DNA Sensors with Diamond as a Promising Alternative Transducer Material

Abstract
:1. Introduction
1.1. Biological Receptor Molecules
1.2. Attachment of Biological Receptor Molecules
1.2.1. Non-Covalent, Physical Adsorption
1.2.2. Covalent Binding
1.3. Types of Transducers
2. The Role of Diamond in Biosensors
2.1. Classification of Diamond
2.1.1. Natural Diamond
Type I diamonds
Type II diamonds
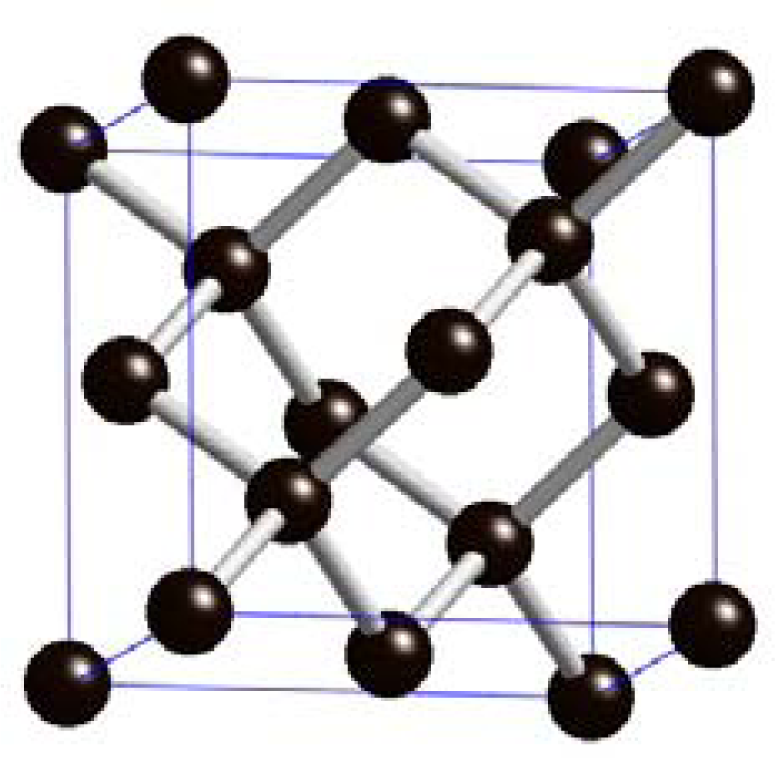
2.1.2. Synthetic Diamond
Single-crystalline diamond (SCD)
Polycrystalline diamond (PCD)
• Microcrystalline diamond (MCD)
• Nanocrystalline diamond (NCD)
• Ultrananocrystalline diamond (UNCD)
2.2. Properties of Diamond
2.2.1. Electronic Properties
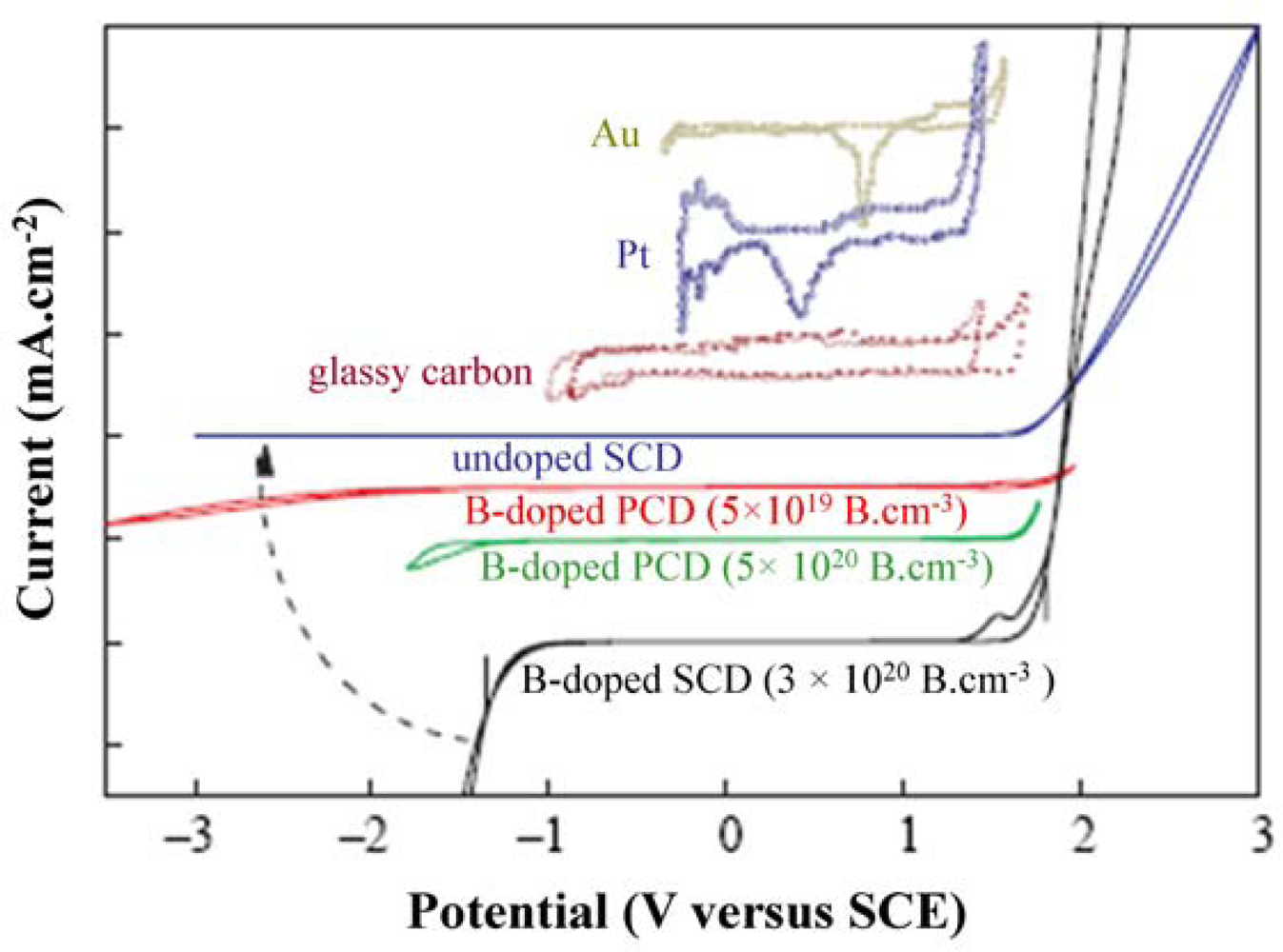
Large electrochemical potential window
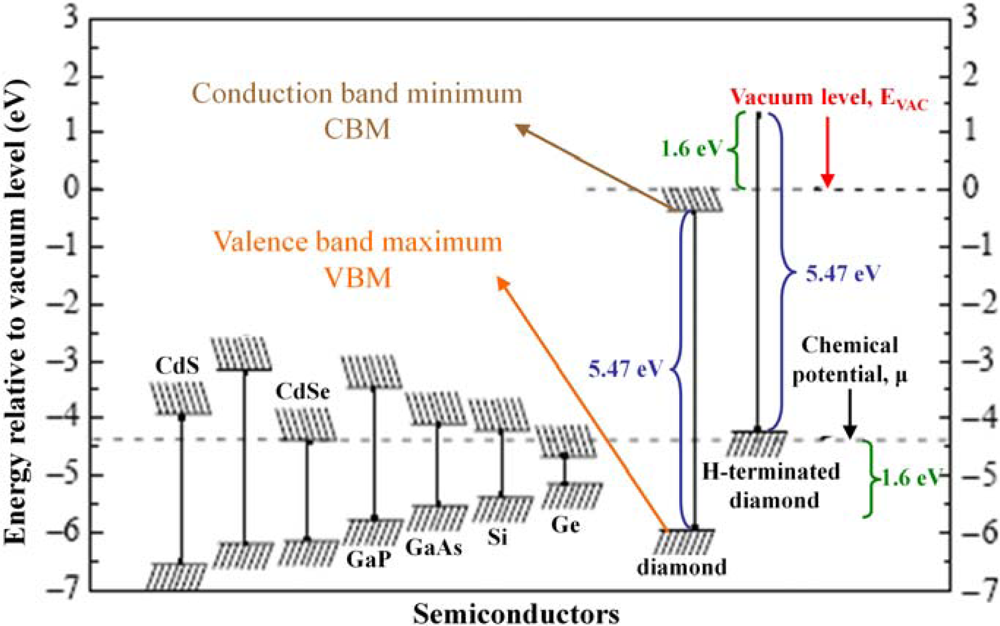
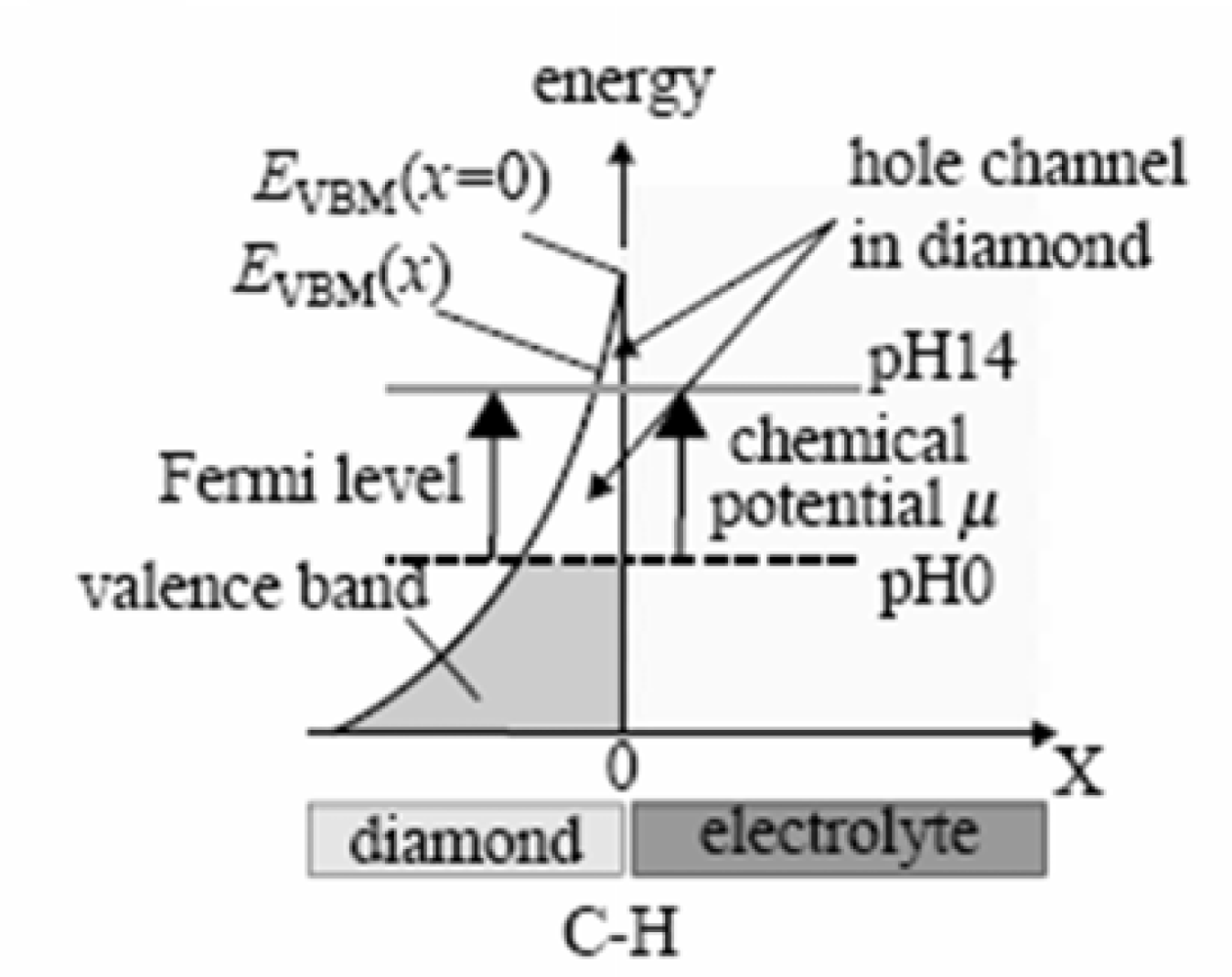
Hydrogen (H)-induced surface conductivity
Diamond doping
• p-type doping
• n-type doping
2.2.2. Physical Properties
Hardness
Thermal conductivity
Optics
2.2.3. Biochemical Properties
Chemical biofunctionalisation
Electrochemical biofunctionalisation
Photochemical biofunctionalisation
3. Biosensor Classification
3.1. Electrochemical Transduction
3.1.1. Amperometric
3.1.2. Coulometric
3.1.3. Potentiometric
3.1.4. Conductimetric
3.1.5. Impedimetric
3.1.6. Field-Effect
- Applying a negative gate voltage to an n-channel FET (NPN FET) causes the positive charge carriers in the p-type semiconducting body electrode to become attracted to the gate electrode. This positively charged channel blocks current flow between source and drain.
- Applying a positive gate voltage to an n-channel FET (NPN FET) will create a conductive channel from source to drain. By attracting electrons from source and drain to the gate electrode and repelling the positive charge carriers from the p-type semiconductor body electrode further into the bulk, the resistance in the space-charge region decreases and current flow between source and drain increases.
