
Glycosaminoglycan Binding and Non-Endocytic Membrane Translocation of Cell-Permeable Octaarginine Monitored by Real-Time In-Cell NMR Spectroscopy

Abstract
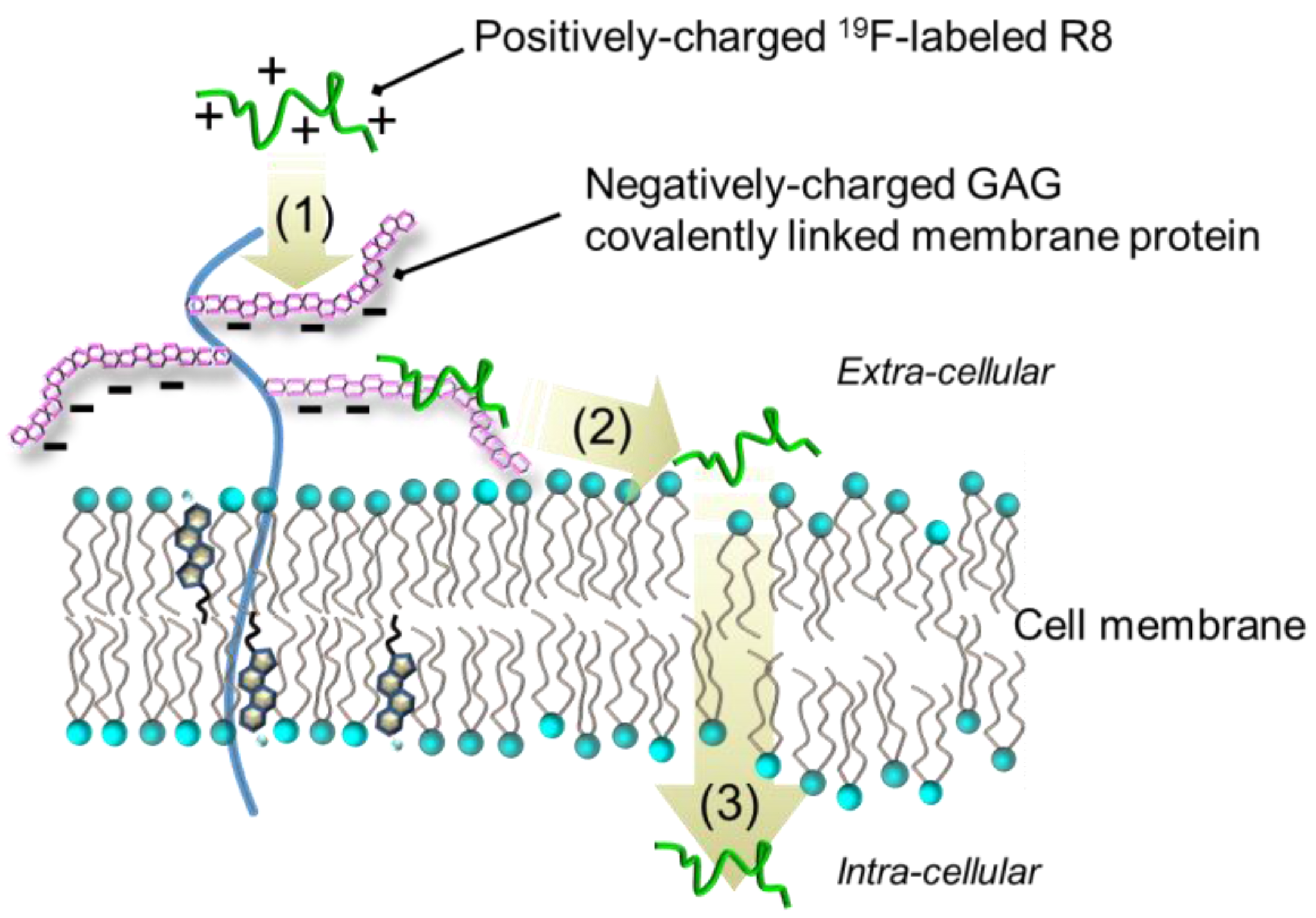
:1. Introduction
2. Results
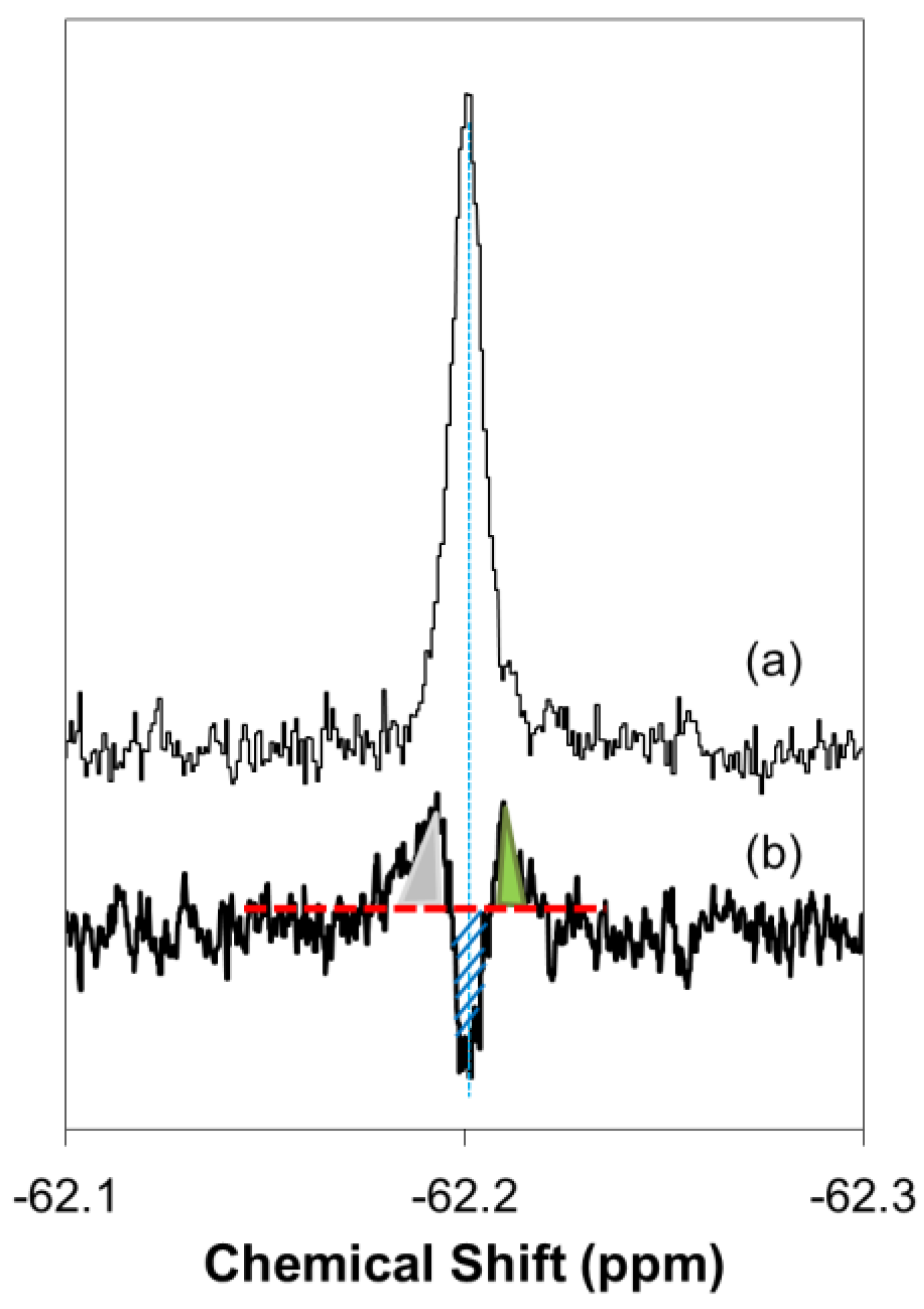
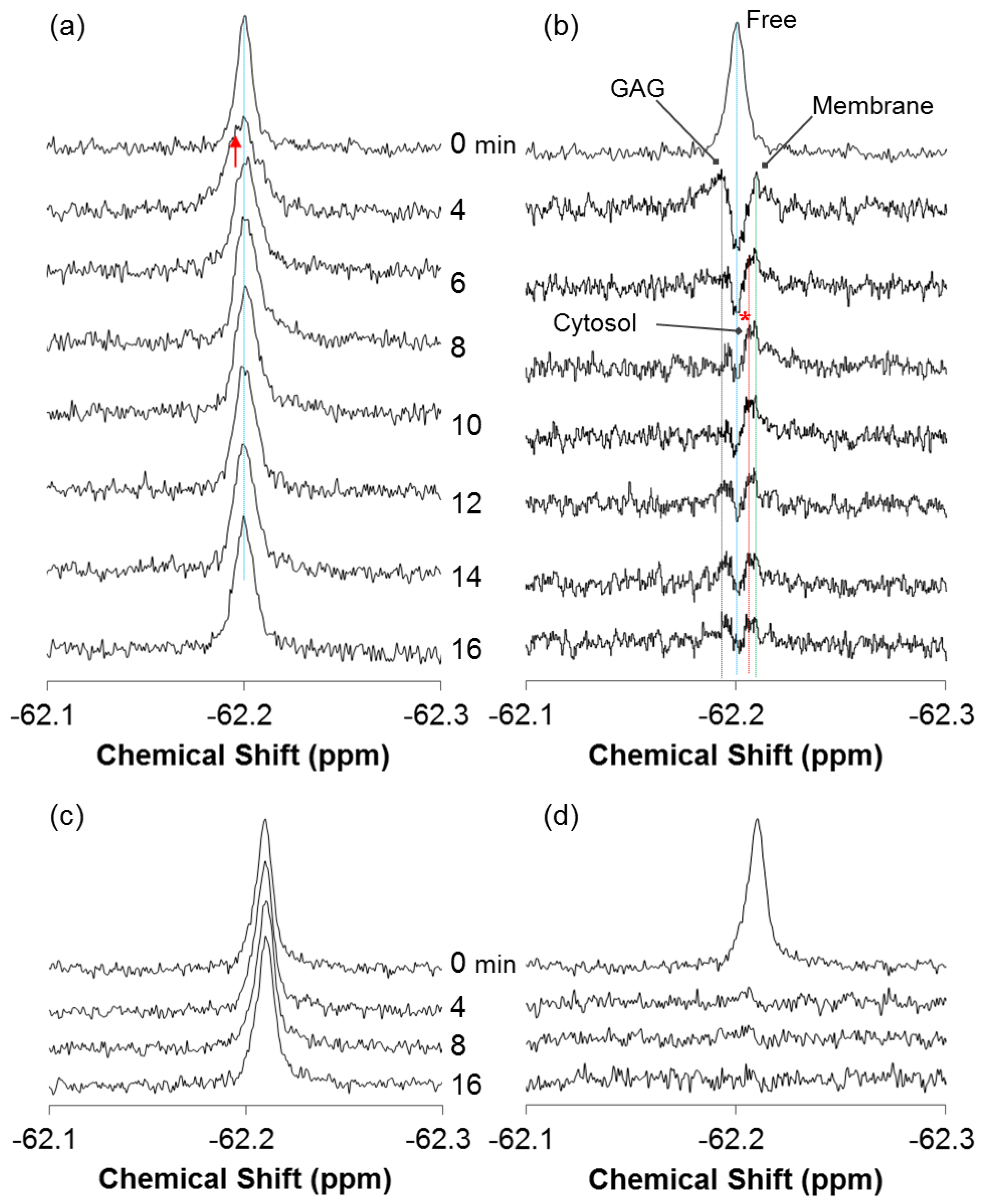
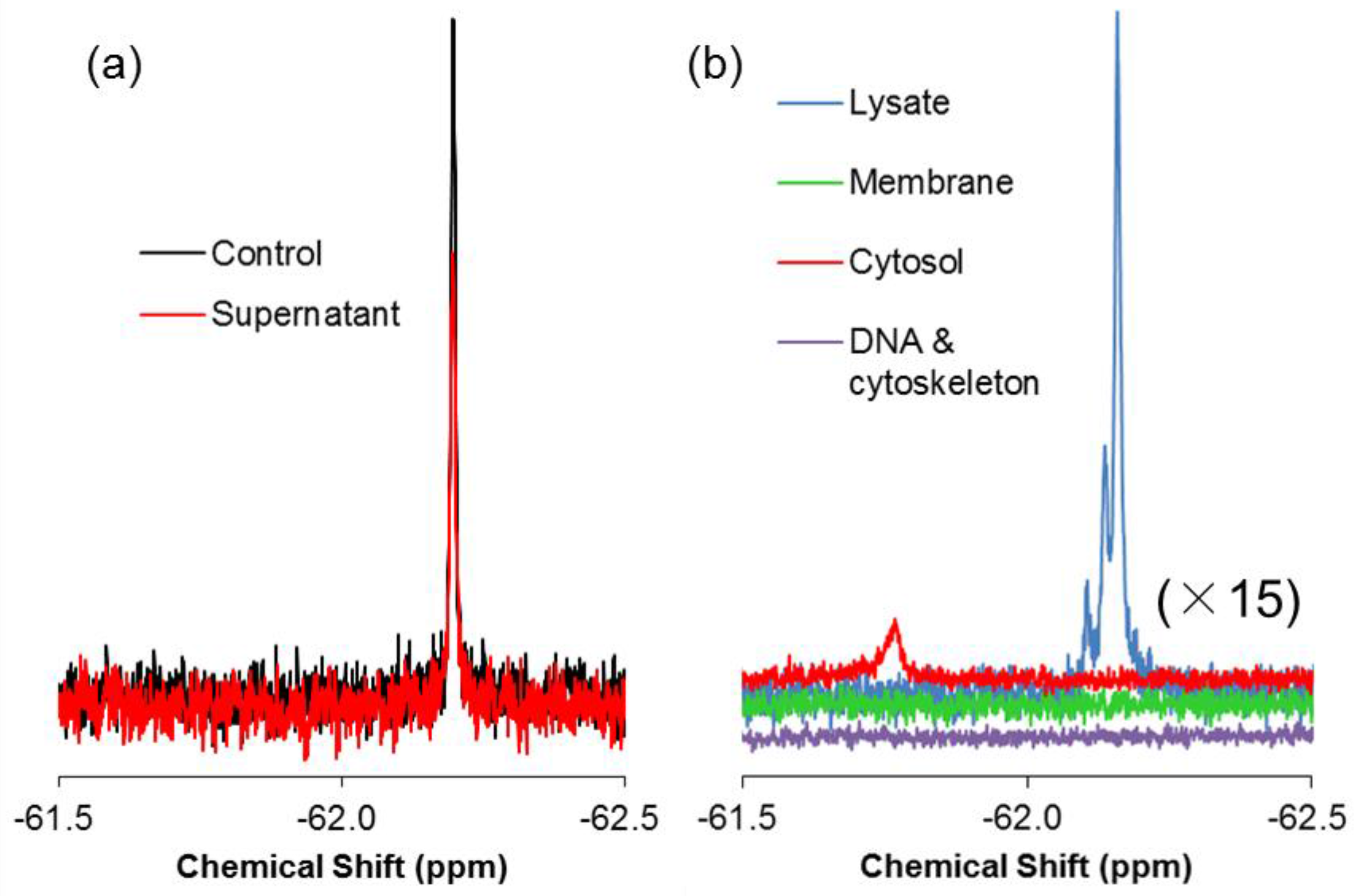
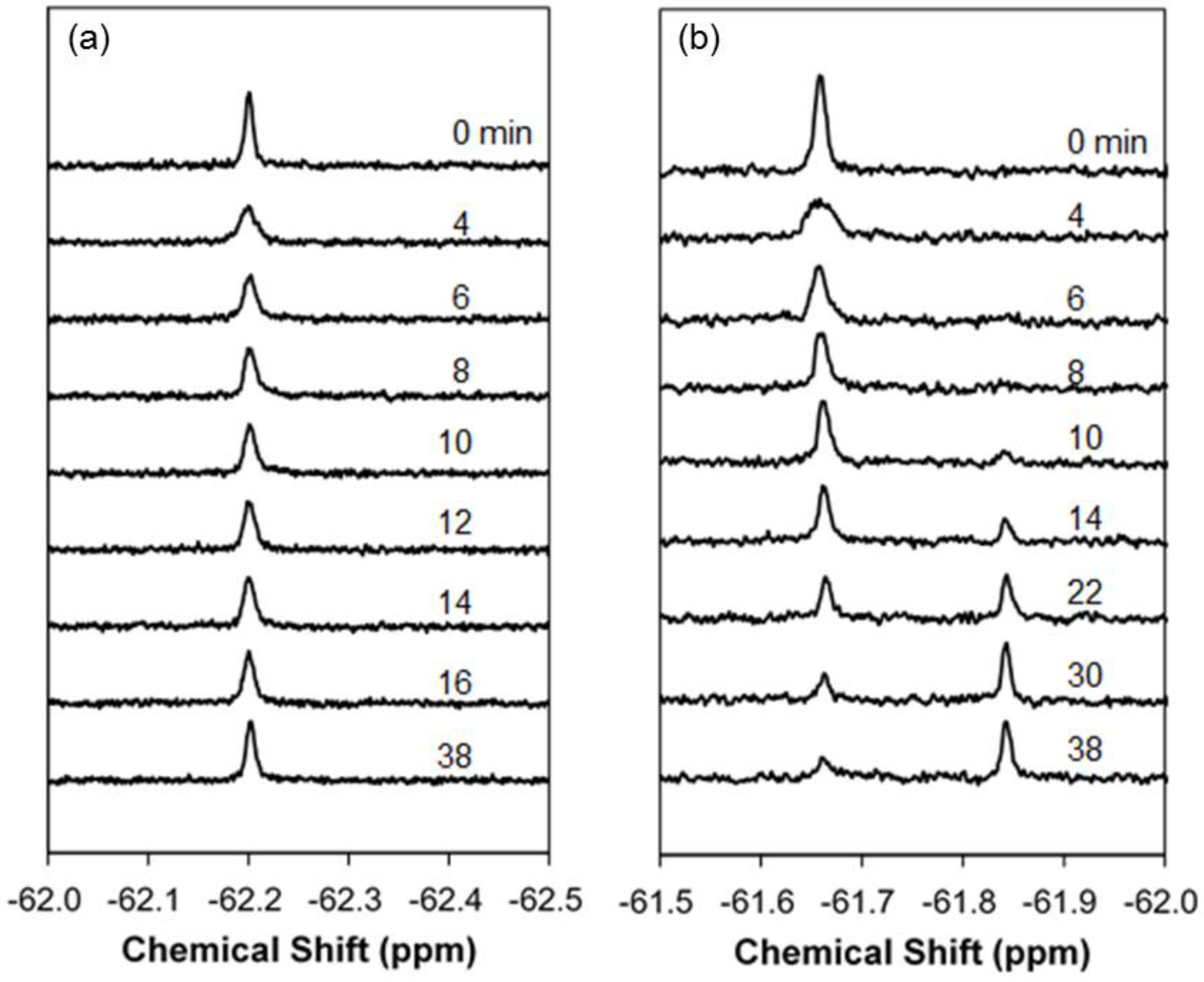
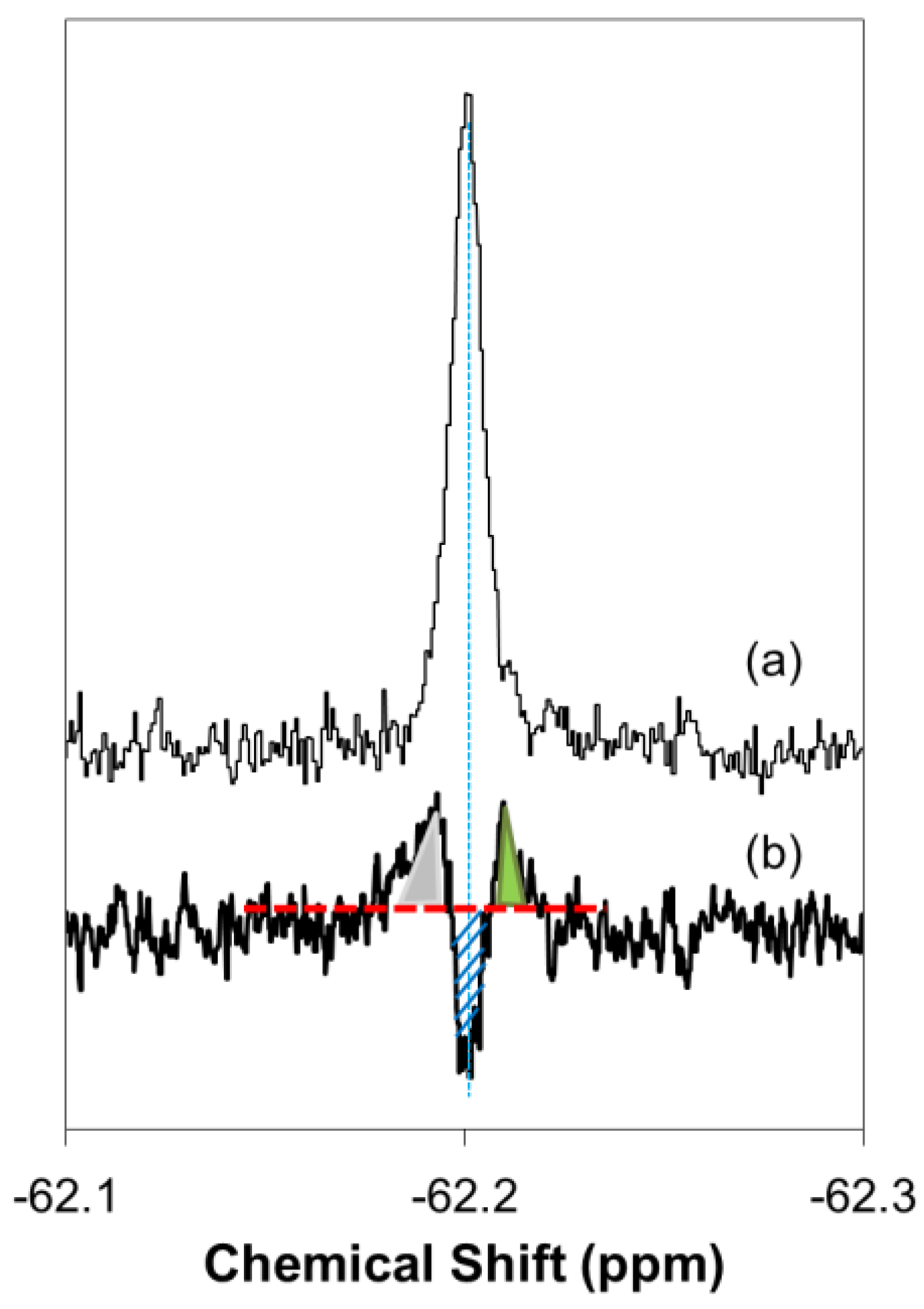
2.1. Real-Time In-Cell 19F NMR Spectra
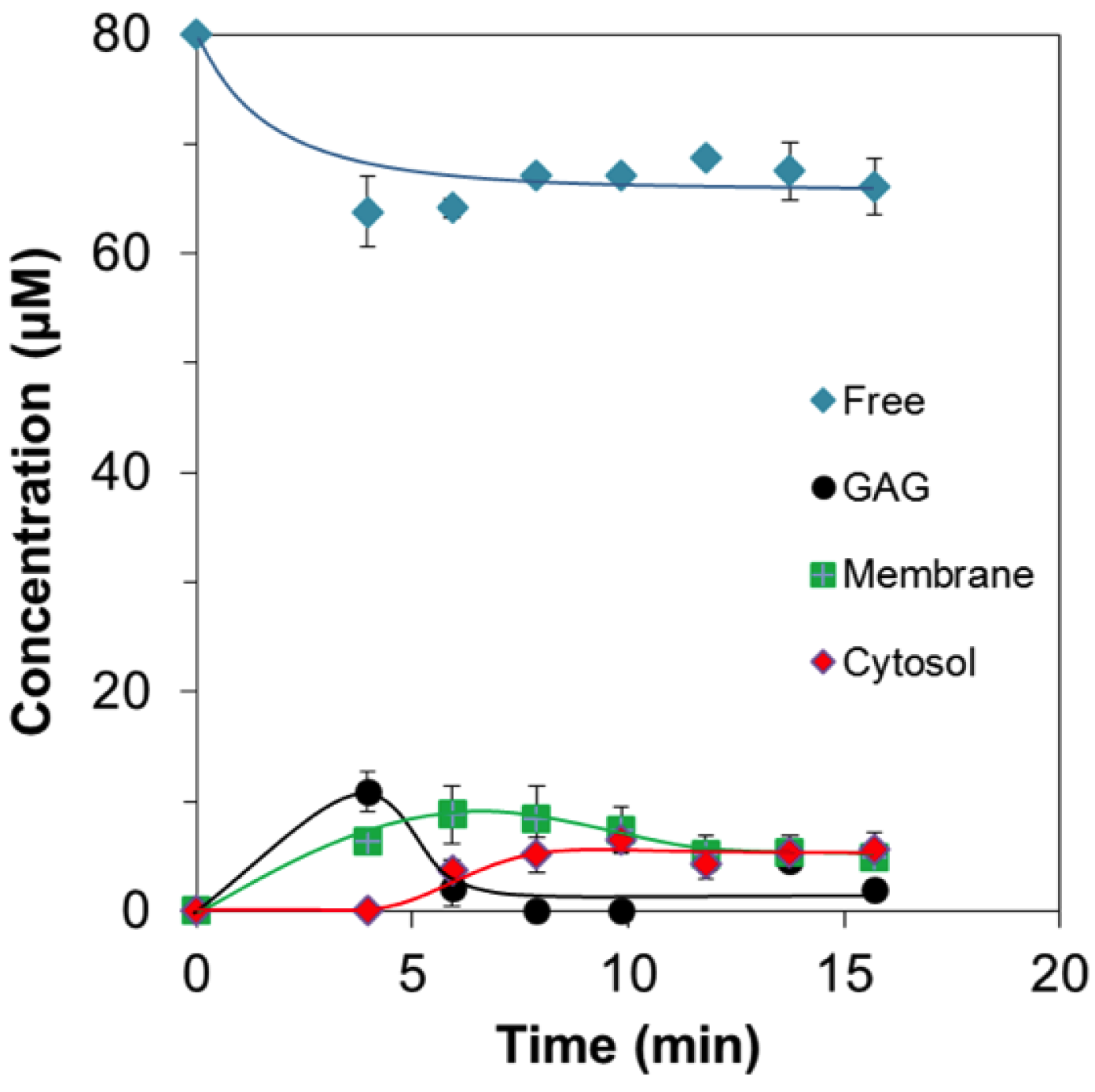
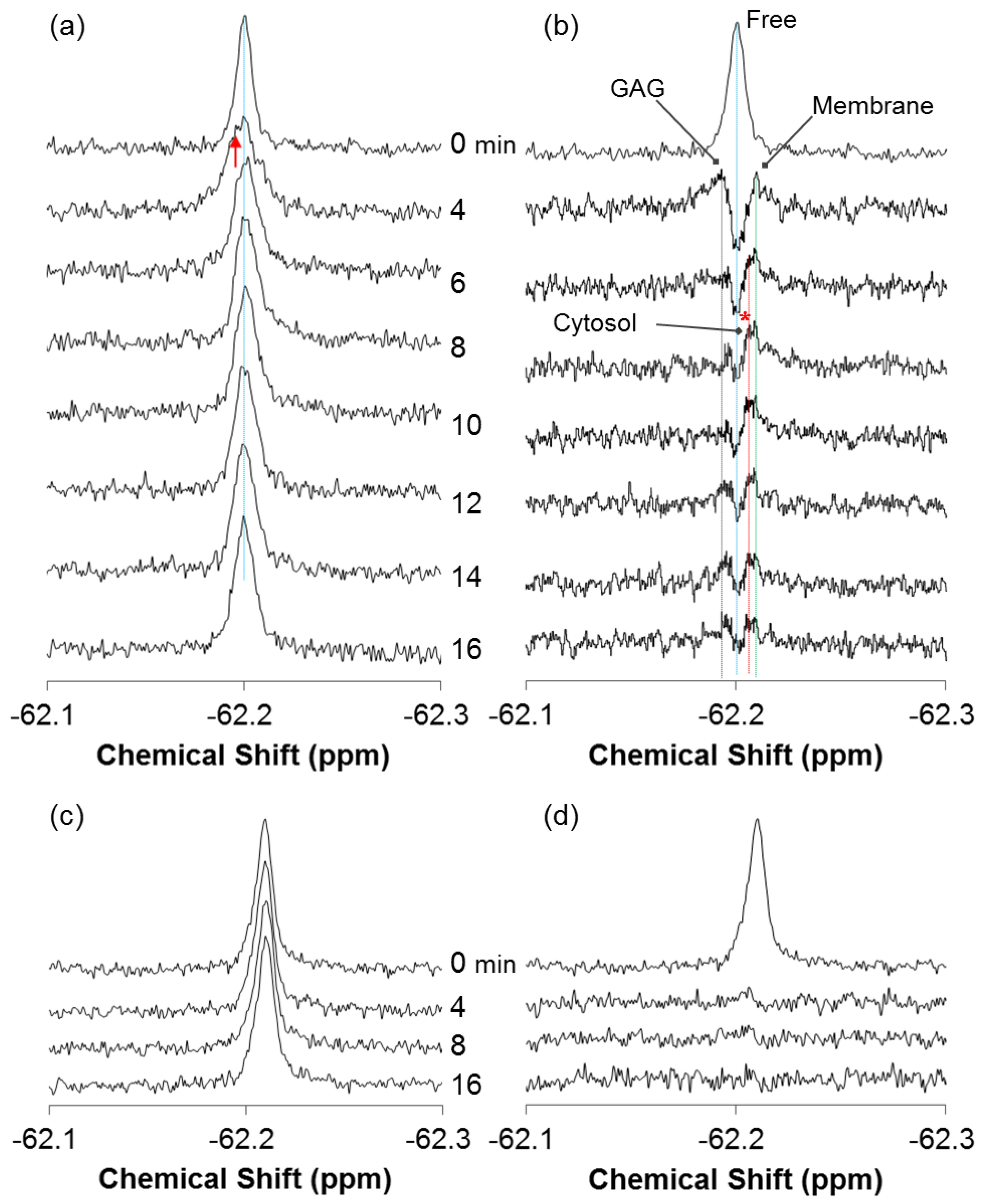
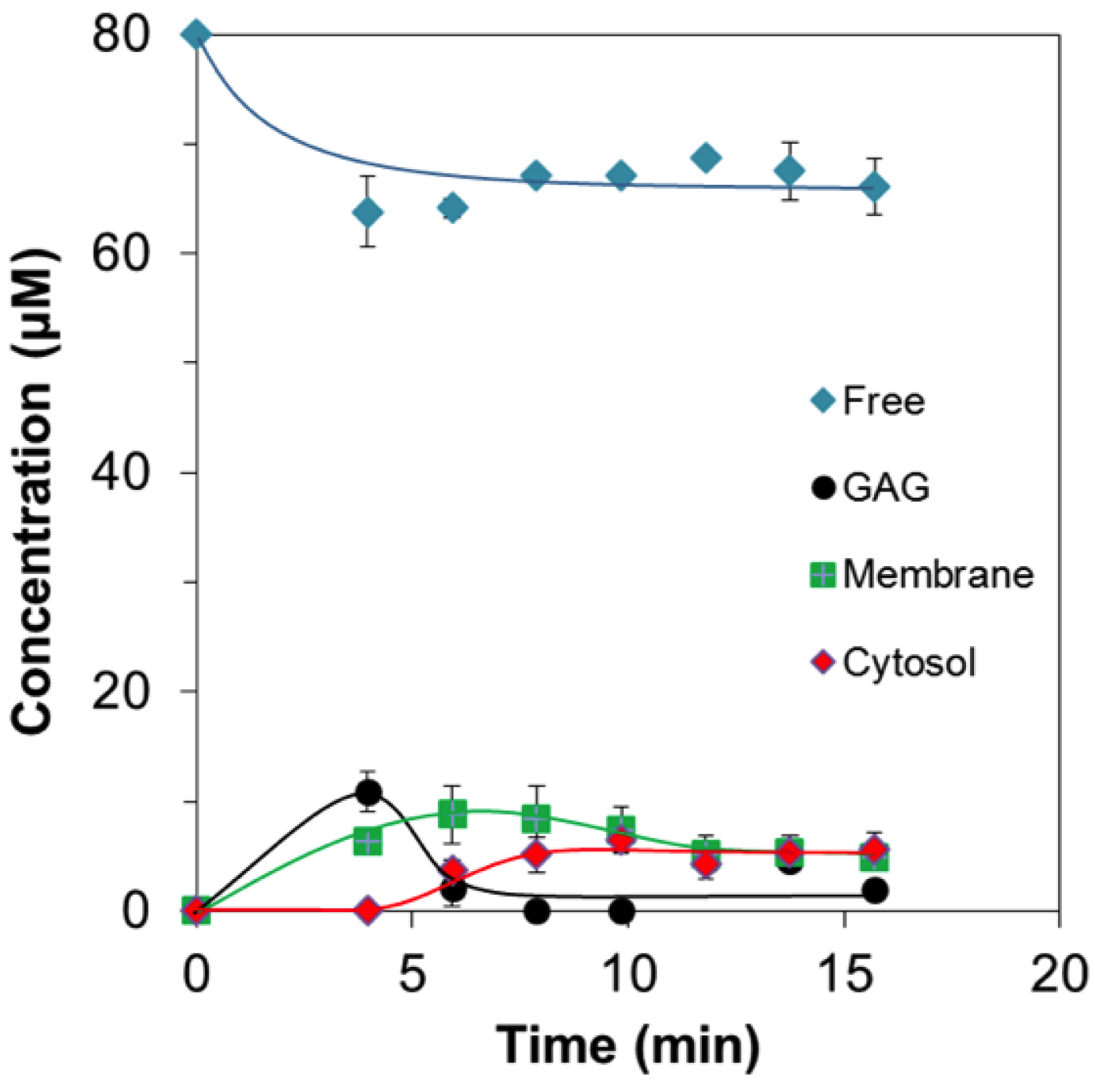
2.2. Quantitative Analysis of Direct Membrane Translocation
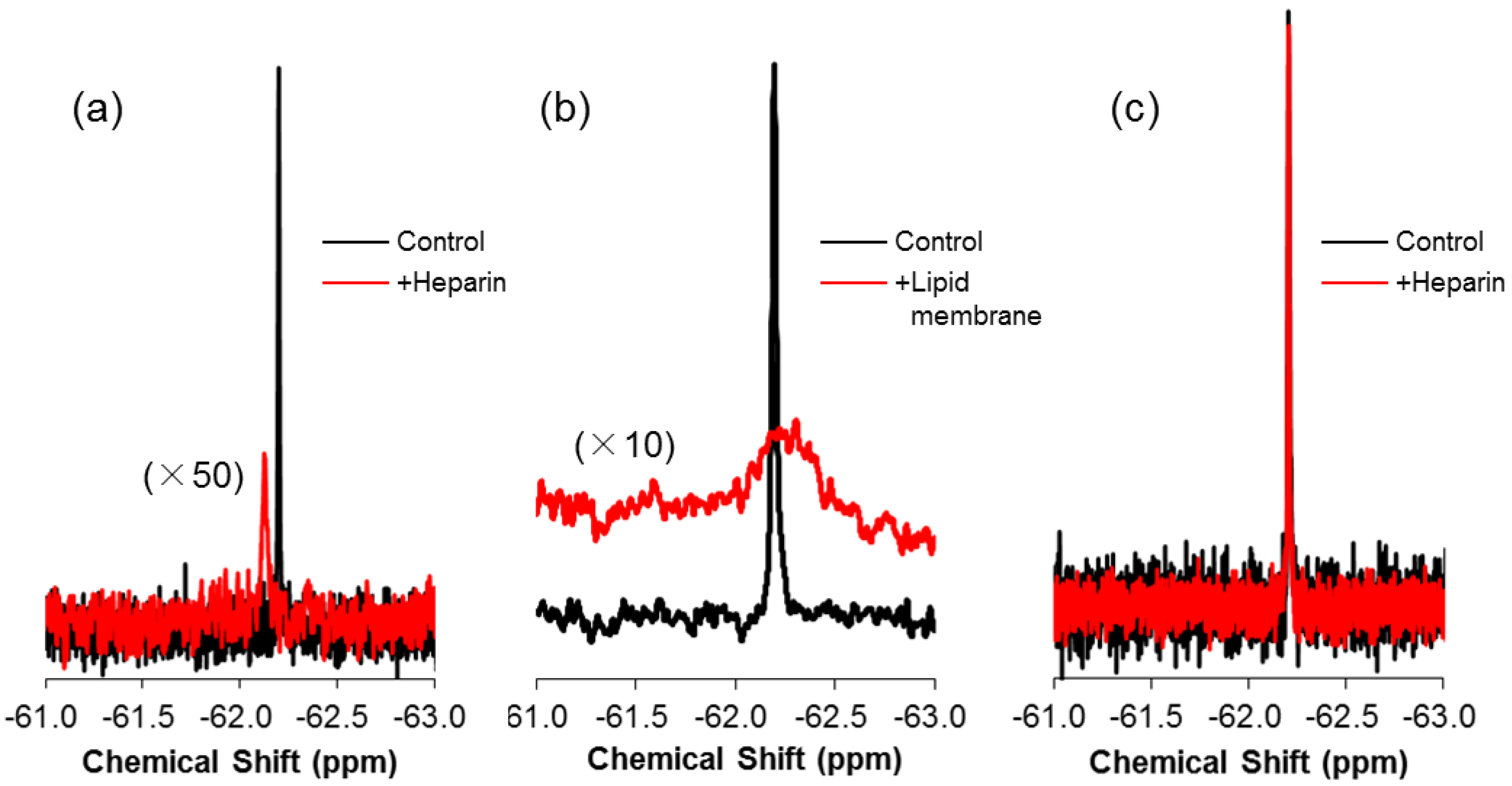
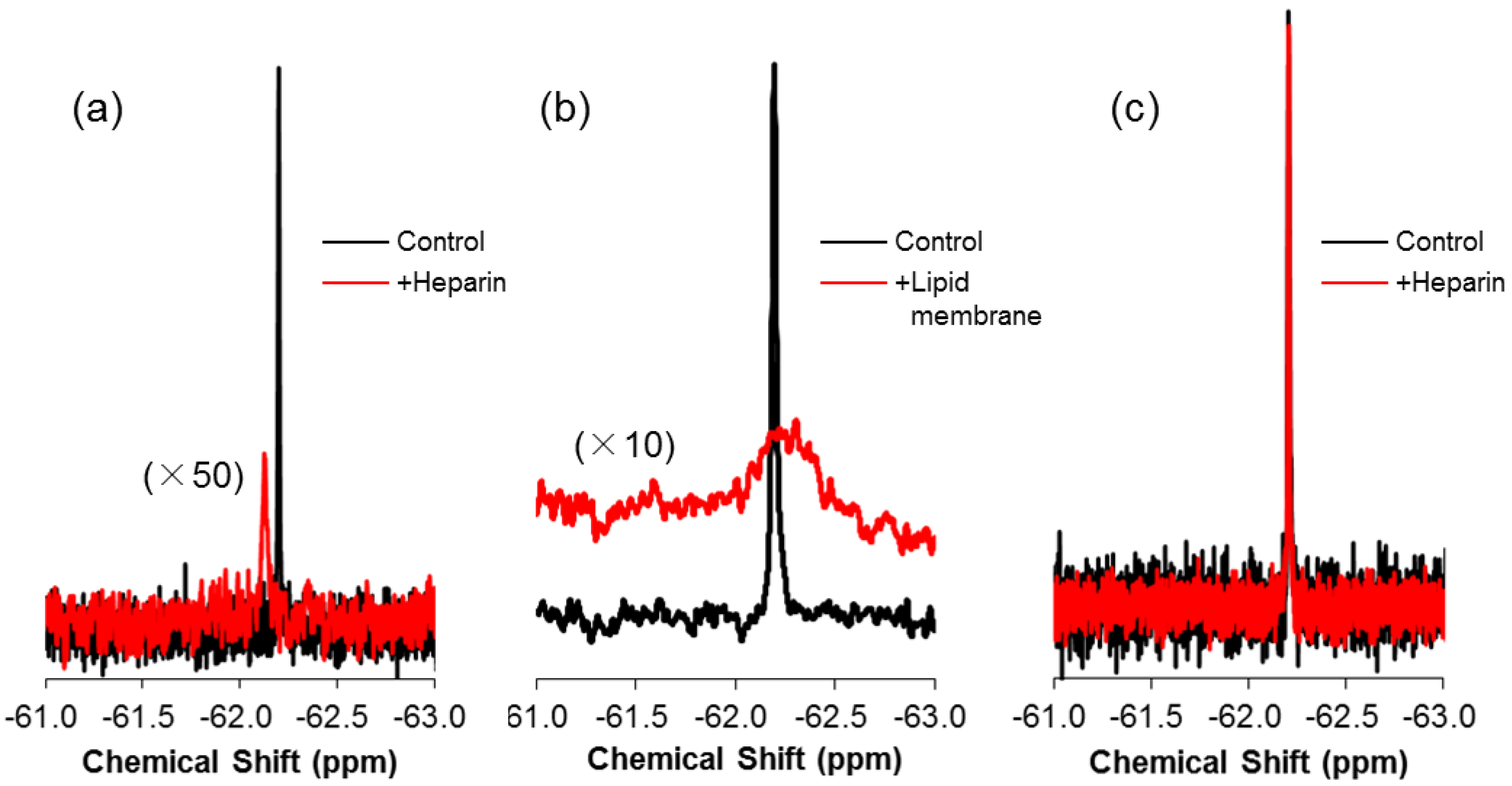
2.3. 19F-R8 Distribution under Equilibrium
3. Discussion
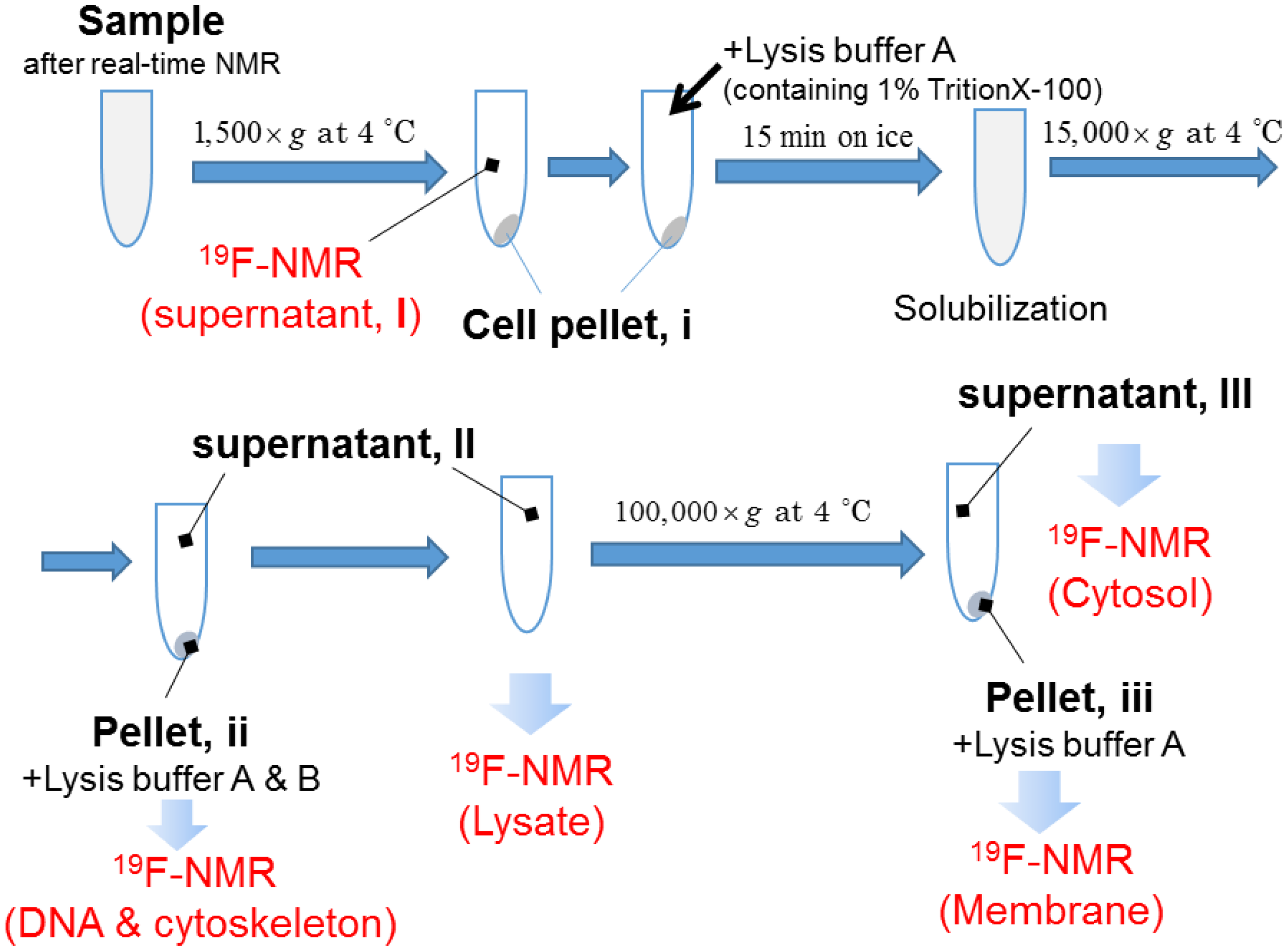
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Preparation of Large Unilamellar Vesicle (LUV)
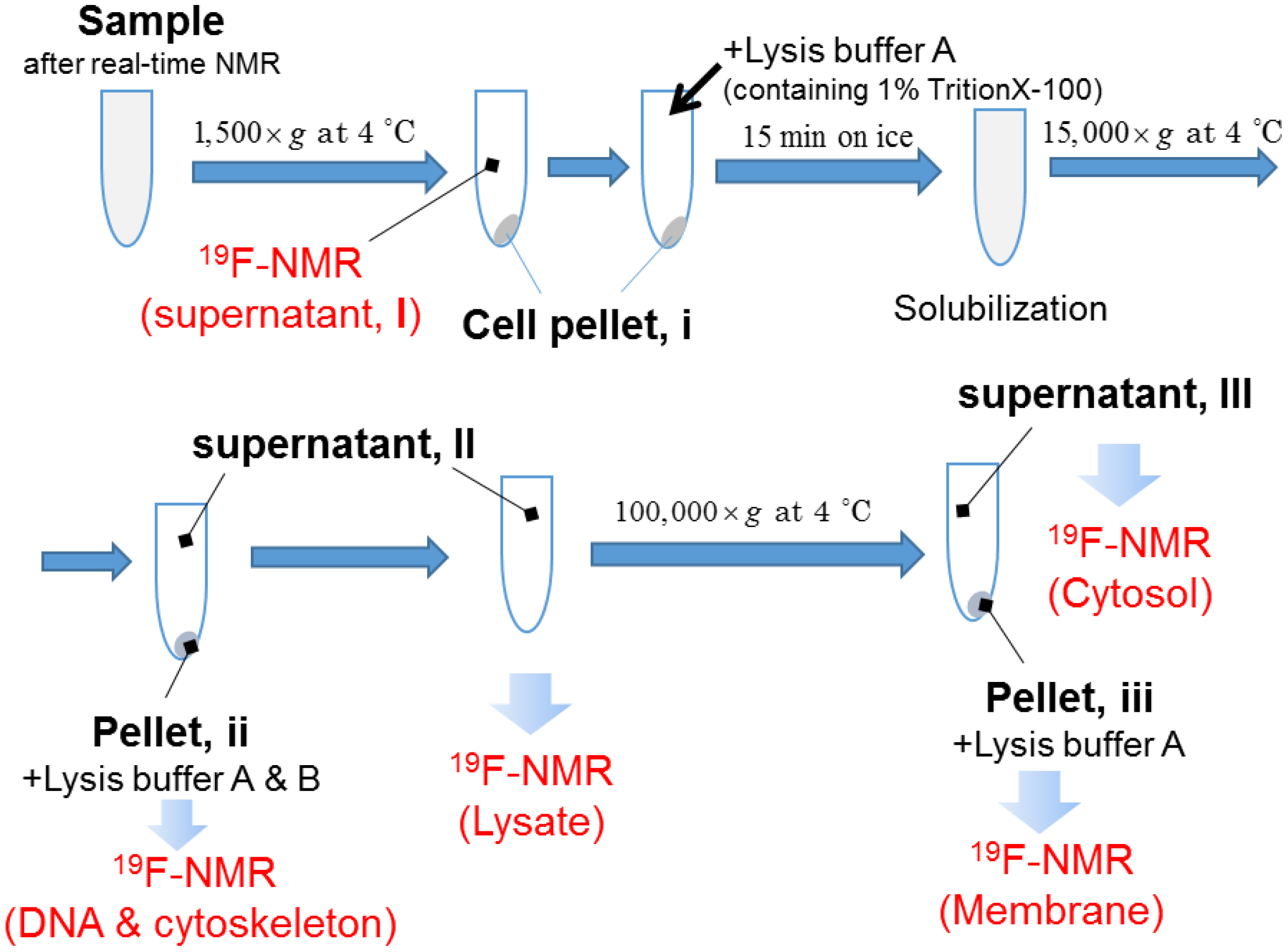
4.4. Real-Time In-Cell 19F NMR Measurement
4.5. Steady State 19F NMR Measurement
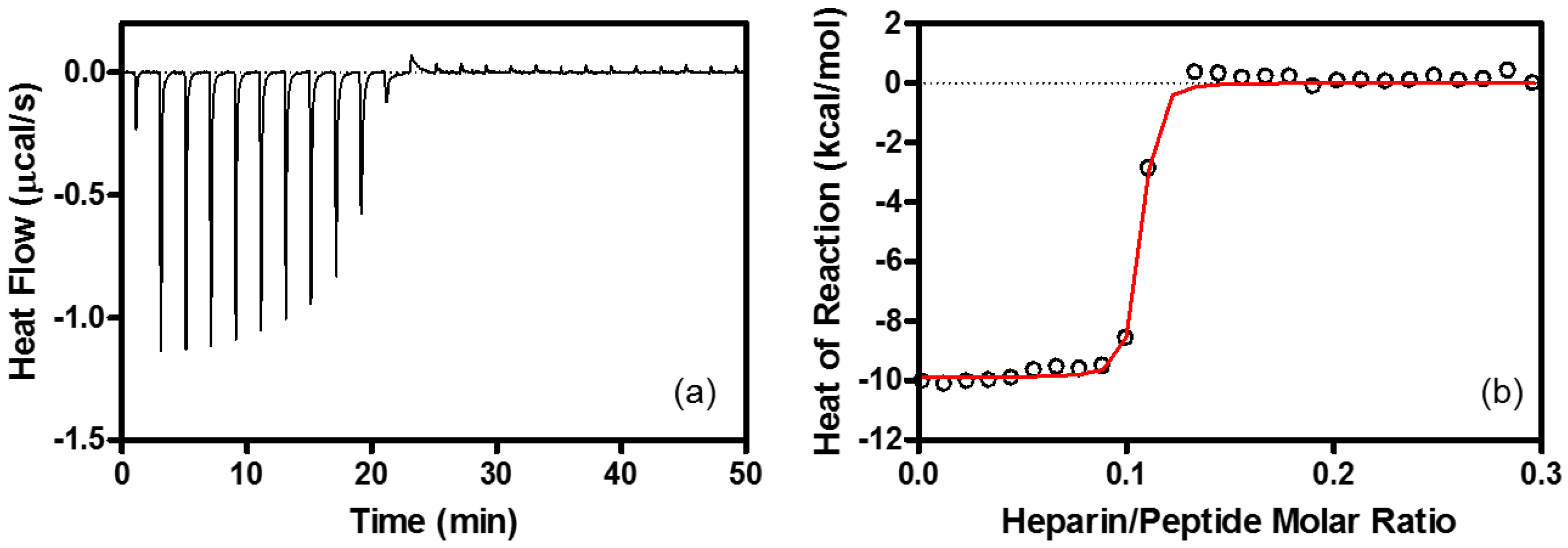
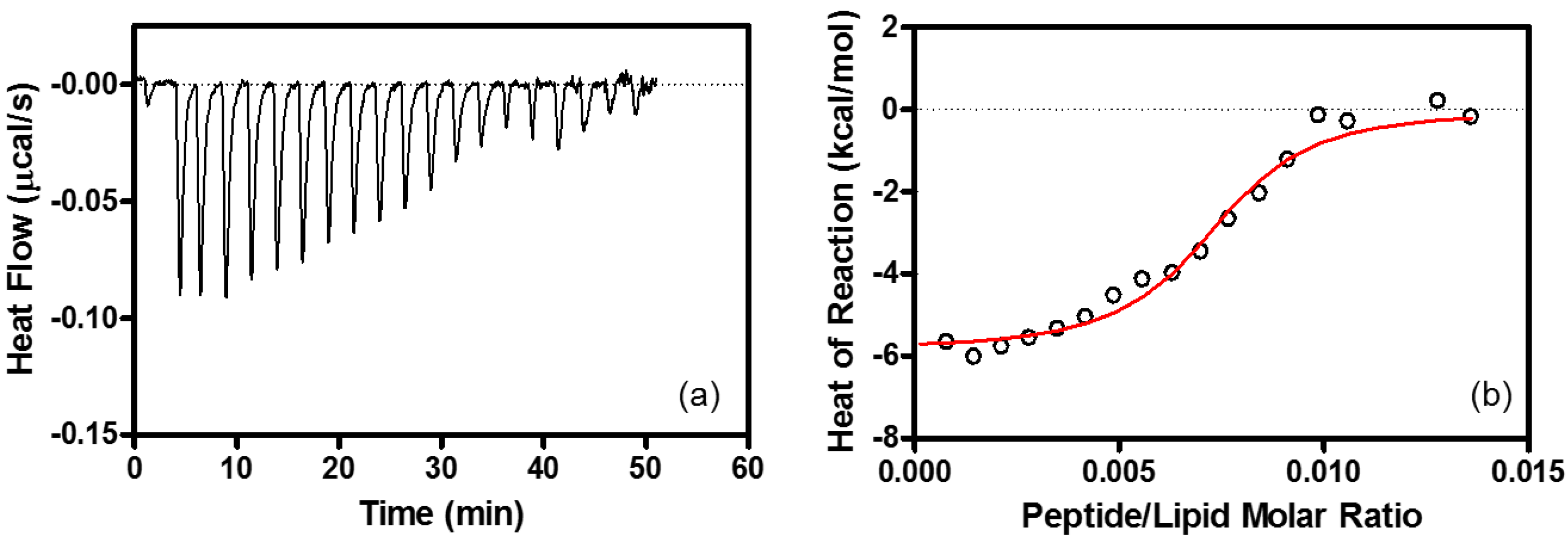
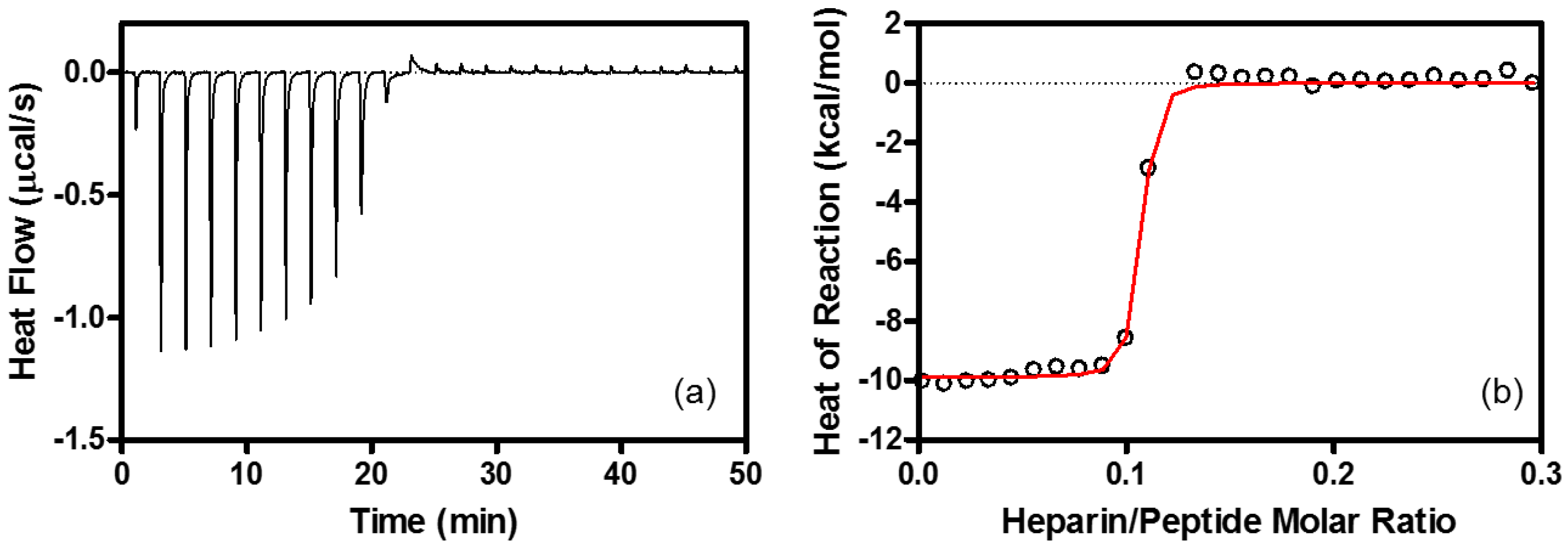
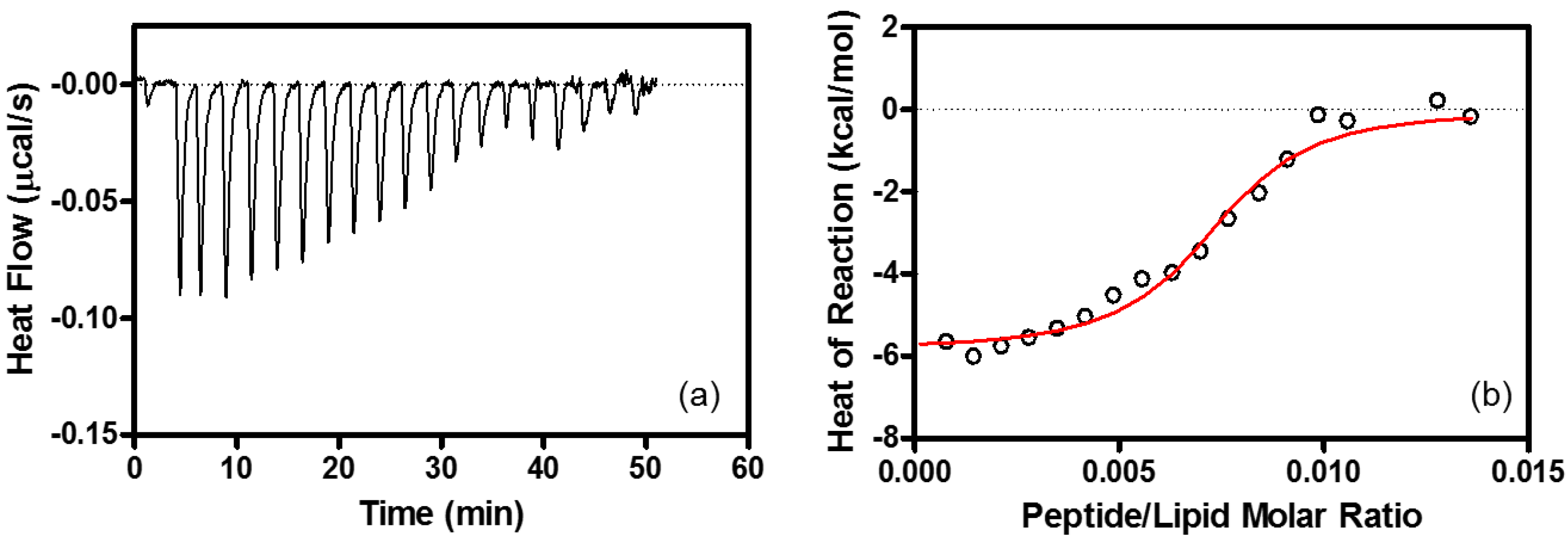
4.6. Isothermal Titration Calorimetry (ITC)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Concentration Analysis
References
- Trabulo, S.; Cardoso, A.L.; Mano, M.; de Lima, M.C.P. Cell-penetrating peptides-mechanisms of cellular uptake and generation of delivery systems. Pharmaceuticals 2010, 3, 961–993. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.A.; Mussbach, F.; Breitling, R.; Hemmerich, P.; Schaefer, B.; Lorkowski, S.; Reissmann, S. Relationships between cargo, cell penetrating peptides and cell type for uptake of non-covalent complexes into live cells. Pharmaceuticals 2013, 6, 184–203. [Google Scholar] [CrossRef] [PubMed]
