Translocation Biosensors – Cellular System Integrators to Dissect CRM1-Dependent Nuclear Export by Chemicogenomics



Abstract
:1. Introduction
1.1. Cellular Biosensors
1.2. Nucleo-Cytoplasmic Transport
1.3. Chemogenomics
2. Results and Discussion
2.1. The RevNES-Biosensor
2.2. Generation and Characterization of Cell Lines Stably Expressing the RevNES-Biosensor
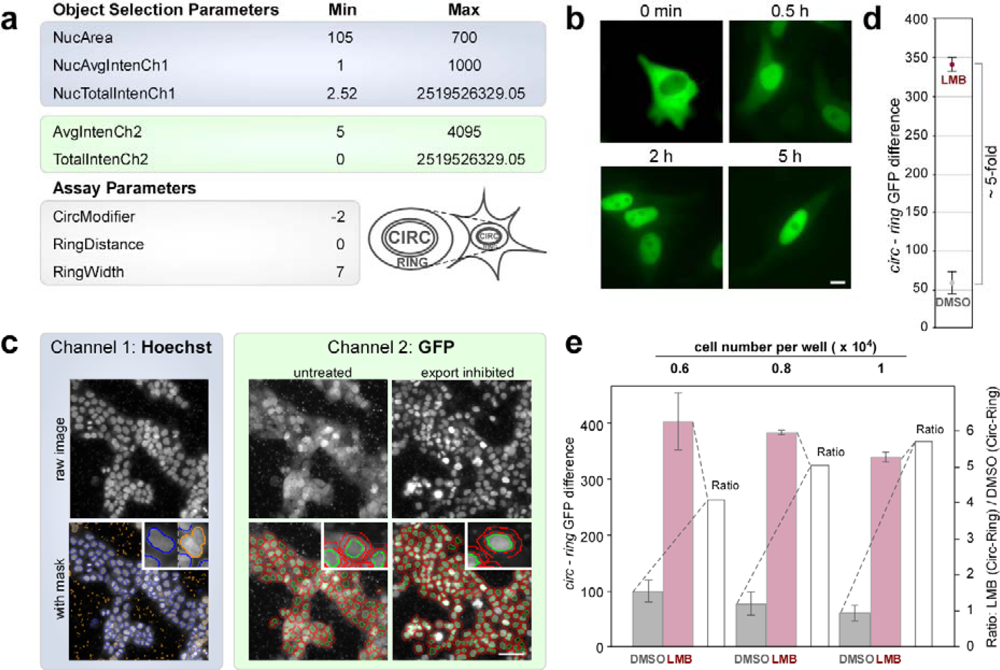
2.3. Assay Development on the Cellomics ArrayScan® VTI Imaging Platform
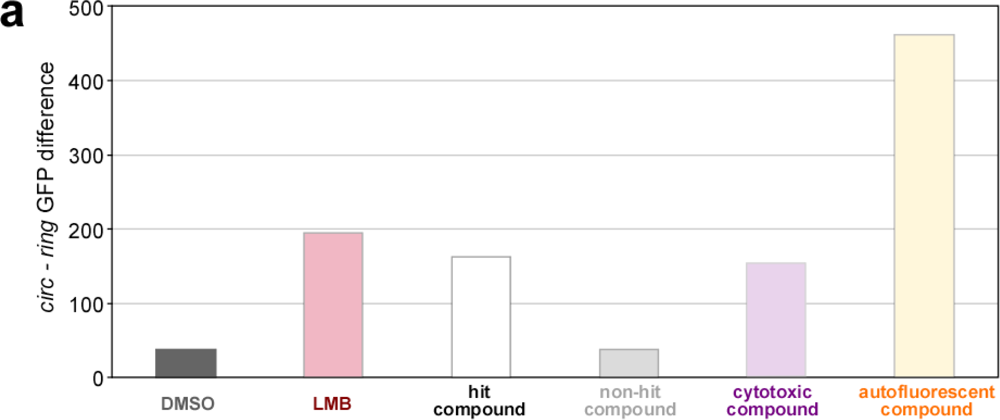
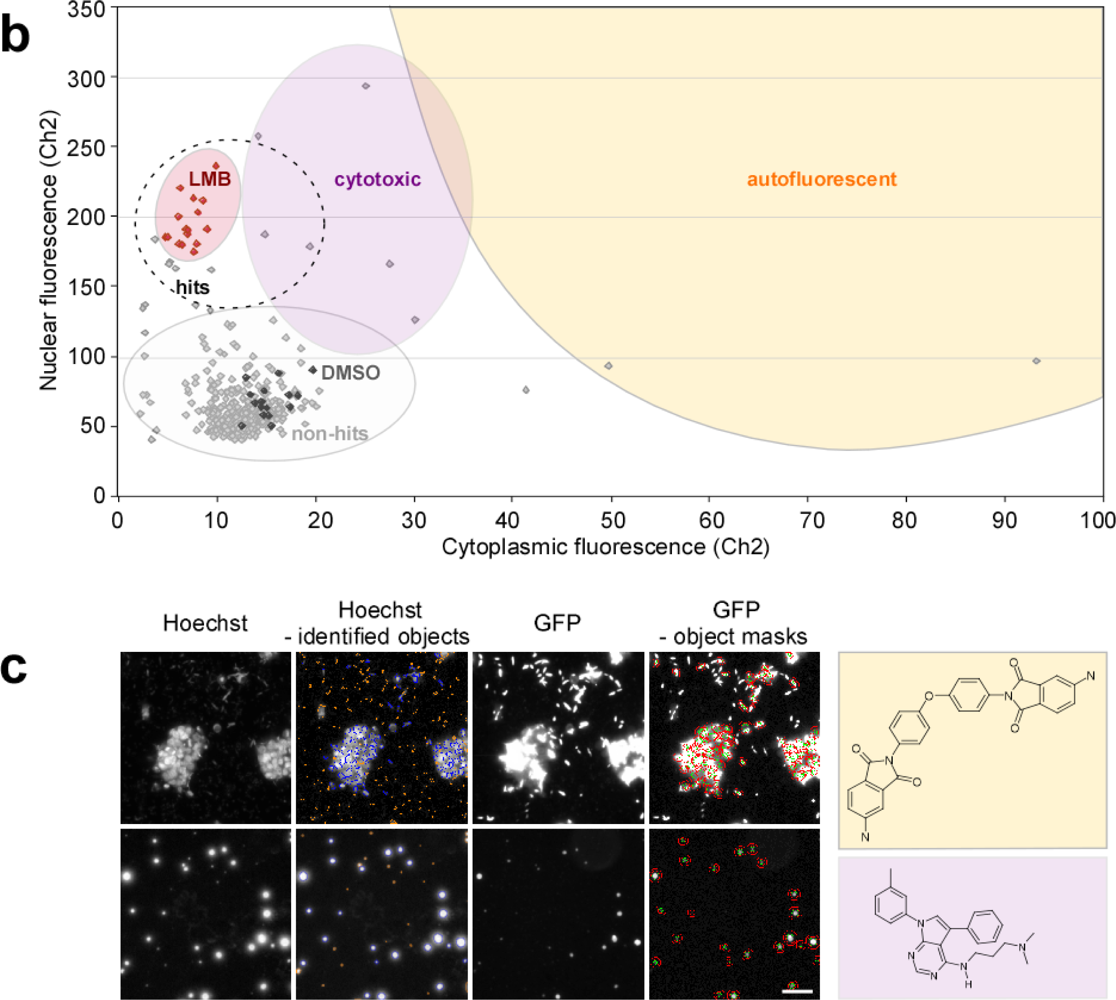
2.4. Biosensor-Based HTS for Export Inhibitors
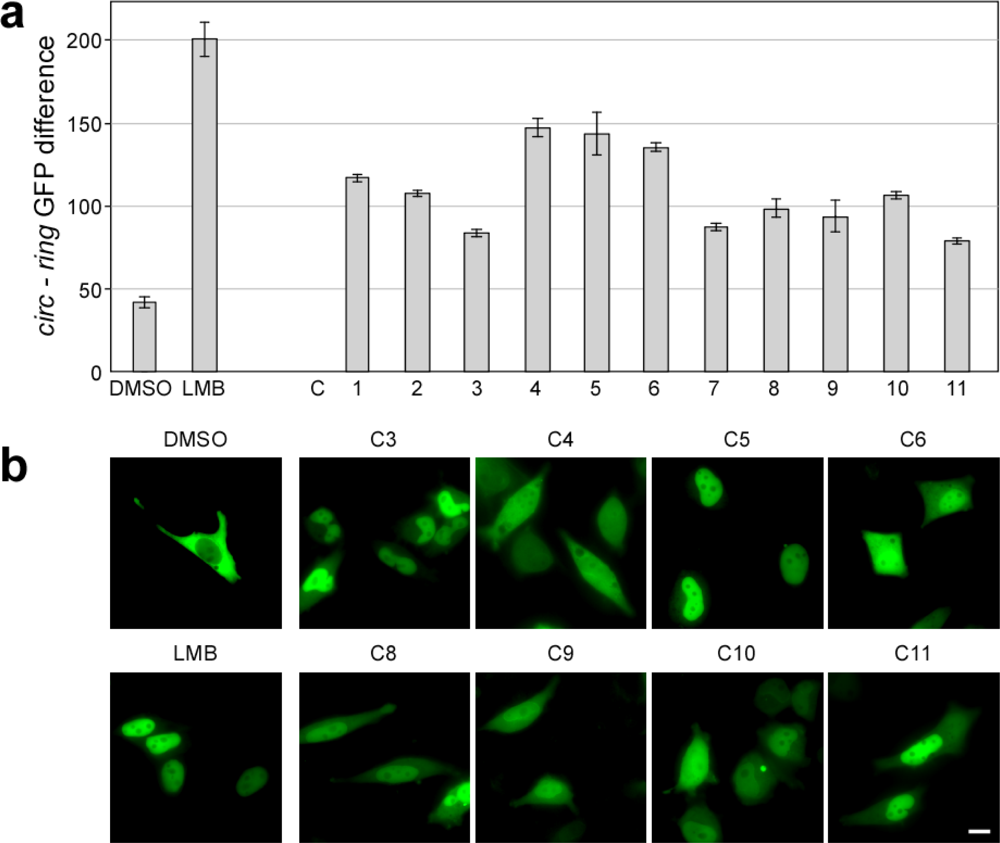
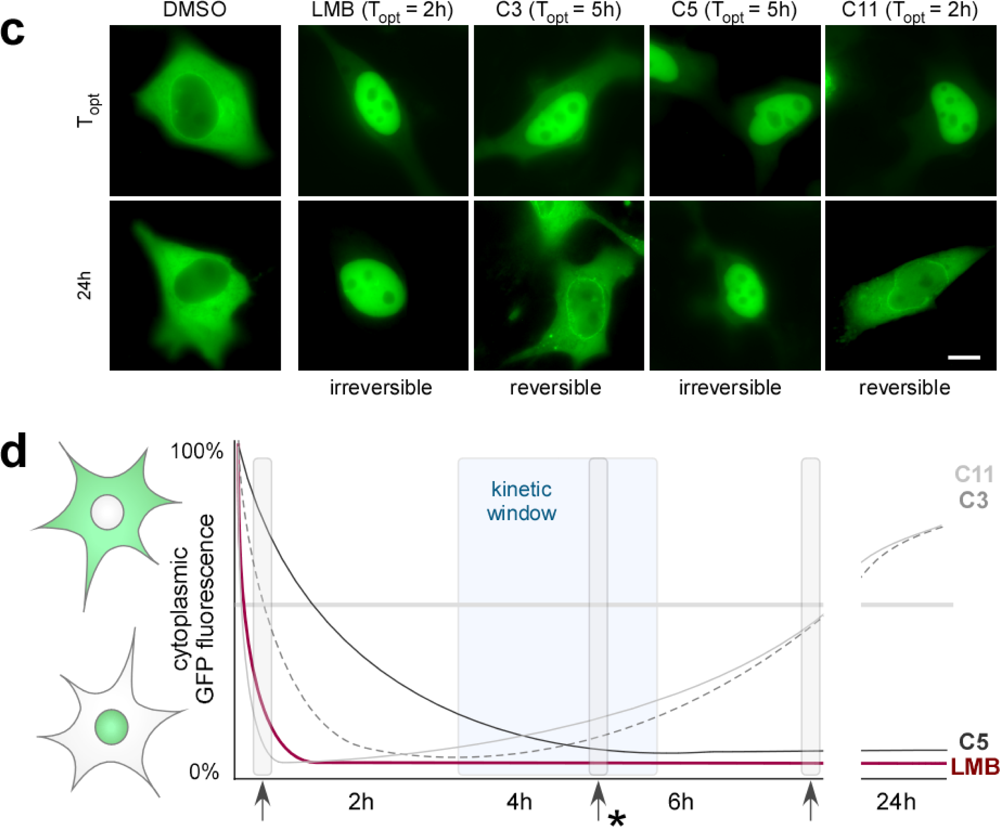
2.5. Functional and Kinetic Profiling of Novel Export Antagonists
3. Experimental Section
3.1. Plasmids
3.2. Cell Culture, Transfection, Retrovirus Production and Transduction
3.3. Generation of Stable Cell Lines
3.4. Automated High Throughput/Content Screening Platform, Assay Preparation and Execution
3.5. Image Acquisition and Analysis
3.6. The ChemBioNet Compound Collection
3.7. Drug Treatment and Fluorescent Imaging of Cells
3.8. Measurement of Cell Viability
3.9. Statistical Analysis
4. Conclusions
(a) Prolonged incubation with LMB affects cell viability. 1×103 A431bio cells/well were seeded into 96-well plates and treated with the indicated concentrations of LMB or were mock treated (DMSO). ATP concentrations in cell lysates, reflecting cell viability, were determined after the indicated time points using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI) as described [39]. Whereas LMB treatment for up to 5h did not show a significant effect, incubation for 24h resulted in a strong reduction in cell viability. No significant dose dependence was observed. RLU, relative light units; columns, mean; bars, SD. (b) The novel export inhibitors are active in cell culture models of different tumor types. Indicated cell lines transiently expressing the RevNES biosensor were treated with the indicated compounds (25 μM). The localization