- Applying a negative gate voltage to a p-channel FET (PNP FET) will create a conductive channel from source to drain. By attracting positive holes from source and drain to the gate electrode and repelling the electrons from the n-type semiconductor body electrode further into the bulk, the resistance in the space-charge region decreases and current flow between source and drain increases.
- Applying a positive gate voltage to a p-channel FET (PNP FET) causes the electrons in the n-type semiconducting body electrode to become attracted to the gate electrode. This negatively charged channel blocks current flow between source and drain.
3.2. Optical Transduction
3.2.1. Indirect
3.2.2. Direct
3.3. Piezo-Electric Transduction
4. Summary and Conclusions
Acknowledgments
References and Notes
- Gorton, L.O. Comprehensive Analytical Chemistry; Elsevier Science: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Häussling, L.; Michel, B.; Ringsdorf, R.; Rohrer, H. Direct observation of streptavidin specifically adsorbed on biotin-functionalized self-assembled monolayers with the scanning tunneling microscope. Angew. Chem. Int. Ed 1991, 30, 569–572. [Google Scholar]
- Rasmussen, S.R.; Larsen, M.R.; Rasmussen, S.E. Covalent immobilization of DNA onto polystyrene microwells: The molecules are only bound at the 5′ end. Anal. Biochem 1991, 198, 138–142. [Google Scholar]
- Hashimoto, K.; Ito, K.; Ishimori, Y. Sequence-specific gene detection with a gold electrode modified with DNA probes and an electrochemically active dye. Anal. Chem 1994, 66, 3830–3833. [Google Scholar]
- Strother, T.; Cai, W.; Zhao, X.S.; Hamers, R.J.; Smith, L.M. Synthesis and characterization of DNA modified silicon (111) surfaces. J. Am. Chem. Soc 2000, 122, 1205–1209. [Google Scholar]
- Kremsky, J.N.; Wooters, J.L.; Dougherty, J.P.; Meyers, R.E.; Collins, M.; Brown, E.L. Immobilization of DNA via oligonucleotides containing an aldehyde or carboxylic acid group at the 5′ terminus. Nucleic Acids Res 1987, 15, 2891–2909. [Google Scholar]
