A Fluorimetric Sensor for Detection of One Living Cell

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and methods
2.1 Chemicals
2.2 Plant cultures
Tobacco suspension
Spruce embryos
2.3 Cell counting
2.4 Cell viability microscope assay
2.5 Localization of esterases in one cell
2.6 Esterase assay
Cell extract
BY-2 Protoplasts
2.7 Native PAGE
2.8 Data analysis
3. Results and discussion
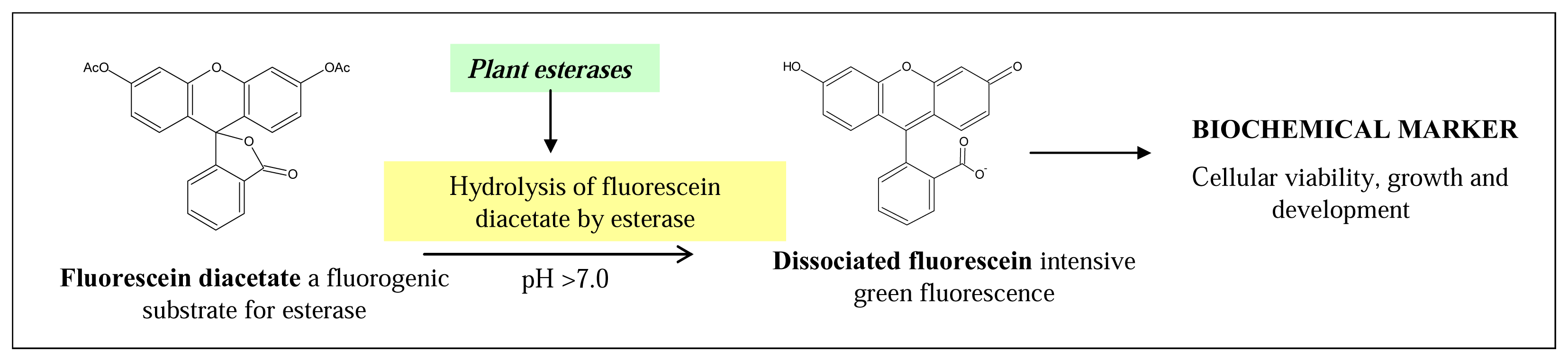
3.1 Direct detection of fluorescein
3.2 Fluorimetric detection of plant esterase activity
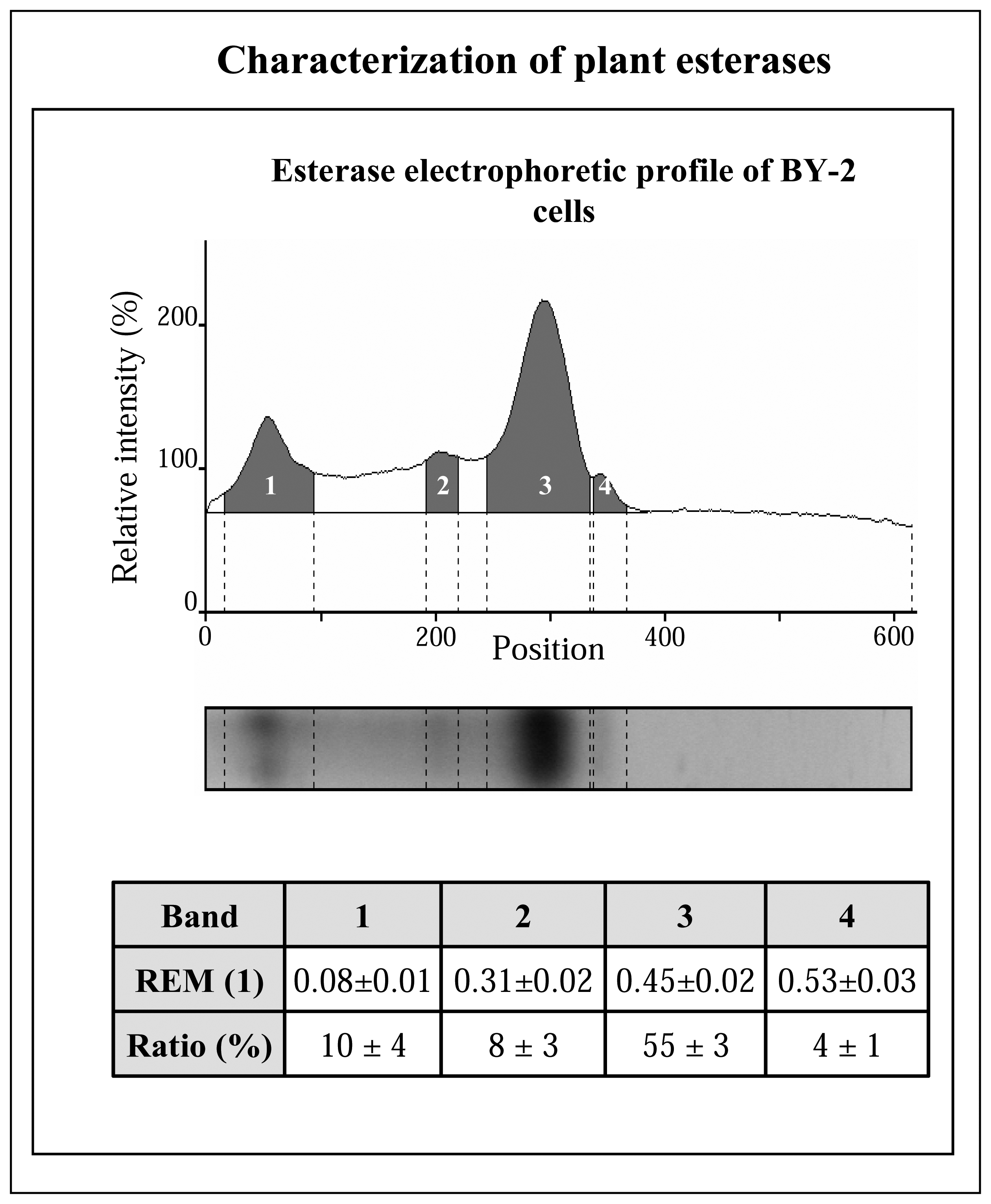
3.2.1 Native electrophoresis of esterases
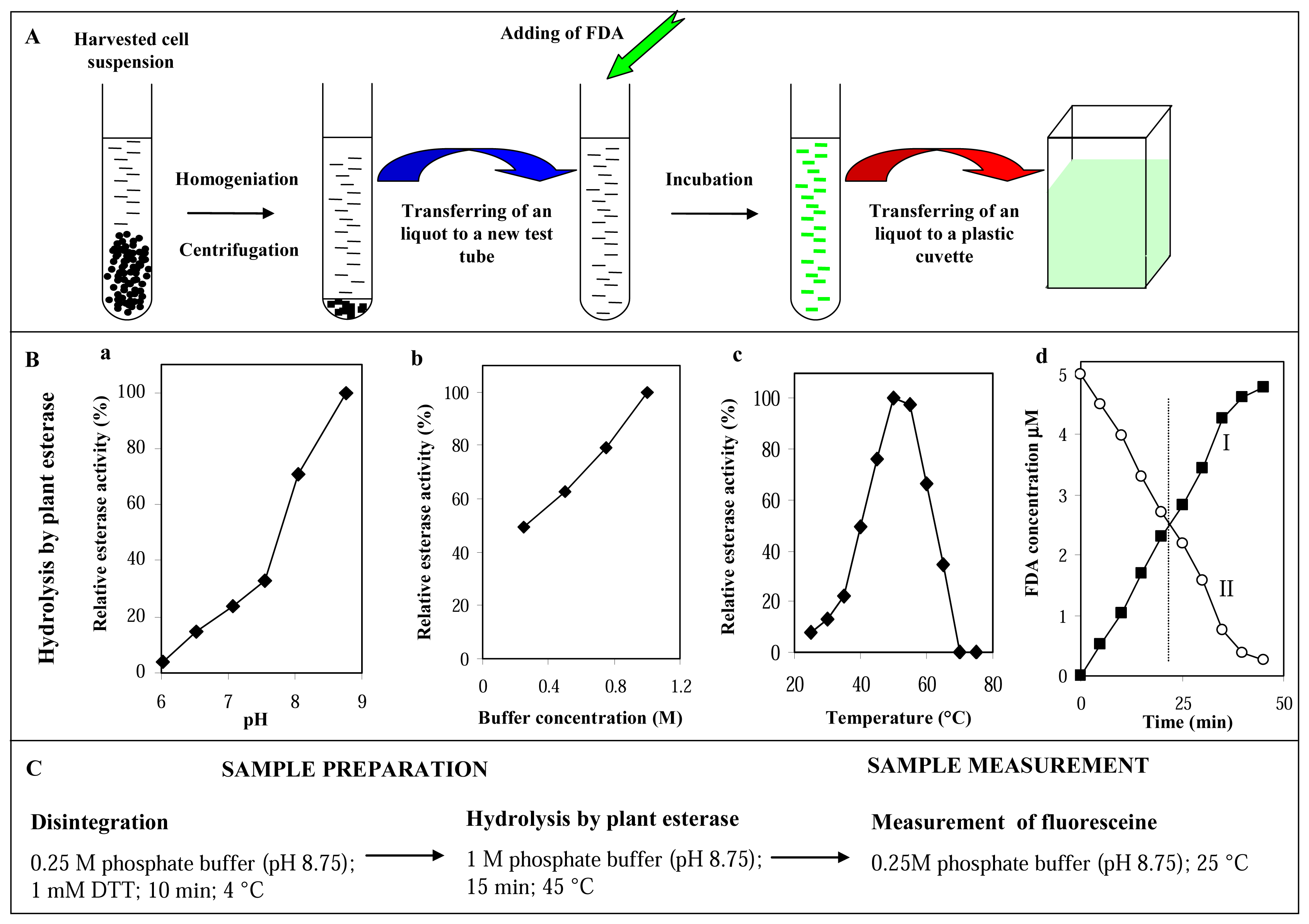
3.2.2 Optimisation of the plant esterase determination
Step 1: disintegration of cells
Step 2: hydrolysis of fluorescein diacetate by plant esterases
Influence of pH and concentration of phosphate buffer
Influence of temperature
Influence of reaction time on detection of plant esterases and spontaneous hydrolysis of FDA
Step 3: measurement of fluorescein after PE hydrolysis of FDA
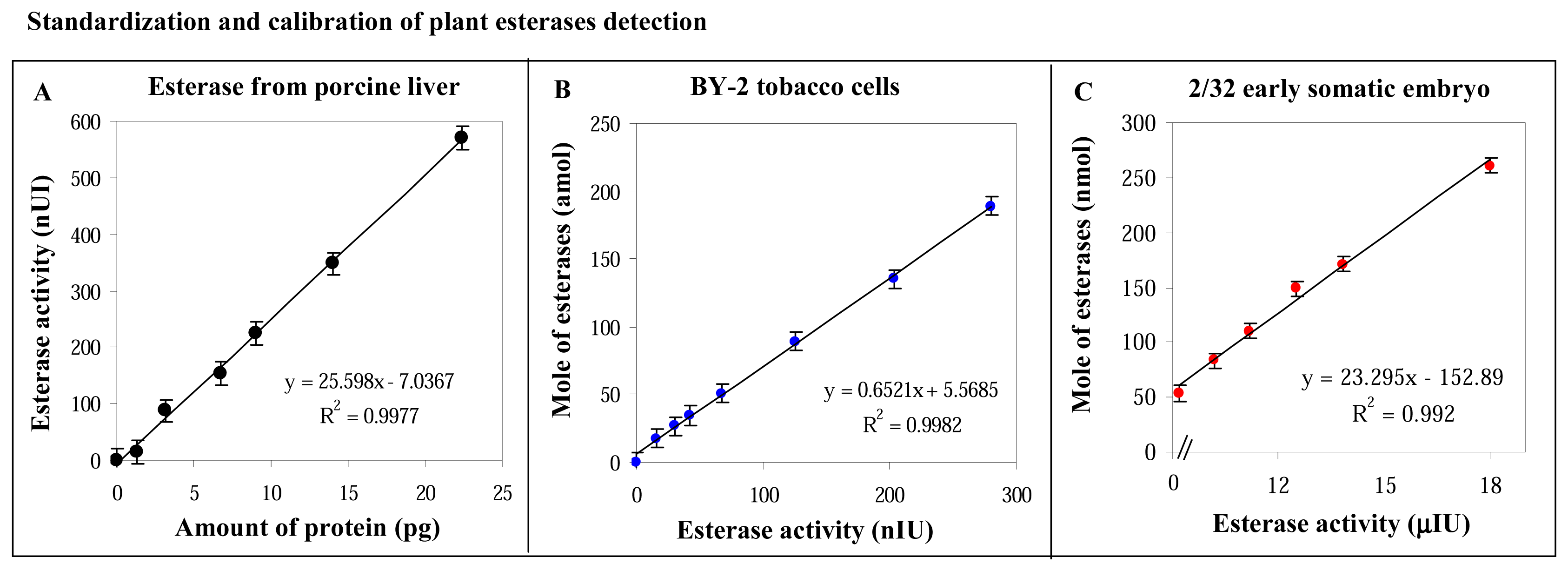
3.3 Standardization and calibration of the fluorimetry detection of plant esterases
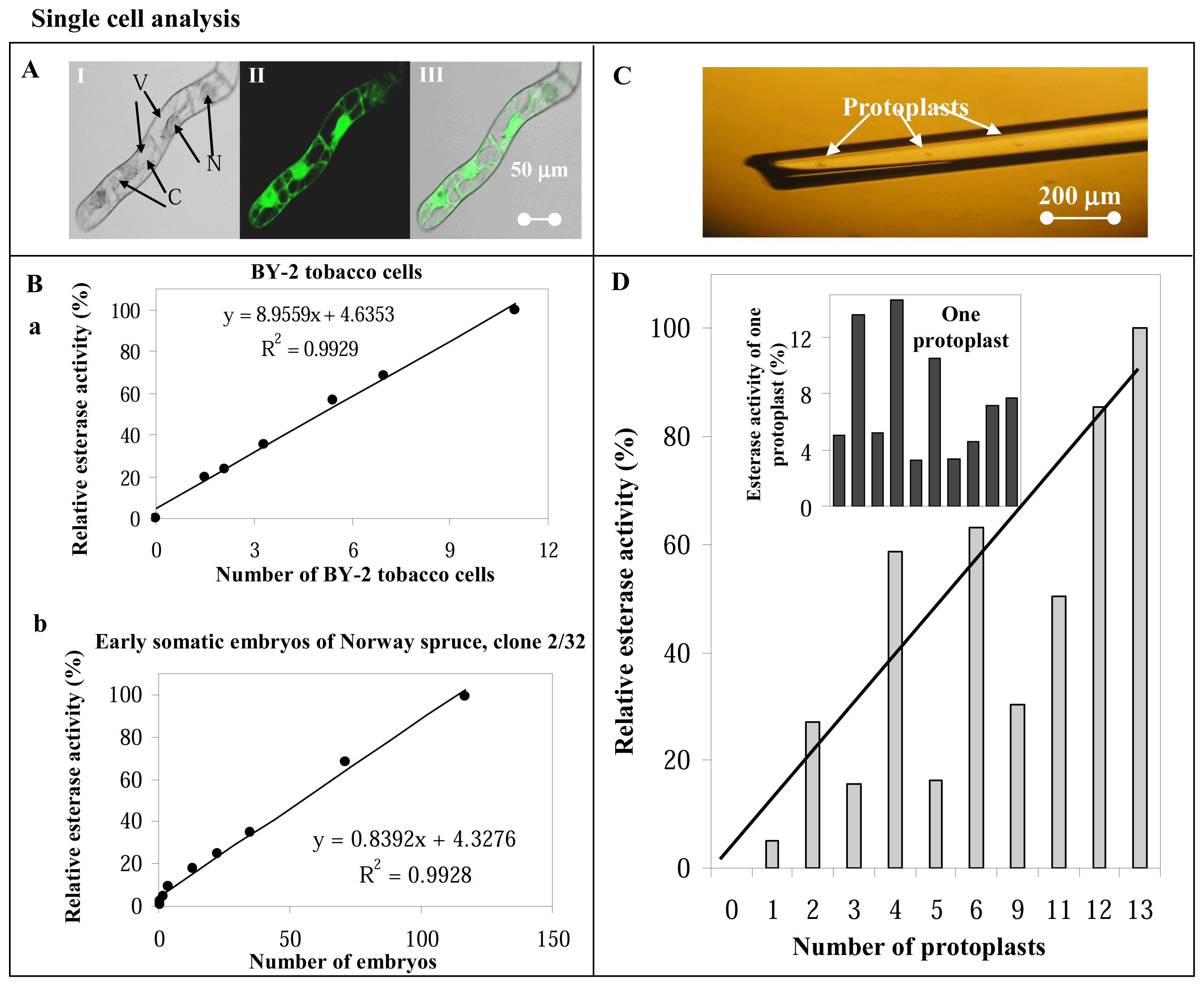
3.4 Single cell analysis
Localization of esterase activity in BY-2 tobacco cell
Fluorimetric detection of esterase activity in cell extracts of BY-2 tobacco cells and spruce embryos (clone 2/32)
Fluorimetric detection of esterase activity in one protoplast
4. Conclusions
Acknowledgments
References
- Lu, X.; Huang, W. -H.; Wang, Z.-L.; Cheng, J.-K. Recent developments in single-cell analysis. Anal. Chim. Acta 2004, 510, 127–138. [Google Scholar]