of the biosensor is visualized in living cells by fluorescence microscopy 5 h after mock (DMSO), 2 h after LMB treatment and at time points when the compounds displayed their maximum inhibitory activity (Topt). All export inhibitors blocked export in FaDu (head and neck cancer cell line), MDA-MB-231 (breast cancer cell line) or RKO (colon cancer cell line) cells. Scale bar, 10 μm.
Acknowledgments
References and Notes
- Giuliano, K.A.; Johnston, P.A.; Gough, A.; Taylor, D.L. Systems cell biology based on high-content screening. Methods Enzymol 2006, 414, 601–619. [Google Scholar]
- Sachs, K.; Perez, O.; Pe'er, D.; Lauffenburger, D.A.; Nolan, G.P. Causal protein-signaling networks derived from multiparameter single-cell data. Science 2005, 308, 523–529. [Google Scholar]
- Knauer, S.K.; Moodt, S.; Berg, T.; Liebel, U.; Pepperkok, R.; Stauber, R.H. Translocation biosensors to study signal-specific nucleo-cytoplasmic transport, protease activity and protein-protein interactions. Traffic 2005, 6, 594–606. [Google Scholar]
- Michael, S.; Auld, D.; Klumpp, C.; Jadhav, A.; Zheng, W.; Thorne, N.; Austin, C.P.; Inglese, J.; Simeonov, A. A robotic platform for quantitative high-throughput screening. Assay Drug Dev. Technol 2008, 6, 637–657. [Google Scholar]
- Simpson, P.B.; Wafford, K.A. New directions in kinetic high information content assays. Drug Discov. Today 2006, 11, 237–244. [Google Scholar]
- Wolff, M.; Kredel, S.; Wiedenmann, J.; Nienhaus, G.U.; Heilker, R. Cell-based assays in practice: cell markers from autofluorescent proteins of the GFP-family. Comb. Chem. High Throughput Screen 2008, 11, 602–609. [Google Scholar]
- Giuliano, K.A.; Taylor, D.L.; Waggoner, A.S. Reagents to measure and manipulate cell functions. Methods Mol. Biol 2007, 356, 141–163. [Google Scholar]
- Giuliano, K.A.; Chen, Y.T.; Taylor, D.L. High-content screening with siRNA optimizes a cell biological approach to drug discovery: defining the role of P53 activation in the cellular response to anticancer drugs. J. Biomol. Screen 2004, 9, 557–568. [Google Scholar]