- Yang, W.; Auciello, O.; Butler, J.E.; Cai, W.; Carlisle, J.A.; Gerbi, J.E.; Gruen, D.M.; Knickerbocker, T.; Lasseter, T.L.; Russell, J.N., Jr.; Smith, L.M.; Hamers, R.J. DNA-modified nanocrystalline diamond thin-films as stable, biologically active substrates. Nat. Mater 2002, 1, 253–257. [Google Scholar]
- Nebel, C.E.; Shin, D.; Rezek, B.; Tokuda, N.; Uetsuka, H.; Watanabe, H. Diamond and biology. J. R. Soc. Interface 2007, 4, 439–461. [Google Scholar]
- Davidson, J.L. Synthetic Diamond: Emerging CVD Science and Technology; Wiley-Interscience: New York, NY, USA, 1994. [Google Scholar]
- Wolfe, C.M.; Holonyak, N., Jr.; Stillman, G.E. Physical properties of semiconductors; Prentice-Hall, Inc: Upper Saddle River, NJ, USA, 2007. [Google Scholar]
- Millan, K.M.; Spurmanis, A.J.; Mikkelsen, S.R. Covalent immobilization of DNA into glassy-carbon electrodes. Electroanalysis 1992, 4, 929–932. [Google Scholar]
- Pierson, H.O. Handbook of Carbon, Graphite, Diamond and Fullerenes; Noyes Publications: Park Ridge, NJ, USA, 1993. [Google Scholar]
- Zaitsev, A.M. Optical Properties of Diamond: A Data Handbook; Springer-Verlag: Berlin, Germany, 2001. [Google Scholar]
- Wilks, J.; Wilks, E. Properties and Applications of Diamond; Butterworth-Heinemann: Oxford, UK, 1991. [Google Scholar]
- Bundy, F.B.; Hall, H.T.; Strong, H.M.; Wentorf, R.H., Jr. Man made diamonds. Nature 1955, 176, 51–55. [Google Scholar]
- Lee, S.T.; Lin, Z.; Jiang, X. CVD diamond films: nucleation and growth. Mater. Sci. Eng 1999, 25, 123–154. [Google Scholar]
- May, P.W. Diamond thin films: a 21st-century material. Phil. Trans. R. Soc. Lond. A 2000, 358, 473–495. [Google Scholar]
- Liu, H.; Dandy, D.S. Diamond Chemical Vapour Deposition: Nucleation and Early Growth Stages; Noyes Publications: Park Ridge, NJ, USA, 1993. [Google Scholar]
- Williams, O.A.; Nesládek, M. Growth and properties of nanocrystalline diamond films. Phys. Stat. Sol 2006, 203, 3375–3386. [Google Scholar]
- Granger, M.C.; Xu, J.; Strojek, J.W.; Swain, G.M. Polycrystalline diamond electrodes: basic properties and applications as amperometric detectors in flow injection analysis and liquid chromatography. Anal. Chim. Acta 1999, 397, 145–161. [Google Scholar]
