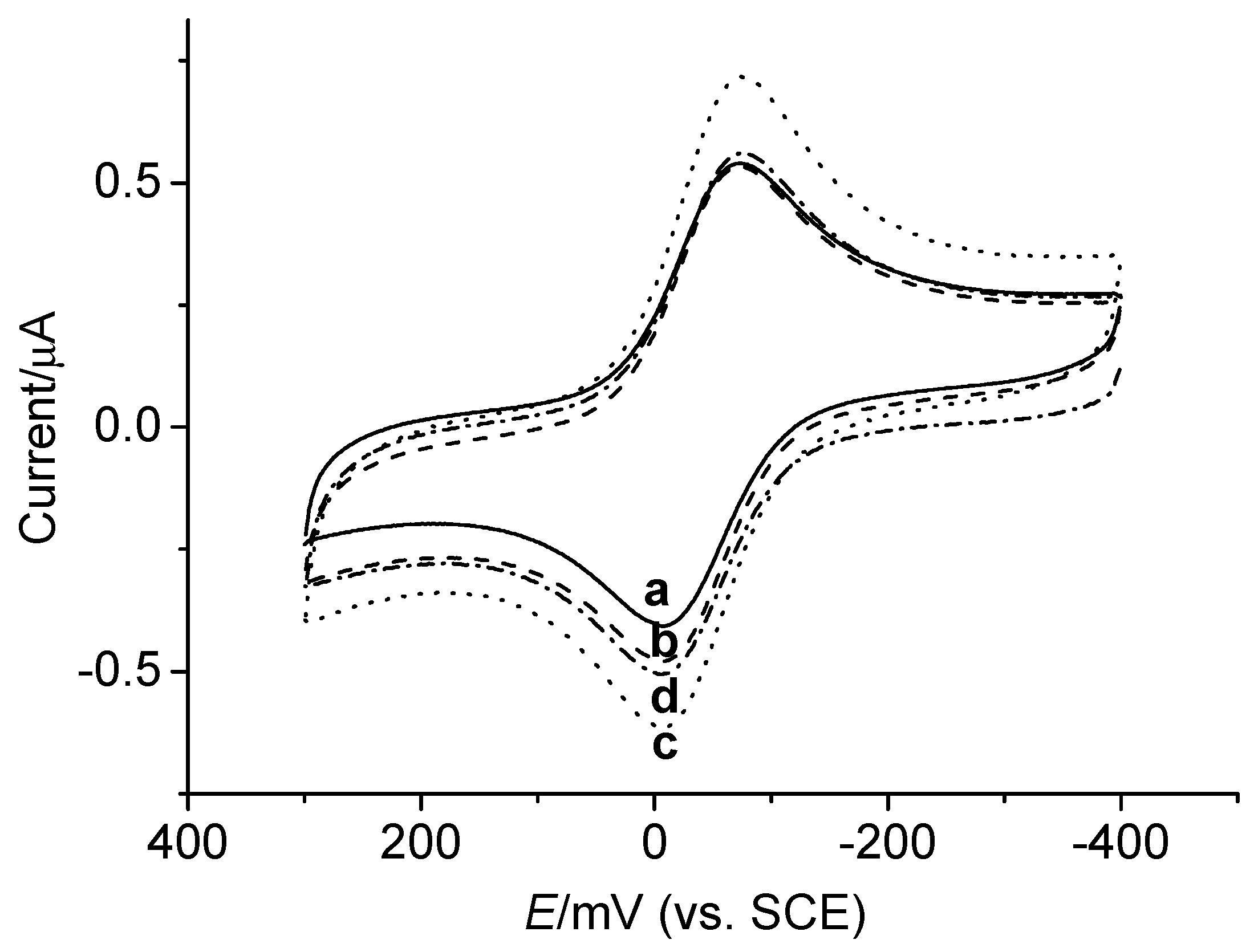
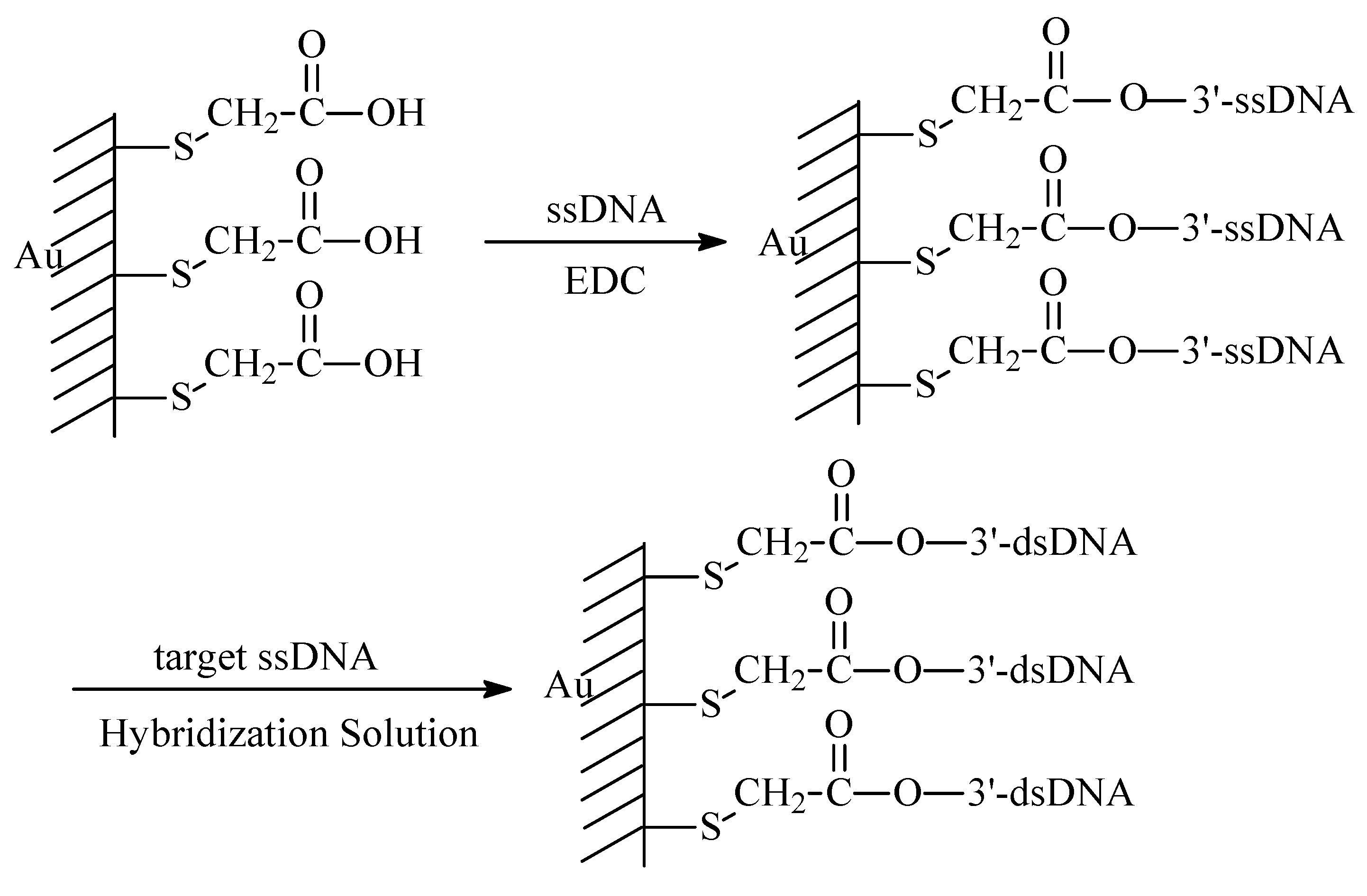
DNA Electrochemical Biosensors Combining With PCR Technique




Conclusions
Acknowledgements
References
- Paleček, E.; Davidson, J.N.; Cohn, W.E. (Eds.) Progress in Nucleic Acids Research and Molecular Biology; Vol. 9, Academic Press: New York, 1969; p. 31.
- Paleček, E.; Fojta, M. Anal. Chem. 2001, 73, 74A.
- Bakker, E.; Telting-Diaz, M. Anal. Chem. 2002, 74, 2781, and references therein.
- Wang, J. Biosen. & Bioelectron. 1998, 13, 757, and references therein.
- Paleček, E.; Fojta, M.; Tomschik, M; Wang, J. Biosen. & Bioelectron. 1998, 13, 621, and references therein.