- Kuo, H.C.; Cheng, C.F.; Clark, R.B.; Lin, J.J.C.; Lin, J.L.C.; Hoshijima, M.; Nguyen-Tran, V.T.B.; Gu, Y.S.; Ikeda, Y.; Chu, P.H.; Ross, J.; Giles, W.R.; Chien, K.R. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I-to and confers susceptibility to ventricular tachycardia. Cell 2001, 107, 801–813. [Google Scholar]
- Roper, S.N.; Yachnis, A.T. Cortical dysgenesis and epilepsy. Neuroscientist 2002, 8, 356–371. [Google Scholar]
- Oguri, S. Electromigration methods for amino acids, biogenic amines and aromatic amines. J. Chrom. B 2000, 747, 1–19. [Google Scholar]
- Ginsberg, S.D.; Che, S.L. RNA amplification in brain tissues. Neurochem. Res. 2002, 27, 981–992. [Google Scholar]
- Mawrin, C.; Kirches, E.; Dietzmann, K. Single-cell analysis of mtDNA in amyotrophic lateral sclerosis: Towards the characterization of individual neurons in neurodegenerative disorders. Pathol. Res. Pract. 2003, 199, 415–418. [Google Scholar]
- Trushina, E.; Heldebrant, M.P.; Perez-Terzic, C.M.; Bortolon, R.; Kovtun, I.V.; Badger, J.D.; Terzic, A.; Estevez, A.; Windebank, A.J.; Dyer, R.B.; Yao, J.; McMurray, C.T. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington's disease. Proc. Natl. Acad. Sci. USA 2003, 100, 12171–12176. [Google Scholar]
- Lin, Y.W.; Chiu, T.C.; Chang, H.T. Laser-induced fluorescence technique for DNA and proteins separated by capillary electrophoresis. J. Chrom. B 2003, 793, 37–78. [Google Scholar]
- Vilkner, T.; Janasek, D.; Manz, A. Micro total analysis systems. Recent developments. Anal. Chem. 2004, 76, 3373–3385. [Google Scholar]
- Andersson, H.; van den Berg, A. Microtechnologies and nanotechnologies for single-cell analysis. Curr. Opin. Biotech. 2004, 15, 44–49. [Google Scholar]