- Maier, F.; Riedel, M.; Mantel, B.; Ristein, J.; Ley, L. Origin of surface conductivity in diamond. Phys. Rev. Lett 2000, 85, 3472–3475. [Google Scholar]
- Nebel, C.E.; Rezek, B.; Shin, D.; Watanabe, H. Surface electronic properties of H-terminated diamond in contact with adsorbates and electrolytes. Phys. Stat. Sol 2006, 203, 3273–3298. [Google Scholar]
- Chakrapani, V.; Angus, J.C.; Anderson, A.B.; Wolter, S.D.; Stoner, B.R.; Sumanasekera, G.U. Charge transfer equilibria between diamond and an aqueous oxygen electrochemical redox couple. Science 2007, 318, 1424–1430. [Google Scholar]
- Koizumi, S.; Kamo, M.; Sato, Y.; Ozaki, H.; Inuzuka, T. Growth and characterization of phosphorous doped (111) homoepitaxial diamond thin films. Appl. Phys. Lett 1997, 71, 1065–1067. [Google Scholar]
- Davis, R.F. Diamond Films and Coatings: Development, Properties and Applications; Noyes Publications: Park Ridge, NJ, USA, 1992. [Google Scholar]
- Mumm, D.R.; Faber, K.T.; Drory, M.D.; Gardinier, C.F. High-temperature hardness of chemically vapor-deposited diamond. J. Am. Ceram. Soc 1993, 76, 238–240. [Google Scholar]
- Takahashi, K.; Tanga, M.; Takai, O.; Okamura, H. DNA bonding to diamond. Bio. Ind 2000, 17, 44–51. [Google Scholar]
- Ushizawa, K.; Sato, Y.; Mitsumori, T.; Machinami, T.; Ueda, T.; Ando, T. Covalent immobilization of DNA on diamond and its verification by diffuse reflectance infrared spectroscopy. Chem. Phys. Lett 2002, 351, 105–108. [Google Scholar]
- Wang, J.; Firestone, M.A.; Auciello, O.; Carlisle, J.A. Functionalization of ultrananocrystalline diamond films by electrochemical reduction of aryldiazonium salts. Langmuir 2004, 20, 450–456. [Google Scholar]
- Christiaens, P.; Vermeeren, V.; Wenmackers, S.; Daenen, M.; Haenen, K.; Nesladek, M.; vandeVen, M.; Ameloot, M.; Michiels, L.; Wagner, P. EDC-mediated DNA attachment to nanocrystalline CVD diamond films. Biosens. Bioelectron 2006, 22, 170–177. [Google Scholar]