- Howl, J.; Matou-Nasri, S.; West, D.C.; Farquhar, M.; Slaninová, J.; Östenson, C.G.; Zorko, M.; Östlund, P.; Kumar, S.; Langel, Ü.; et al. Bioportide: An emergent concept of bioactive cell-penetrating peptides. Cell. Mol. Life Sci. 2012, 69, 2951–2966. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.R.; Lin, M.D.; Chiang, H.J.; Lee, H.; Revon, B. Arginine-rich cell-penetrating peptides deliver gene into living human cells. Gene 2012, 505, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides and tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, M.; Takeuchi, T.; Nakase, I.; Jones, A.T.; Futaki, S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug. Chem. 2008, 19, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.Y.; Delaroche, D.; Burlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and Endocytosis for Cell-Penetrating Peptide Internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef] [PubMed]
- Watkins, C.L.; Schmaljohann, D.; Futaki, S.; Jones, A.T. Low concentration thresholds of plasma membranes for rapid energy-independent translocation of a cell-penetrating peptide. Biochem. J. 2009, 420, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Broc, R. The uptake of arginine-rich cell-penetrating peptides: Putting the puzzle together. Bioconjug. Chem. 2014, 25, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A. Thermodynamic studies and binding mechanisms of cell-penetrating peptides with lipids and glycosaminoglycans. Adv. Drug Deliv. Rev. 2008, 60, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Amand, H.L.; Rydberg, H.A.; Fornander, L.H.; Lincoln, P.; Nordén, B.; Esbjörner, E.K. Cell surface binding and uptake of arginine- and lysine-rich penetratin peptides in absence and presence of proteoglycans. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.J.; Chatterjee, A.; Ganguli, M. Different roles of cell surface and exogenous glycosaminoglycans in controlling gene delivery by arginine-rich peptides with varied distribution of arginines. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Pallerla, M.; Zaltsman, Y.; Burlina, F.; Alves, I.D.; Lequin, O.; Sagan, S. Tryptophan within basic peptide sequences triggers glycosaminoglycan-dependent endocytosis. FASEB J. 2013, 27, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Hwee, G.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Lai, G.H.; Cheng, J.; Deming, T.J.; et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 16883–16888. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Seelig, J. Contributions of glycosaminoglycan binding and clustering to the biological uptake of the nonamphipathic cell-penetrating peptide WR9. Biochemistry 2011, 50, 4650–4664. [Google Scholar] [CrossRef] [PubMed]