- Zhang, H.; Jin, W.R. Analysis of amino acids in individual human erythrocytes by capillary electrophoresis with electroporation for intracellular derivatization and laser-induced fluorescence detection. Electrophoresis 2004, 25, 480–486. [Google Scholar]
- Tamaki, E.; Sato, K.; Tokeshi, M.; Sato, K.; Aihara, M.; Kitamori, T. Single-cell analysis by a scanning thermal lens microscope with a microchip: Direct monitoring of cytochrome c distribution during apoptosis process. Anal. Chem. 2002, 74, 1560–1564. [Google Scholar]
- Yavuz, A.S.; Lipsky, P.E.; Yavuz, S.; Metcalfe, D.D.; Akin, C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood 2002, 100, 661–665. [Google Scholar]
- Sun, X.M.; Jin, W.R.; Li, D.; Bai, Z.L. Measurement of alkaline phosphatase isoenzymes in individual mouse bone marrow fibroblast cells based on capillary electrophoresis with on-capillary enzyme-catalyzed reaction and electrochemical detection. Electrophoresis 2004, 25, 1860–1866. [Google Scholar]
- Sun, X.M.; Jin, W.R. Catalysis-electrochemical determination of zeptomole enzyme and its application for single-cell analysis. Anal. Chem. 2003, 75, 6050–6055. [Google Scholar]
- Lynch, M.; Mosher, C.; Huff, J.; Nettikadan, S.; Johnson, J.; Henderson, E. Functional protein nanoarrays for biomarker profiling. Proteomics 2004, 4, 1695–1702. [Google Scholar]
- Kleparnik, K.; Horky, M. Detection of DNA fragmentation in a single apoptotic cardiomyocyte by electrophoresis on a microfluidic device. Electrophoresis 2003, 24, 3778–3783. [Google Scholar]
- Woods, L.A.; Roddy, T.P.; Ewing, A.G. Capillary electrophoresis of single mammalian cells. Electrophoresis 2004, 25, 1181–1187. [Google Scholar]