- Vermeeren, V.; Wenmackers, S.; Daenen, M.; Haenen, K.; Williams, O.A.; Ameloot, M.; Vande, V.M.; Wagner, P.; Michiels, L. Topographical and functional characterization of the ssDNA probe layer generated through EDC-mediated covalent attachment to nanocrystalline diamond using fluorescence microscopy. Langmuir 2008, 24, 9125–9134. [Google Scholar]
- Gu, H.; Su, X.; Loh, K.P. Electrochemical impedance sensing of DNA hybridization on conducting polymer film-modified diamond. J. Phys. Chem. B 2005, 109, 13611–13618. [Google Scholar]
- Nichols, B.M.; Butler, J.E.; Russell, J.N., Jr.; Hamers, R.J. Photochemical functionalization of hydrogen-terminated diamond surfaces: a structural and mechanistic study. J. Phys. Chem. B 2005, 109, 20938–20947. [Google Scholar]
- Yang, W.; Butler, J.E.; Russell, J.N., Jr.; Hamers, R.J. Interfacial electrical properties of DNA-modified diamond thin films: intrinsic response and hybridization-induced field effects. Langmuir 2004, 20, 6778–6787. [Google Scholar]
- Vermeeren, V.; Bijnens, N.; Wenmackers, S.; Daenen, M.; Haenen, K.; Williams, O.A.; Ameloot, M.; van de Ven, M.; Wagner, P.; Michiels, L. Towards a real-time, label-free, diamond-based DNA sensor. Langmuir 2007, 23, 13193–13202. [Google Scholar]
- D'Orazio, P. Biosensors in clinical chemistry. Clin. Chim. Acta 2003, 334, 41–69. [Google Scholar]
- Ye, Y.K.; Zhao, J.H.; Yan, F.; Zhu, Y.L.; Ju, H.X. Electrochemical behavior and detection of hepatitis B virus DNA PCR production at gold electrode. Biosens. Bioelectron 2003, 18, 1501–1508. [Google Scholar]
- Wang, J.; Kawde, A.N.; Jan, M.R. Carbon-nanotube-modified electrodes for amplified enzyme-based electrical detection of DNA hybridization. Biosens. Bioelectron 2004, 20, 995–1000. [Google Scholar]
- Park, S.J.; Taton, T.A.; Mirkin, C.A. Array-based electrical detection of DNA with nanoparticle probes. Science 2002, 295, 1503–1506. [Google Scholar]
- Cai, W.; Peck, J.R.; van der Weide, D.W.; Hamers, R.J. Direct electrical detection of hybridization at DNA-modified silicon surfaces. Biosens. Bioelectron 2004, 19, 1013–1019. [Google Scholar]