- Suntharalingam, M.; Wente, S.R. Peering through the pore: nuclear pore complex structure, assembly, and function. Dev. Cell 2003, 4, 775–789. [Google Scholar]
- Brown, C.R.; Silver, P.A. Transcriptional regulation at the nuclear pore complex. Curr. Opin. Genet. Dev 2007, 17, 100–106. [Google Scholar]
- Komili, S.; Silver, P.A. Coupling and coordination in gene expression processes: a systems biology view. Nat. Rev. Genet 2008, 9, 38–48. [Google Scholar]
- Lim, R.Y.; Fahrenkrog, B.; Koser, J.; Schwarz-Herion, K.; Deng, J.; Aebi, U. Nanomechanical basis of selective gating by the nuclear pore complex. Science 2007, 318, 640–643. [Google Scholar]
- Maco, B.; Fahrenkrog, B.; Huang, N.P.; Aebi, U. Nuclear pore complex structure and plasticity revealed by electron and atomic force microscopy. Methods Mol. Biol 2006, 322, 273–288. [Google Scholar]
- D'Angelo, M.A.; Hetzer, M.W. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol 2008, 18, 456–466. [Google Scholar]
- Frey, S.; Gorlich, D. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell 2007, 130, 512–523. [Google Scholar]
- Mosammaparast, N.; Pemberton, L.F. Karyopherins: from nuclear-transport mediators to nuclear-function regulators. Trends Cell Biol 2004, 14, 547–556. [Google Scholar]
- Terry, L.J.; Shows, E.B.; Wente, S.R. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 2007, 318, 1412–1416. [Google Scholar]
- Pemberton, L.F.; Paschal, B.M. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 2005, 6, 187–198. [Google Scholar]
- Meissner, T.; Krause, E.; Vinkemeier, U. Ratjadone and leptomycin B block CRM1-dependent nuclear export by identical mechanisms. FEBS Lett 2004, 576, 27–30. [Google Scholar]
- Kau, T.R.; Way, J.C.; Silver, P.A. Nuclear transport and cancer: from mechanism to intervention. Nat. Rev. Cancer 2004, 4, 106–117. [Google Scholar]
- Schweitzer, A.; Knauer, S.K.; Stauber, R.H. Therapeutic potential of nuclear receptors. Expert Opin. Ther. Patents 2008, 18, 861–888. [Google Scholar]