- Hu, K.; Zarrine-Afsar, A.; Ahmadzadeh, H.; Krylov, S.N. Single-cell analysis by chemical cytometry combined with fluorescence microscopy. Instrum. Sci. Technol. 2004, 32, 31–41. [Google Scholar]
- Harris, D.C. Quantitative chemical analysis; W. H. Freeman and Company, 2003. [Google Scholar]
- Goddard, J.P.; Reymond, J.L. Recent advances in enzyme assays. Trends Biotech. 2004, 22, 363–370. [Google Scholar]
- Durdux, M.; Grunwald, D.; Gautier, T.; Ronot, X.; Boutonnat, J. Fluorescence-based assessment of LRP activity: A comparative study. Anticancer Res. 2004, 24, 725–732. [Google Scholar]
- Bornemann, C.; Burggraef, T.; Heimbuchel, G.; Hanisch, F.G.; Winkels, S. Fluorescence-labelled antigen-binding fragments (Fab) from monoclonal antibody 5F12 detect human erythropoietin in immunoaffinity capillary electrophoresis. Anal. Bioanal. Chem. 2003, 376, 1074–1080. [Google Scholar]
- Stiborova, M.; Rupertova, M.; Hodek, P.; Frei, E.; Schmeiser, H.H. Monitoring of DNA adducts in humans and P-32-postlabelling methods. A review. Collect. Czech. Chem. C. 2004, 69, 476–498. [Google Scholar]
- Svensson, R.; Greno, C.; Johansson, A.S.; Mannervik, B.; Morgenstern, R. Synthesis and characterization of 6-chloroacetyl-2-dimethylaminonaphthalene as a fluorogenic substrate and a mechanistic probe for glutathione transferases. Anal. Biochem. 2002, 311, 171–178. [Google Scholar]
- Vitecek, J.; Adam, V.; Petrek, J.; Babula, P.; Novotna, P.; Kizek, R.; Havel, J. Application of fluorimetric determination of esterases in plant material. Chem. Listy 2005, 99, 496–501. [Google Scholar]