- Keighley, S.D.; Estrela, P.; Li, P.; Migliorato, P. Optimization of label-free DNA detection with electrochemical impedance spectroscopy using PNA probes. Biosens. Bioelectron 2008, 24, 912–917. [Google Scholar]
- Young, H.D.; Freedman, R.A. University Physics, 10th ed; Addison Wesley: Boston, MA, USA, 2000. [Google Scholar]
- Ingebrandt, S.; Offenhäusser, A. Label-free detection of DNA using field-effect transistors. Phys. Stat. Sol 2006, 203, 3399–3411. [Google Scholar]
- Song, K.S.; Zhang, G.J.; Nakamura, Y.; Furukawa, K.; Hiraki, T.; Yang, J.H.; Funatsu, T.; Ohdomari, I.; Kawarada, H. Label-free DNA sensors using ultrasensitive diamond field-effect transistors in solution. Phys. Rev. E. Stat. Nonlin. Soft. Matter Phys 2006, 74, 041919. [Google Scholar]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar]
- Wu, C.F.; Valdes, J.J.; Bentley, W.E.; Sekowski, J.W. DNA microarray for discrimination between pathogenic 0157:H7 EDL933 and non-pathogenic Escherichia Coli strains. Biosens. Bioelectron 2003, 19, 1–8. [Google Scholar]
- Schwonbeck, S.; Krause-Griep, A.; Gajovic-Eichelmann, N.; Ehrentreich-Forster, E.; Meinl, W.; Glatt, H.; Bier, F.F. Cohort analysis of a single nucleotide polymorphism on DNA chips. Biosens. Bioelectron 2004, 20, 956–966. [Google Scholar]
- Bernhard, B.; Lengeler, B. Electronic Structure of Noble Metals and Polariton-Mediated Light Scattering; Springer-Verlag: Berlin, Germany, 1978. [Google Scholar]
- Flanagan, M.T.; Pantell, R.H. Surface plasmon resonance and immunosensors. Electron. Lett 1984, 20, 968–970. [Google Scholar]
- Wang, R.; Tombelli, S.; Minunni, M.; Spiriti, M.M.; Mascini, M. Immobilisation of DNA probes for the development of SPR-based sensing. Biosens. Bioelectron 2004, 20, 967–974. [Google Scholar]
- Sauerbrey, G. Verwendung von schwingquarzen zur wägung dünner schichten und zur mikrowägung. Z. Phys 1959, 155, 206–222. [Google Scholar]