- Wang, J. J. of Pharm. and Biomed. Anal. 1999, 19, 47, and references therein.
- Paleček, E. Talanta 2002, 56, 809, and references therein.
- Homs, W.C.I. Anal. Lett. 2002, 35, 1875, and references therein.
- Wang, J. Anal. Chim. Acta 2002, 469, 63, and references therein.
- Lu, X.J.; Ju, H.X. Chin. J. of Anal. Chem. 2003, 31, 110, and references therein.
- Zhang, J.L.; Fan, H.Z.; Pan, J.H. Acta Pharm. Sinica 1997, 32, 314.
- Wang, H.F.; Yang, P.; Li, Q.S.; Wu, Q. Chem. J. Chin. Univ. 1997, 18, 671.
- Wang, J.; Grant, D.H.; Ozsoz, M.; Cai, X.H.; Tian, B.M.; Fernandes, J.R. Anal. Chim. Acta 1997, 349, 77.
- Li, M.X.; Li, N.Q. Chem. J. Chin. Univ. 2001, 22, 1230.
- Fojta, M.; Staňková, V.; Paleček, E.; Koscielniak, P.; Mitáš, J. Talanta 1998, 46, 155. [PubMed]
- Jelen, F.; Tomschik, M.; Paleček, E. J. Electroanal. Chem. 1997, 423, 141.
- Jelen, F.; Erdem, A.; Paleček, E. Bioelectrochem. 2002, 55, 165.
- Teijeiro, C.; Perez, P.; Marín, D.; Paleček, E. Bioelectrochem. and Bioenerg. 1995, 38, 77.
- Kizek, R.; Havran, L.; Fojta, M.; Paleček, E. Bioelectrochem. 2002, 55, 119.
- Zhang, H.M.; Li, N.Q. J. of Pharm. and Biomed. Anal. 2000, 22, 67.
- Fojta, M.; Harran, L.; Kubiěárová, T.; Paleček, E. Bioelectrochem. 2002, 55, 25.
- Fojta, M.; Kubiěárová, T.; Paleček, E. Biosen. & Bioelectron. 2000, 15, 107.
- Tomschik, M.; Jelen, F.; Harran, L.; Trnková, L.; Nielsen, P.E.; Paleček, E. J. Electroanal. Chem. 1999, 476, 71.
- Jelen, F.; Fojta, M.; Paleček, E. J. Electroanal. Chem. 1997, 427, 49.
- Wu, J.T.; Zhou, J.Z.; Huang, Y.; Luo, J.; Lin, Z.H. Chin. J. Anal. Chem. 1998, 26, 819.
- Wu, J.T.; Huang, Y.; Zhou, J.Z.; Luo, J.; Lin, Z.H. Bioelectrochem. and Bioenerg. 1997, 44, 151.
- Jelen, F.; Yosypchuk, B.; Kourilová, A.; Novotný, L.; Paleček, E. Anal. Chem. 2002, 74, 4788. [PubMed]
- Paleček, E.; Kizek, R.; Havran, L.; Billova, S.; Fojta, M. Anal. Chim. Acta. 2002, 469, 73.
- Paleček, E.; Fojta, M.; Jelen, F. Bioelectrochem. 2002, 55, 85.
- Luo, J.W.; Zhang, M.; Zhang, R.Y.; Pang, D.W. J. Anal. Sci. 2002, 18, 1.
- Millan, K. M.; Saraullo, A.; Mikkelsen, S.R. Anal. Chem. 1994, 66, 2943. [PubMed]
- Wang, J.; Rivas, G.; Ozsoz, M.; Grant, D.H.; Cai, X.H.; Parrado, C. Anal. Chem. 1997, 69, 1457. [PubMed]
- Ikebukuro, K.; Kohiki, Y.; Sode, K. Biosen. & Bioelectron. 2002, 17, 1075.
- Wang, J.; Rivas, G.; Cai, X.H.; Chicharro, M.; Parrado, C.; Dontha, N.; Begleiter, A.; Mowat, M.; Paleček, E.; Nielsen, P.E. Anal. Chim. Acta 1997, 344, 111.