- Silhol, M.; Tyagi, M.; Giacca, M.; Lebleu, B.; Vivès, E. Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. Eur. J. Biochem. 2002, 269, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; June, R.K.; Dowdy, S.F. Revised role of glycosaminoglycans in TAT protein transduction domain-mediated cellular transduction. J. Biol. Chem. 2010, 285, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Takechi-Haraya, Y.; Nadai, R.; Kimura, H.; Nishitsuji, K.; Uchimura, K.; Sakai-Kato, K.; Kawakami, K.; Shigenaga, A.; Kawakami, T.; Otaka, A.; et al. Enthalpy-driven interactions with sulfated glycosaminoglycans promote cell membrane penetration of arginine peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Meerovich, I.; Muthukrishnan, N.; Johnson, G.A.; Erazo-Oliveras, A.; Pellois, J.P. Photodamage of lipid bilayers by irradiation of a fluorescently labeled cell-penetrating peptide. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Okamura, E.; Ninomiya, K.; Futaki, S.; Nagai, Y.; Kimura, T.; Wakai, C.; Matubayasi, N.; Sugiura, Y.; Nakahara, M. Real-time in-cell 19F NMR study on uptake of fluorescent and nonfluorescent 19F-octaarginines into human jurkat cells. Chem. Lett. 2005, 34, 1064–1065. [Google Scholar] [CrossRef]
- Szeto, H.H.; Schiller, P.W.; Zhao, K.; Luo, G. Fluorescent dyes alter intracellular targeting and function of cell-penetrating tetrapeptides. FASEB J. 2005, 19, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Puckett, C.A.; Barton, J.K. Fluorescein redirects a ruthenium-octaarginine conjugate to the nucleus. J. Am. Chem. Soc. 2009, 131, 8738–8739. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.R.; Malvy, C.; Auguste, T.; Tóth, G.K.; Kiss-Ivánkovits, O.; Illyés, E.; Hollósi, M.; Bottka, S.; Laczkó, I. Synthesis and studies on cell-penetrating peptides. Bioconjug. Chem. 2009, 20, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Aubry, S.; Aussedat, B.; Delaroche, D.; Jiao, C.Y.; Bolbach, G.; Lavielle, S.; Chassaing, G.; Sagan, S.; Burlina, F. MALDI-TOF mass spectrometry: A powerful tool to study the internalization of cell-penetrating peptides. Biochim. Biophys. Acta Biomembr. 2010, 1798, 2182–2189. [Google Scholar] [CrossRef] [PubMed]