- Knauer, S.K.; Carra, G.; Stauber, R.H. Nuclear export is evolutionarily conserved in CVC paired-like homeobox proteins and influences protein stability, transcriptional activation, and extracellular secretion. Mol. Cell. Biol 2005, 25, 2573–2582. [Google Scholar]
- Hutten, S.; Kehlenbach, R.H. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 2007, 17, 193–201. [Google Scholar]
- Arnaoutov, A.; Azuma, Y.; Ribbeck, K.; Joseph, J.; Boyarchuk, Y.; Karpova, T.; McNally, J.; Dasso, M. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat. Cell Biol 2005, 7, 626–632. [Google Scholar]
- Kramer, O.H.; Knauer, S.K.; Greiner, G.; Jandt, E.; Reichardt, S.; Guhrs, K.H.; Stauber, R.H.; Bohmer, F.D.; Heinzel, T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 2009, 23, 223–235. [Google Scholar]
- Heger, P.; Lohmaier, J.; Schneider, G.; Schweimer, K.; Stauber, R.H. Qualitative highly divergent nuclear export signals can regulate export by the competition for transport cofactors in vivo. Traffic 2001, 2, 544–555. [Google Scholar]
- Bredel, M.; Jacoby, E. Chemogenomics: an emerging strategy for rapid target and drug discovery. Nat. Rev. Genet 2004, 5, 262–275. [Google Scholar]
- Allen, J.J.; Shokat, K.M. Chemical genomics: dialed in transcriptional network control with non-steroidal glucocorticoid receptor modulators. ACS Chem. Biol 2006, 1, 139–140. [Google Scholar]
- Stauber, R.H.; Kratzer, F.; Schneider, G.; Hirschmann, N.; Hauber, J.; Rosorius, O. Investigation of nucleo-cytoplasmic transport using UV-guided microinjection. J. Cell Biochem 2001, 80, 388–396. [Google Scholar]
- Petosa, C.; Schoehn, G.; Askjaer, P.; Bauer, U.; Moulin, M.; Steuerwald, U.; Soler-Lopez, M.; Baudin, F.; Mattaj, I.W.; Muller, C.W. Architecture of CRM1/Exportin1 suggests how cooperativity is achieved during formation of a nuclear export complex. Mol. Cell 2004, 16, 761–775. [Google Scholar]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar]