- Vitecek, J.; Adam, V.; Petrek, J.; Vacek, J.; Kizek, R.; Havel, L. Esterases as a marker for growth of BY-2 tobacco cells and early somatic embryos of the Norway spruce. Plant Cell Tiss. Org. 2004, 79, 195–201. [Google Scholar]
- Vitecek, J.; Petrlova, J.; Adam, V.; Petrek, J.; Havel, L.; Kramer, K.J.; Kizek, R. Application of fluorimetric analysis of plant esterases to study of programmed cell death and effects of cadmium(II) ions. Biol. Plant. 2007, in press. [Google Scholar]
- Aldridge, W.N. The esterases: pespectives and problems. Chem.-Biol. Interact. 1993, 87, 5–13. [Google Scholar]
- Nagata, T.; Nemoto, Y.; Hasezawa, S. Tobacco BY-2 cell line as the “HeLa” cell line in the cell biology of higher plants. Int Rev Cytol 1992, 132, 1–30. [Google Scholar]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plant 1962, 15, 473. [Google Scholar]
- Jokinen, K.J.; Durzan, D.J. Properties of rescued embryonal suspensor mass of Norway spruce (Picea abies L. Karst.) determined by the genotype and environment in vitro. Silva. Fenn. 1994, 28, 95–106. [Google Scholar]
- Durzan, D.J.; Jokinen, K.; Guerra, M.P.; Santerre, A.; Chalupa, V.; Havel, L. Latent diploid parthenogenesis and parthenote cleavage in egg-equivalents of norway spruce. Int. J. Plant Sci. 1994, 155, 677–688. [Google Scholar]
- von Arnold, S.J. Improved efficiency of somatic embryogenesis in mature embryos of Picea abies (L.). Plant Physiol. 1987, 127, 233–244. [Google Scholar]
- Havel, L.; Durzan, D.J. Apoptosis during diploid parthenogenesis and early somatic embryogenesis of Norway spruce. Int. J. Plant Sci. 1996, 157, 8–16. [Google Scholar]
- Havel, L.; Durzan, D.J. Apoptosis in plants. Bot. Acta 1996, 109, 268–277. [Google Scholar]