- Lécorché, P.; Walrant, A.; Burlina, F.; Dutot, L.; Sagan, S.; Mallet, J.; Desbat, B.; Chassaing, G.; Alves, I.D.; Lavielle, S. Cellular uptake and biophysical properties of galactose and/or tryptophan containing cell-penetrating peptides. Biochim. Biophys. Acta Biomembr. 2012, 1818, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Matheron, L.; Cribier, S.; Chaignepain, S.; Jobin, M.L.; Sagan, S.; Alves, I.D. Direct translocation of cell penetrating peptides in liposomes: A combined mass spectrometry quantification and fluorescence detection study. Anal. Biochem. 2013, 438, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Aki, K.; Okamura, E. D-β-aspartyl residue exhibiting uncommon high resistance to spontaneous peptide bond cleavage. Sci. Rep. 2016, 6, 21594. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.L.; Murphy, C.D. 19F NMR applications in chemical biology. J. Fluor. Chem. 2009, 130, 132–143. [Google Scholar] [CrossRef]
- Kitevski-LeBlanc, J.L.; Prosser, R.S. Current applications of 19F NMR to studies of protein structure and dynamics. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 62, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Serber, B.Z.; Corsini, L.; Durst, F.; Serber, Z.; Dötsch, V. In-cell NMR spectroscopy. Methods Enzymol. 2005, 394, 17–41. [Google Scholar] [PubMed]
- Suzuki, Y.; Brender, J.J.R.; Hartman, K.; Ramamoorthy, A.; Marsh, E.N.G. Alternative pathways of human islet amyloid polypeptide aggregation distinguished by 19F NMR-detected kinetics of monomer consumption. Biochemistry 2012, 51, 8154–8162. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Li, D.; Chen, H.; Xiong, Y.; Wang, Y.; Tian, C. In situ 19F NMR studies of an E. coli membrane protein. Protein Sci. 2012, 21, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Kitevski-Leblanc, J.L.; Hoang, J.; Thach, W.; Larda, S.T.; Prosser, R.S. 19F NMR studies of a desolvated near-native protein folding intermediate. Biochemistry 2013, 52, 5780–5789. [Google Scholar] [CrossRef] [PubMed]