- Wang, J.; Rivas, G.; Parrado, C.; Cai, X.H.; Flair, M.N. Talanta 1997, 44, 2003. [PubMed]
- Lucarelli, F.; Kicela, A.; Palchetti, I.; Marrazza, G.; Mascini, M. Bioelectrochem. 2002, 58, 113.
- Gil, E.S.; Kubota, L.T. Bioelectrochem. 2000, 51, 145.
- Wang, J.; Rivas, G.; Fernandes, J.R.; Paz, J.L.L.; Jiang, M; Waymire, R. Anal. Chim. Acta. 1998, 375, 197.
- Motonaka, J.; Mishima, Y.; Maruyama, K.; Minagawa, K.; Ikeda, S. Sensors and Actuators B. 2000, 66, 234.
- Wang, J.; Ozsoz, M.; Cai, X.H.; Rivas, G.; Shiraishi, H.; Grant, D.H.; Chicharro, M.; Fernandes, J.; Paleček, E. Bioelectrochem. and Bioenerg. 1998, 45, 33.
- Wang, J.; Kawde, A.N. Electrochem. Comm. 2002, 4, 349.
- Wang, J.; Rivas, G.; Cai, X.H.; Dontha, N.; Shiraishi, H.; Luo, D.B.; Valera, F.S. Anal. Chim. Acta 1997, 337, 41.
- Sun, X.Y.; Xu, C.; Liu, S.H.; He, P.G.; Fang, Y.Z. Chem. J. Chin. Univ. 1998, 19, 1393.
- Sun, X.Y.; He, P.G.; Xu, C.; Liu, X.P.; Liu, S.H.; Fang, Y.Z. J. Anal. Sci. 1999, 15, 279.
- Xu, C.; Cai, H.; He, P.G.; Fang, Y.Z. Chem. J. Chin. Univ. 2001, 22, 1492.
- Zhou, J.Z.; Wu, L.L.; Luo, J.; Lin, Z.H. Electrochem. 1999, 5, 186.
- Xu, C.; He, P.G.; Fang, Y.Z. Chem. J. Chin. Univ. 2000, 21, 1187.
- Xu, C.; He, P.G.; Fang, Y.Z. Anal. Chim. Acta. 2000, 411, 31.
- Hashimoto, K.; Ito, K.; Ishimori, Y. Anal. Chim. Acta 1994, 286, 219.
- Wang, J.; Zhang, X.J.; Parrado, C.; Rivas, G. Electrochem. Comm. 1999, 1, 197.
- Korbut, O.; Bučková; Tarapčik, P.; Labuda, J.; Gründler, P. J. Electroanal. Chem. 2001, 506, 143.
- Brabec, V. Electrochim. Acta 2000, 45, 2929.
- Wang, J.; Cai, X.H.; Fernandes, J.R.; Grant, D.H.; Ozsoz, M. J. Electroanal. Chem. 1998, 441, 167.
- Hassmann, J.; Misch, A.; Schülein, J.; Krause, J.; Graβl, B.; Müller, P.; Bertling, W.M. Biosen. & Bioelectron. 2001, 16, 857.
- Schülein, J.; Graβl, B.; Krause, J.; Schulze, C.; Kugler, C.; Müller, P.; Bertling, W.M.; Hassmann, J. Talanta 2002, 56, 875.
- Cheng, X.D.; Wang, H.E.; Sun, J.; Wen, X.M.; Chen, Z.D.; Hou, S.F.; Sun, Q.S. J. Jining Med. College 1999, 22, 7.
- Miao, Q.; Jin, B.K.; Lin, X.Q. Chem. J. Chin. Univ. 2000, 21, 27.
- Bao, X.Y.; Chen, J.G.; Chen, X.; Dang, Y.L. Chin. J. Anal. Lab. 2001, 20, 1.
- Carter, M.T.; Rodriguez, M.; Bard, A.J. J. Am. Chem. Soc. 1989, 111, 8901.
- Rodriguez, M.; Bard, A.J. Anal. Chem. 1990, 62, 2658. [PubMed]
- Lin, X.Q.; Miao, Q.; Jin, B.K. Chin. Chem. Lett. 1999, 10, 157.
- Cai, H.; Cao, X.; Jiang, Y.; He, P.G.; Fang, Y.Z. Anal. Bioanal. Chem. 2003, 375, 287. [PubMed]
- Xu, C.; Cai, H.; He, P.G.; Fang, Y.Z. Analyst 2001, 126, 62. [PubMed]
- Lee, T.Y.; Shim, Y.B. Anal. Chem. 2001, 73, 5629.