- Feng, K.; Li, J.; Jiang, J.H.; Shen, G.L.; Yu, R.Q. QCM detection of DNA targets with single-base mutation based on DNA ligase reaction and biocatalyzed deposition amplification. Biosens. Bioelectron 2007, 22, 1651–1657. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Diamond | Silicon | Germanium |
|---|---|---|---|
| Thermal expansion (×10−6.K−1) | 1.1 | 2.6 | 5.57 |
| Band gap (eV) | 5.47 | 1.12 | 0.66 |
| Carrier mobility (cm2.V−1.s−1) | |||
| → electron | 2,200 | 1,500 | 3,900 |
| → hole | 1,600 | 475 | 1,900 |
| Breakdown voltage (× 105.V.cm−1) | 100 | 3 | 1 |
| Dielectric constant | 5.5 | 11.9 | 16.2 |
| Resistivity (Ω.cm) | 1013 | 103 | 46–60 |
| Thermal conductivity (W.cm−1.K−1) | 9–23 | 1.68 | 0.599 |
| Refractive index | 2.42 | 3.5 | 4 |
| Hardness (kg.mm−2) | 8,000 | 1,150 | 780 |
| Biomolecule | Type biosensor |
|---|---|
| Enzyme | Catalytic or enzyme biosensor |
| Affinity-complex forming biomolecules (membrane receptor, aptamer, protein, antibody, …) | Affinity-based biosensor |
| → antibody | → immunosensor |
| → DNA | → DNA-based biosensor |
| Cell | Whole-cell biosensor |
| Measured parameter | Type biosensor |
|---|---|
| Electrochemistry | Electrochemical biosensor |
| → current | → amperometric biosensor |
| → charge | → coulometric biosensor |
| → voltage | → potentiometric biosensor |
| → conductivity | → conductometric biosensor |
| → impedance | → impedimetric biosensor |
| → field-effect | → field-effect transistor-based biosensor |
| Optics | Optical biosensors |
| → absorbtion | → colorimetric biosensor |
| → chemiluminescence | → chemiluminescent biosensor |
| → fluorescence (FRET*, reporter genes) | → fluorescent biosensor (cell-, array-based) |
| → refractive index | → Surface Plasmon Resonance biosensor |
| Mass | Piezo-electric biosensor |
| Bioreceptor | Transduction | Substrate | Target | SNP detection? | Limit of Detection | Ref. |
|---|---|---|---|---|---|---|
| Electrochemical | ||||||
| HBV ssDNA | amperometric (CV) | Au | HBV dsDNA amplicons | No | 2 fM | [37] |