- Beissert, T.; Hundertmark, A.; Kaburova, V.; Travaglini, L.; Mian, A.A.; Nervi, C.; Ruthardt, M. Targeting of the N-terminal coiled coil oligomerization interface by a helix-2 peptide inhibits unmutated and imatinib-resistant BCR/ABL. Int. J. Cancer 2008, 122, 2744–2752. [Google Scholar]
- Engels, K.; Knauer, S.K.; Metzler, D.; Simf, C.; Struschka, O.; Bier, C.; Mann, W.; Kovacs, A.F.; Stauber, R.H. Dynamic intracellular survivin in oral squamous cell carcinoma: underlying molecular mechanism and potential as an early prognostic marker. J. Pathol 2007, 211, 532–540. [Google Scholar]
- Fetz, V.; Bier, C.; Habtemichael, N.; Schuon, R.; Schweitzer, A.; Kunkel, M.; Engels, K.; Kovacs, A.F.; Schneider, S.; Mann, W.; Stauber, R.H.; Knauer, S.K. Inducible NO synthase confers chemoresistance in head and neck cancer by modulating survivin. Int. J. Cancer 2008, 124, 2033–2041. [Google Scholar]
- Stauber, R.H.; Horie, K.; Carney, P.; Hudson, E.A.; Tarasova, N.I.; Gaitanaris, G.A.; Pavlakis, G.N. Development and Applications of Enhanced Green Fluorescent Protein Mutants. BioTechniques 1998, 24, 462–471. [Google Scholar]
- Ridler, T.W.; Calvard, S. Picture thresholding using an iterative selection method. IEEE Trans. Syst. Man Cybern 1978, 8, 630–635. [Google Scholar]
- Stauber, R.H.; Mann, W.; Knauer, S.K. Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential. Cancer Res 2007, 67, 5999–6002. [Google Scholar]
- Knauer, S.K.; Bier, C.; Habtemichael, N.; Stauber, R.H. The Survivin-Crm1 interaction is essential for chromosomal passenger complex localization and function. EMBO Rep 2006, 7, 1259–1265. [Google Scholar]
- Knauer, S.K.; Bier, C.; Schlag, P.; Fritzmann, J.; Dietmaier, W.; Rodel, F.; Klein-Hitpass, L.; Kovacs, A.F.; Doring, C.; Hansmann, M.L.; Hofmann, W.K.; Kunkel, M.; Brochhausen, C.; Engels, K.; Lippert, B.M.; Mann, W.; Stauber, R.H. The survivin isoform survivin-3B is cytoprotective and can function as a chromosomal passenger complex protein. Cell Cycle 2007, 6, 1502–1509. [Google Scholar]
- van Neck, T.; Pannecouque, C.; Vanstreels, E.; Stevens, M.; Dehaen, W.; Daelemans, D. Inhibition of the CRM1-mediated nucleocytoplasmic transport by N-azolylacrylates: structure-activity relationship and mechanism of action. Bioorg. Med. Chem 2008, 16, 9487–9497. [Google Scholar]