- Brett, C.M.A.; Brett, A.M.O.; Serrano, A.H.P. Electrochim. Acta 1999, 44, 4233.
- Li, M.X.; Li, N.Q.; Gu, Z.N.; Zhou, X.H.; Sun, Y.L.; Wu, Y.Q. Microchem. J. 1999, 61, 32.
- Siontorou, C.G.; Brett, A.M.O.; Nikolelis, D.P. Talanta. 1996, 43, 1137.
- Pedano, M.L.; Rivas, G.A. Biosen. & Bioelectron. 2003, 18, 269.
- Mahadevan, S.; Palaniandavar, M. Inorg. Chim. Acta. 1997, 254, 291.
- Chen, S.M.; Chen, S.V. Electrochim. Acta. 2003, 48, 513.
- Liu, S.Q.; Xu, J.J.; Chen, H.Y. Bioelectrochem. 2002, 57, 149.
- Marrazza, G.; Tombelli, S.; Mascini, M.; Manzoni, A. Clin. Chim. Acta 2001, 307, 241. [PubMed]
- Buckova, M.; Labuda, J.; Sandula, J.; Krizkova, L.; Stepanek, I.; Durackova, Z. Talanta 2002, 56, 939. [PubMed]
- Azek, F.; Grossiord, C.; Joannes, M.; Limoges, B.; Brossier, P. Anal. Biochem. 2000, 284, 107. [PubMed]
- Wang, J.; Cai, X.H.; Rivas, G.; Shiraishi, H.; Dontha, N. Biosen. & Bioelectron. 1997, 12, 587.
- Wang, J.; Kawde, A.N. Anal. Chim. Acta 2001, 431, 219.
- Zhou, J.Z.; Wu, L.L.; Dong, L.Q.; Lin, Z.Y.; Yan, J.W.; Dong, P.; Bao, Y.; Lin, Z.H. Electrochem. 2001, 7, 276.
- Liu, J.; Li, Q.W.; Luo, G.A.; Sun, H.W.; Feng, J. J. Instru. Anal. 2002, 21, 13.
- Zhou, J.H.; Jiang, H.J.; Feng, Y.Y.; Yang, H.; Xing, W.; Lu, T.H. Chin. J. Appl. Chem. 2001, 18, 994.
- Zhao, Y.D.; Pang, D.W.; Wang, Z.L.; Cheng, J.K.; Qi, Y.P. J. Electroanal. Chem. 1997, 431, 203.
- Hashimoto, K.; Ito, K.; Ishimori, Y. Sensors and Actuators B 1998, 46, 220.
- Zhao, Y.D.; Pang, D.W.; Hu, S.; Wang, Z.L.; Cheng, J.K.; Dai, H.P. Talanta 1999, 49, 751. [PubMed]
- Pang, D.W.; Adruna, H.D. Anal. Chem. 1998, 70, 3162. [PubMed]
- Maruyama, K.; Mishima, Y.; Minagawa, K.; Motonaka, J. J. Electroanal. Chem. 2001, 510, 96.
- Takenaka, S.; Uto, Y.; Takagi, M.; Kondo, H. Chem. Lett. 1998, 989.
- Zhao, Y.D.; Pang, D.W.; Zhang, M.; Cheng, J.K. F’ J. Anal. Chem. 1999, 363, 708.
- Yang, M.; Yau, H. C.M.; Chan, H.L. Langmuir 1998, 14, 6121.
- Zhao, Y.D.; Pang, D.W.; Hu, S.; Wang, Z.L.; Cheng, J.K.; Qi, Y.P.; Dai, H.P.; Mao, B.W.; Tian, Z.Q.; Luo, J.; Lin, Z.H. Anal. Chim. Acta. 1999, 388, 93.
- Xu, D.K.; Huang, K.; Liu, Z.H.; Liu, Y.Q.; Ma, L.R. Electroanlysis 2001, 13, 882.
- Maruyama, K.; Motonaka, J.; Mishima, Y.; Matsuzaki, Y.; Nakabayashi, I.; Nakabayashi, Y. Sensors and Actuators B 2001, 76, 215.