- Jones, K.H.; Senft, J.A. An improved method to determine cell viability by simultaneous staining with fluorescein diacetate propidium iodide. J. Histochem. Cytochem. 1985, 33, 77–79. [Google Scholar]
- Petrek, J.; Vitecek, J.; Vlasinova, H.; Kizek, R.; Kramer, K.J.; Adam, V.; Klejdus, B.; Havel, L. Application of computer imaging, stripping voltammetry and mass spectrometry for study of the effect of lead (Pb-EDTA) on growth and viability of early somatic embryos of Norway spruce (Picea abies /L./ Karst.). Anal. Bioanal. Chem. 2005, 383, 576–586. [Google Scholar]
- Mlejnek, P.; Prochazka, S. Activation of caspase-like proteases and induction of apoptosis by isopentenyladenosine in tobacco BY-2 cells. Planta 2002, 215, 158–166. [Google Scholar]
- Pinard, C.L.; Weiss, M.L.; Brightman, A.H.; Fenwick, B.W.; Davidson, H.J. Evaluation of lysozyme and lactoferrin in lacrimal and other ocular glands of bison and cattle and in tears of bison. Am. J. Vet. Res. 2003, 64, 104–108. [Google Scholar]
- Lomolino, G.; Lante, A.; Crapisi, A.; Spettoli, P.; Curioni, A. Detection of Saccharomyces cerevisiae cerbosylesterase activity after native and sodium dodecyl sulfate electrophoresis by using fluorescein diacetate as substrate. Electrophoresis 2001, 22, 1021–1023. [Google Scholar]
- Vallejos, E. Enzyme activity staining; Elsevier Science Publishers B.V., 1983; pp. 513–516. [Google Scholar]
- Leach, A.M.; Wheeler, A.R.; Zare, R.N. Flow injection analysis in a microfluidic format. Anal. Chem. 2003, 75, 967–972. [Google Scholar]
- Shimazaki, Y.; Hiraka, Y.; Uesugi, M.; Manabe, T. Simultaneous analysis of esterase and transferase activities in cytosol proteins from the bovine retina by using microscale non-denaturing two-dimensional electrophoresis. BBA - Proteins Proteom. 2004, 1696, 51–57. [Google Scholar]