- De la Vega, M.; Marin, M.; Kondo, N.; Miyauchi, K.; Kim, Y.; Epand, R.F.; Emapnd, R.M.; Melikyan, G.B.; de la Vega, M. Inhibition of HIV-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology 2011, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Eckenrode, H.M.; Dounce, S.M.; Dai, H. Time-resolved molecular transport across living cell membranes. Biophys. J. 2013, 104, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Palm, C.; Jayamanne, M.; Kjellander, M.; Hällbrink, M. Peptide degradation is a critical determinant for cell-penetrating peptide uptake. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; VanderNoot, V.A.; Mealy, T.R.; Seaton, B.A.; Linhardt, R.J. Interaction of heparin with annexin V. FEBS Lett. 1999, 446, 327–330. [Google Scholar] [CrossRef]
- Ziegler, A.; Seelig, J. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: Binding mechanism and thermodynamic parameters. Biophys. J. 2004, 86, 254–263. [Google Scholar] [CrossRef]
- Futamura, M.; Dhanasekaran, P.; Handa, T.; Phillips, M.C.; Lund-Katz, S.; Saito, H. Two-step mechanism of binding of apolipoprotein E to heparin: Implications for the kinetics of apolipoprotein E-heparan sulfate proteoglycan complex formation on cell surfaces. J. Biol. Chem. 2005, 280, 5414–5422. [Google Scholar] [CrossRef] [PubMed]
- Blaum, B.S.; Deakin, J.A.; Johansson, C.M.; Herbert, A.P.; Barlow, P.N.; Lyon, M.; Uhrín, D. Lysine and arginine side chains in glycosaminoglycan-protein complexes investigated by NMR, cross-linking, and mass spectrometry: A case study of the factor H-heparin interaction. J. Am. Chem. Soc. 2010, 132, 6374–6381. [Google Scholar] [CrossRef] [PubMed]
- Noborn, F.; Callaghan, P.O.; Hermansson, E.; Zhang, X.; Ancsin, J.B.; Damas, A.M.; Dacklin, I.; O’Callaghan, P.; Presto, J.; Johansson, J.; et al. Heparan sulfate/heparin promotes transthyretin fibrillization through selective binding to a basic motif in the protein. Proc. Natl. Acad. Sci. USA 2011, 108, 5584–5589. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Bourgault, S.; Powers, E.T.; Kelly, J.W. Heparin binds 8 kDa gelsolin cross-β-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 2011, 50, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, E.; Kitas, E.; Seelig, J. Binding of oligoarginine to membrane lipids and heparan sulfate: Structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry 2005, 44, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Okamura, E.; Yoshii, N. Drug binding and mobility relating to the thermal fluctuation in fluid lipid membranes. J. Chem. Phys. 2008, 129, 215102. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Simeonova, M.; Gameiro, P.; Rangel, M.; Ivanova, G. Interaction of 5-fluorouracil loaded nanoparticles with 1,2-dimyristoyl-sn-glycero-3-phosphocholine liposomes used as a cellular membrane model. J. Phys. Chem. B 2012, 116, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Esbjörner, E.K.; Lincoln, P.; Nordén, B. Counterion-mediated membrane penetration: Cationic cell-penetrating peptides overcome Born energy barrier by ion-pairing with phospholipids. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Vorobyov, I.; Allen, T.W. On the role of anionic lipids in charged protein interactions with membranes. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Wider, G.; Dreier, L. Measuring protein concentrations by NMR spectroscopy. J. Am. Chem. Soc. 2006, 128, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Deleu, M.; Crowet, J.M.; Nasir, M.N.; Lins, L. Complementary biophysical tools to investigate lipid specificity in the interaction between bioactive molecules and the plasma membrane: A review. Biochim. Biophys. Acta Biomembr. 2014, 1838, 3171–3190. [Google Scholar] [CrossRef] [PubMed]
- Belting, M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem. Sci. 2003, 28, 145–151. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Perret, F.; Nishihara, M.; Takeuchi, T.; Futaki, S.; Lazar, A.N.; Coleman, A.W.; Sakai, N.; Matile, S. Anionic fullerenes, calixarenes, coronenes, and pyrenes as activators of oligo/polyarginines in model membranes and live cells. J. Am. Chem. Soc. 2005, 127, 1114–1115. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J. Am. Chem. Soc. 2014, 136, 17459–17467. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Jessop, T.C.; Wender, P.A. Adaptive translocation: The role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv. Drug Deliv. Rev. 2005, 57, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Waring, A.J.; Hong, M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Waring, A.J.; Ruchala, P.; Hong, M. Membrane-bound dynamic structure of an arginine-rich cell-penetrating peptide, the protein transduction domain of HIV TAT, from solid-state NMR. Biochemistry 2010, 49, 6009–6020. [Google Scholar] [CrossRef] [PubMed]
- Takechi, Y.; Saito, H.; Okamura, E. Slow tumbling but large protrusion of phospholipids in the cell sized giant vesicle. Chem. Phys. Lett. 2013, 570, 136–140. [Google Scholar] [CrossRef]
- Bobone, S.; Piazzon, A.; Orioni, B.; Pedersen, J.Z.; Nan, Y.H.; Hahm, K.S.; Shin, S.Y.; Stella, L. The thin line between cell-penetrating and antimicrobial peptides: The case of Pep-1 and Pep-1-K. J. Pept. Sci. 2011, 17, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.; Melo, M.; Castanho, M. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.S.; Svetlovics, J.; McKeown, A.N.; Huskins, L.; Almeida, P.F. What determines the activity of antimicrobial and cytolytic peptides in model membranes. Biochemistry 2011, 50, 7919–7932. [Google Scholar] [CrossRef] [PubMed]
- Last, N.B.; Schlamadinger, D.E.; Miranker, A.D. A common landscape for membrane-active peptides. Protein Sci. 2013, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, S.; Järver, P.; Johansson, H.J.; Langel, U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: A comparative study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.R.; Patel, N.R.; Torchilin, V.P. Therapeutic delivery using cell-penetrating peptides. Eur. J. Nanomed. 2013, 5, 141–158. [Google Scholar] [CrossRef]
- Jo, J.; Hong, S.; Choi, W.Y.; Lee, D.R. Cell-penetrating peptide (CPP)-conjugated proteins is an efficient tool for manipulation of human mesenchymal stromal cells. Sci. Rep. 2014, 4, 4378. [Google Scholar] [CrossRef] [PubMed]
- Aki, K.; Fujii, N. Kinetics of isomerization and inversion of aspartate 58 of αA-crystallin peptide mimics under physiological conditions. PLoS ONE 2013, 8, e58515. [Google Scholar] [CrossRef] [PubMed]