- Liu, H.H.; Lu, J.L.; Zhang, M.; Pang, D.W.; Abruna, H.D. J. Electroanal. Chem. 2003, 544, 93.
- Zhang, Q.L.; Liu, J.G.; Liu, j.; Xue, G.Q.; Li, H.; Liu, J.Z.; Zhou, H.; Qu, L.H.; Ji, L.N. J. Inorg. Biochem. 2001, 85, 291. [PubMed]
- Shao, Y.; Jin, Y.D.; Dong, S.J. Electrochem. Comm. 2002, 4, 773.
- Lisdat, F.; Ge, B.; Scheller, F.W. Electrochem. Comm. 1999, 1, 65.
- Neumann, E.; Tonsing, K.; Siemens, P. Bioelectrochem. 2000, 51, 125.
- Kertesz, V.; Whitemore, N.A.; Chambers, J.Q.; McKinney, M.S.; Baker, D.C. J. Electroanal. Chem. 2000, 493, 28.
- Mishima, Y.; Motonaka, J.; Maruyama, K.; Minagawa, K.; Ikeda, S. Sensors and Actuators B 2000, 65, 340.
- Moser, I.; Schalkhammer, T.; Pittner, F.; Urban, G. Biosen. & Bioelectron. 1997, 12, 729.
- Trnkova, L. Talanta 2002, 56, 887. [PubMed]
- Trnkova, L.; Kizek, R.; Dracka, O. Bioelectrochem. 2002, 55, 131.
- Fan, C.H.; Song, H.Y.; Hu, X.F.; Li, G.X.; Zhu, J.Q.; Xu, X.X.; Zhu, D.X. Anal. Biochem. 1999, 271, 1. [PubMed]
- Johnson, D.H.; Throp, H.H. J. Phys. Chem. 1996, 100, 13837.
- Xu, J.Z.; Zhu, J.J.; Huang, Q.; Chen, H.Y. Electrochem. Comm. 2001, 3, 665.
- Yang, Q.J.; Xu, D.K.; Jiang, X.P.; He, W.; Liu, Z.H.; Wang, Y.; Ma, L.R. Chin. J. Anal. Chem. 2002, 30, 56.
- Hu, J.B.; Shang, J.; Li, Q.L. Chem. J. Chin. Univ. 2001, 22, 749.
- Pike, A.R.; Lie, L.H.; Eagling, R.A.; Ryder, L.C.; Patole, S.N.; Connolly, B.A.; Horrocks, B.R.; Houlton, A. Angew. Chem. Int. Ed. 2002, 41, 615.
- Fang, P.F.; Zhao, Y.D.; Pang, D.W.; Chen, Y.Y. Acta Sci. Natu. Univ. Peking 2001, 37, 153.
- Zhao, Y.D.; Pang, D.W.; Zhang, M.; Cheng, J.K.; Dai, H.P. Chem. J. Chin. Univ. 2001, 22, 744.
- Zhang, L.R.; Zhu, J.J.; Zhao, G.C.; Chen, H.Y. Chem. J. Chin. Univ. 1998, 19, 1363.
- Stemp, E. D.A.; Holmlin, R.E.; Barton, J.K. Inorg. Chim. Acta 2000, 297, 88.
- Meric, B.; Kerman, K.; Ozkan, D.; Kara, P.; Erensoy, S.; Akarca, U.S.; Mascini, M.; Ozsoz, M. Talanta 2002, 56, 837. [PubMed]
- Lucarelli, F.; Palchetti, I.; Marrazza, G.; Mascini, A. Talanta 2002, 56, 949. [PubMed]
- Fojta, M.; Havran, L.; Kizek, R.; Billova, S. Talanta 2002, 56, 867. [PubMed]
- Wang, J.; Xu, D.K.; Erdem, A.; Polsky, R.; Salazar, M.A. Talanta 2002, 56, 931. [PubMed]
- Kawanishi, S.; Hiraku, Y.; Oikawa, S. Mutation Res. 2001, 488, 65. [PubMed]
- Henke, L.; Krull, U.J. Canadian J. of Anal. Sci. and Spec. 1999, 44, 61, and references therein.
- Wang, B.Q.; Cheng, G.J.; Dong, S.J. Chin. J. of Anal. Chem. 1999, 27, 982, and references therein.