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Frenkel-Morgenstern, M.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; Cohen, L.; Danon, T.; Perzov, N.; Alon, U. Dynamic proteomics of individual cancer cells in response to a drug. Science 2008, 322, 1511–1516. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound parameters
| ||
| N° compounds | % of total | |
| Screened | 16,671 | 100% |
| Rescreened | 120 | 0.72% |
| Validated | 11 | 0.07% |
| Screening parameters
| ||
| Parameter | Value | |
| # plates | 48 | |
| Total scan time | 56h | |
| Scan time/well | 11s | |
| Fields/well | 4 | |
| Average valid objects/well | 470 | |
| Z' factor calculation
| ||
| Formula | Z' factor | |
| CIRCGFP | 0.71 | |
| RingGFP | −0.22 | |
| CIRCGFP/ RingGFP | 0.37 | |
| CIRCGFP - RingGFP | 0.77 | |
© 2009 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fetz, V.; Knauer, S.K.; Bier, C.; Von Kries, J.P.; Stauber, R.H. Translocation Biosensors – Cellular System Integrators to Dissect CRM1-Dependent Nuclear Export by Chemicogenomics. Sensors 2009, 9, 5423-5445. https://doi.org/10.3390/s90705423
Fetz V, Knauer SK, Bier C, Von Kries JP, Stauber RH. Translocation Biosensors – Cellular System Integrators to Dissect CRM1-Dependent Nuclear Export by Chemicogenomics. Sensors. 2009; 9(7):5423-5445. https://doi.org/10.3390/s90705423
Chicago/Turabian StyleFetz, Verena, Shirley K. Knauer, Carolin Bier, Jens Peter Von Kries, and Roland H. Stauber. 2009. "Translocation Biosensors – Cellular System Integrators to Dissect CRM1-Dependent Nuclear Export by Chemicogenomics" Sensors 9, no. 7: 5423-5445. https://doi.org/10.3390/s90705423
APA StyleFetz, V., Knauer, S. K., Bier, C., Von Kries, J. P., & Stauber, R. H. (2009). Translocation Biosensors – Cellular System Integrators to Dissect CRM1-Dependent Nuclear Export by Chemicogenomics. Sensors, 9(7), 5423-5445. https://doi.org/10.3390/s90705423