- Takechi, Y.; Yoshii, H.; Tanaka, M.; Kawakami, T.; Aimoto, S.; Saito, H. Physicochemical mechanism for the enhanced ability of lipid membrane penetration of polyarginine. Langmuir 2011, 27, 7099–7107. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar] [PubMed]
- Tohyama, Y.; Yanagi, S.; Sada, K.; Yamamura, H. Translocation of p72syk to the cytoskeleton in thrombin-stimulated platelets. J. Biol. Chem. 1994, 269, 32796–32799. [Google Scholar] [PubMed]
- Ziegler, A.; Seelig, J. High affinity of the cell-penetrating peptide HIV-1 Tat-PTD for DNA. Biochemistry 2007, 46, 8138–8145. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Seelig, J. Binding and Clustering of Glycosaminoglycans: A Common Property of Mono- and Multivalent Cell-Penetrating Compounds. Biophys. J. 2008, 94, 2142–2149. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Stoichiometry (n) | K (M−1) | ΔG° (kcal/mol) | ΔH° (kcal/mol) | TΔS° (kcal/mol) | |
|---|---|---|---|---|---|
| Heparin | R8/heparin = 11 ± 1.1 | (1.3 ± 0.22) × 108 | −10.9 ± 0.10 | −9.6 ± 0.53 | 1.3 ± 0.54 |
| EPC/EPG LUV | lipid/R8 = 100 ± 10 | (1.5 ± 0.25) × 106 | −8.4 ± 0.10 | −6.3 ± 0.10 | 2.1 ± 0.10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takechi-Haraya, Y.; Aki, K.; Tohyama, Y.; Harano, Y.; Kawakami, T.; Saito, H.; Okamura, E. Glycosaminoglycan Binding and Non-Endocytic Membrane Translocation of Cell-Permeable Octaarginine Monitored by Real-Time In-Cell NMR Spectroscopy. Pharmaceuticals 2017, 10, 42. https://doi.org/10.3390/ph10020042
Takechi-Haraya Y, Aki K, Tohyama Y, Harano Y, Kawakami T, Saito H, Okamura E. Glycosaminoglycan Binding and Non-Endocytic Membrane Translocation of Cell-Permeable Octaarginine Monitored by Real-Time In-Cell NMR Spectroscopy. Pharmaceuticals. 2017; 10(2):42. https://doi.org/10.3390/ph10020042
Chicago/Turabian StyleTakechi-Haraya, Yuki, Kenzo Aki, Yumi Tohyama, Yuichi Harano, Toru Kawakami, Hiroyuki Saito, and Emiko Okamura. 2017. "Glycosaminoglycan Binding and Non-Endocytic Membrane Translocation of Cell-Permeable Octaarginine Monitored by Real-Time In-Cell NMR Spectroscopy" Pharmaceuticals 10, no. 2: 42. https://doi.org/10.3390/ph10020042