- Walcarius, A. Electroanalysis 1998, 10, 1217, and references therein.
- Alegret, S. Analyst 1996, 121, 1751, and references therein.
- Tess, M.E.; Cox, J.A. J. of Pharm. and Biomed. Anal. 1999, 19, 55.
- Lin, J.; Brown, C.W. Trac-Trends in Anal. Chem. 1997, 16, 200.
- Tien, H.T.; Barish, R.H.; Gu, L.Q.; Ottova, A.L. Anal. Sci. 1998, 14, 3.
- Wink, T.; van Zuilen, S.J.; Bult, A.; van Bennekom, W.P. Analyst 1997, 122, R43.
- Paleček, E.; Jelen, F. Critical Rev. in Anal. Chem. 2002, 32, 261, and references therein.
- Gu, J.Y.; Lu, X.J.; Ju, H.X. Electroanalysis 2002, 14, 949.
- Ju, H.X.; Ye, Y.K.; Zhu, Y.L. Bioconj. Chem. in reviewed.
- Paleček, E. Electroanalysis 1996, 8, 7.
- Wang, J.; Cai, X. H.; Jonsson, C.; Balakrishnan, M. Electronalysis 1996, 8, 20.
- Paleček, E. Bioelectrochem. Bioenerg. 1988, 20, 171.
- Paleček, E. Anal. Biochem. 1988, 170, 421.
- Paleček, E.; Jelen, F.; Teijeiro, C.; Fucik, V.; Jovin, T. M. Anal. Chim. Acta 1993, 272, 175.
- Jelen, F.; Tomschik, M.; Paleček, E. J. Electroanal. Chem. 1997, 423, 141.
- Wang, H.S.; Ju, H.X.; Chen, H.Y. Electroanalysis 2001, 13, 1105.
- Wang, H.S.; Ju, H.X.; Chen, H.Y. Anal. Chim. Acta 2002, 461, 243.
- Wang, H.S.; Ju, H.X.; Chen, H.Y. Electroanalysis 2002, 14, 1615.
- Paleček, E.; Billová, S.; Havran, L.; Kizek, R.; Miculková, A.; Jelen, F. Talanta 2002, 56, 919.
- Souteyrand, E.; Cloarec, J.; Martin, J.R.; Wilson, C.; Lawrence, I.; Mikkelsen, S.; Lawrence, M.F. J. Phys. Chem. B 1997, 101, 2980.
- Napier, M.E.; Loomis, C.R.; Sistare, M.F.; Kim, J.; Eckhardt, A.E.; Thorp, H.H. Bioconjugate Chem. 1997, 8, 906.
- Welch, T.W.; Ciftan, S.A.; White, P.S.; Thorp, H.H. Inorg. Chem. 1997, 36, 4812. [PubMed]
- Paleček, E.; Fojta, M. Anal. Chem. 2001, 73, 74A.
- Ye, Y.K.; Zhao, J.H.; Yan, F.; Zhu, Y.L.; Ju, H.X. Biosen. & Bioelectron. in press.
- Ju, H.X.; Ye, Y.K.; Zhao, J.H.; Zhu, Y.L. Anal. Biochem. 2003, 312, 255.
- Mishima, Y.; Motonaka, J.; Ikeda, S. Anal. Chim. Acta. 1997, 345, 45.
- Sample Availability: Available from the authors.
© 2003 by MDPI (http://www.mdpi.net). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ye, Y.; Ju, H. DNA Electrochemical Behaviors, Recognition and Sensing by Combining with PCR Technique. Sensors 2003, 3, 128-145. https://doi.org/10.3390/s30600128
Ye Y, Ju H. DNA Electrochemical Behaviors, Recognition and Sensing by Combining with PCR Technique. Sensors. 2003; 3(6):128-145. https://doi.org/10.3390/s30600128
Chicago/Turabian StyleYe, Yongkang, and Huangxian Ju. 2003. "DNA Electrochemical Behaviors, Recognition and Sensing by Combining with PCR Technique" Sensors 3, no. 6: 128-145. https://doi.org/10.3390/s30600128
APA StyleYe, Y., & Ju, H. (2003). DNA Electrochemical Behaviors, Recognition and Sensing by Combining with PCR Technique. Sensors, 3(6), 128-145. https://doi.org/10.3390/s30600128