- Steward, N.; Martin, R.; Engasser, J.M.; Goergen, J.L. A new methodology for plant cell viability assessment using intracellular esterase activity. Plant Cell Rep. 1999, 19, 171–176. [Google Scholar]
- Kizek, R.; Vacek, J.; Trnkova, L.; Jelen, F. Cyclic voltammetric study of the redox system of glutathione using the disulfide bond reductant tris(2-carboxyethyl)phosphine. Bioelectrochemistry 2004, 63, 19–24. [Google Scholar]
- Bornscheuer, U.T. Microbial carboyl esterases: clasification, properties and application in biocatalysis. FEMS Microbiol. Rev. 2002, 26, 73–81. [Google Scholar]
- Voet, D.; Voet, J.G. Biochemistry; John Wiley & Sons, Inc., 1995. [Google Scholar]
- Joga, M.A.; Font, X.; Gordillo, M.A.; Valero, F. Esterase activity assay by flow injection analysis (FIA). Biotechnol. Lett. 2001, 23, 943–948. [Google Scholar]





© 2007 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Vitecek, J.; Petrlova, J.; Adam, V.; Havel, L.; Kramer, K.J.; Babula, P.; Kizek, R. A Fluorimetric Sensor for Detection of One Living Cell. Sensors 2007, 7, 222-238. https://doi.org/10.3390/s7030222
Vitecek J, Petrlova J, Adam V, Havel L, Kramer KJ, Babula P, Kizek R. A Fluorimetric Sensor for Detection of One Living Cell. Sensors. 2007; 7(3):222-238. https://doi.org/10.3390/s7030222
Chicago/Turabian StyleVitecek, Jan, Jitka Petrlova, Vojtech Adam, Ladislav Havel, Karl J. Kramer, Petr Babula, and Rene Kizek. 2007. "A Fluorimetric Sensor for Detection of One Living Cell" Sensors 7, no. 3: 222-238. https://doi.org/10.3390/s7030222