| Biotinylated BRCA 1 ssDNA | potentiometric | magnetic beads | AP-BRCA 1 ssDNA | No | 6.6 pM | [38] |
| SH-ssDNA | conductimetric | Si/SiO2 | Au-ssDNA | No | 50 nM | [39] |
| SH-ssDNA | impedimetric | p-type NCD | ssDNA | No | Undetermined (5 μM used) | [7] |
| SH-ssDNA | impedimetric | n-type Si | ssDNA | No | Undetermined (3 μM used) | [40] |
| NH2-ssDNA | impedimetric | p-type PCD | ssDNA | Yes | 20 nM | [32] |
| NH2-ssDNA | impedimetric | p-type NCD | ssDNA | Yes | Undetermined (4 μM used) | [35] |
| SH-ssPNA | impedimetric | Au | ssDNA + ferri/ferrocyanide | No | 1 nM | [41] |
| NH2-ssDNA | field-effect | n-type Si | ssDNA | No | Undetermined (3 μM used) | [43] |
| NH2-ssDNA | p-type PCD | ssDNA | Yes | 100 pM | [44] | |
| Optical | ||||||
| E. Coli ssDNA | fluorescent | oligonucleotide array | - O157:H7 Cy5- ssDNA - K12 Cy 3- ssDNA | No | Undetermined (2–3 μg used) | [46] |
| SULT1A1*2 ssDNA | fluorescent | oligonucleotide array | SULT1A1*2 ssDNA | No | Undetermined (500 nM used) | [47] |
| SH-ssDNA | SPR | Au | ssDNA | No | - BIAcore™: 2.5 nM - SPREETA: 10 nM | [50] |
| Piezo-electric | ||||||
| SH-ssDNA | QCM | Au coated quartz | ssDNA + detection probe | Yes | 100 pM | [52] |
© 2009 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vermeeren, V.; Wenmackers, S.; Wagner, P.; Michiels, L. DNA Sensors with Diamond as a Promising Alternative Transducer Material. Sensors 2009, 9, 5600-5636. https://doi.org/10.3390/s90705600
Vermeeren V, Wenmackers S, Wagner P, Michiels L. DNA Sensors with Diamond as a Promising Alternative Transducer Material. Sensors. 2009; 9(7):5600-5636. https://doi.org/10.3390/s90705600
Chicago/Turabian StyleVermeeren, Veronique, Sylvia Wenmackers, Patrick Wagner, and Luc Michiels. 2009. "DNA Sensors with Diamond as a Promising Alternative Transducer Material" Sensors 9, no. 7: 5600-5636. https://doi.org/10.3390/s90705600
APA StyleVermeeren, V., Wenmackers, S., Wagner, P., & Michiels, L. (2009). DNA Sensors with Diamond as a Promising Alternative Transducer Material. Sensors, 9(7), 5600-5636. https://doi.org/10.3390/s90705600