In the overwhelming body of literature on peptides or proteins able to cross biological membranes and to promote the delivery of drugs into cells, few studies have been reported in which different vectors have been applied to the same delivery problem and, in particular, on the correlation between conformation and internalization properties. In this paper, we estimated the ability of different CPPs to interact with simplified model membranes to obtain information, on a molecular level, on CPP-lipid interactions by complementary methods based on the fluorescent properties of carboxyfluoresceinyl-labeled derivatives and on the chiroptical properties of peptides. The variables addressed in this study comprise charge and composition of the membrane-mimicking environment in addition to the vector composition. In particular, to investigate the specificity of peptides towards lipid vesicles, depending on lipid head groups, anionic and zwitterionic lipids were used.
The ability of kFGF, modified Nle
54-Antp, Tat, and oligoarginine peptides to deliver into cell has been demonstrated by different authors [
4,
5,
6,
7,
8,
9,
10,
11]. However, the application of these CPPs to the same cargo and its influence on lipid-interactions has not been completely elucidated. In this work, we used as cargo the phosphopeptide, corresponding to the sequence 392-401pTyr
396 of the HS1 protein (HS1pY), which previously we demonstrated to be a potent inhibitor of the secondary HS1 phosphorylation, acting at level of the Src-homology 2 domains (SH2) mediate protein recruitment [
10,
14].
2.1. Peptide binding to phospholipid
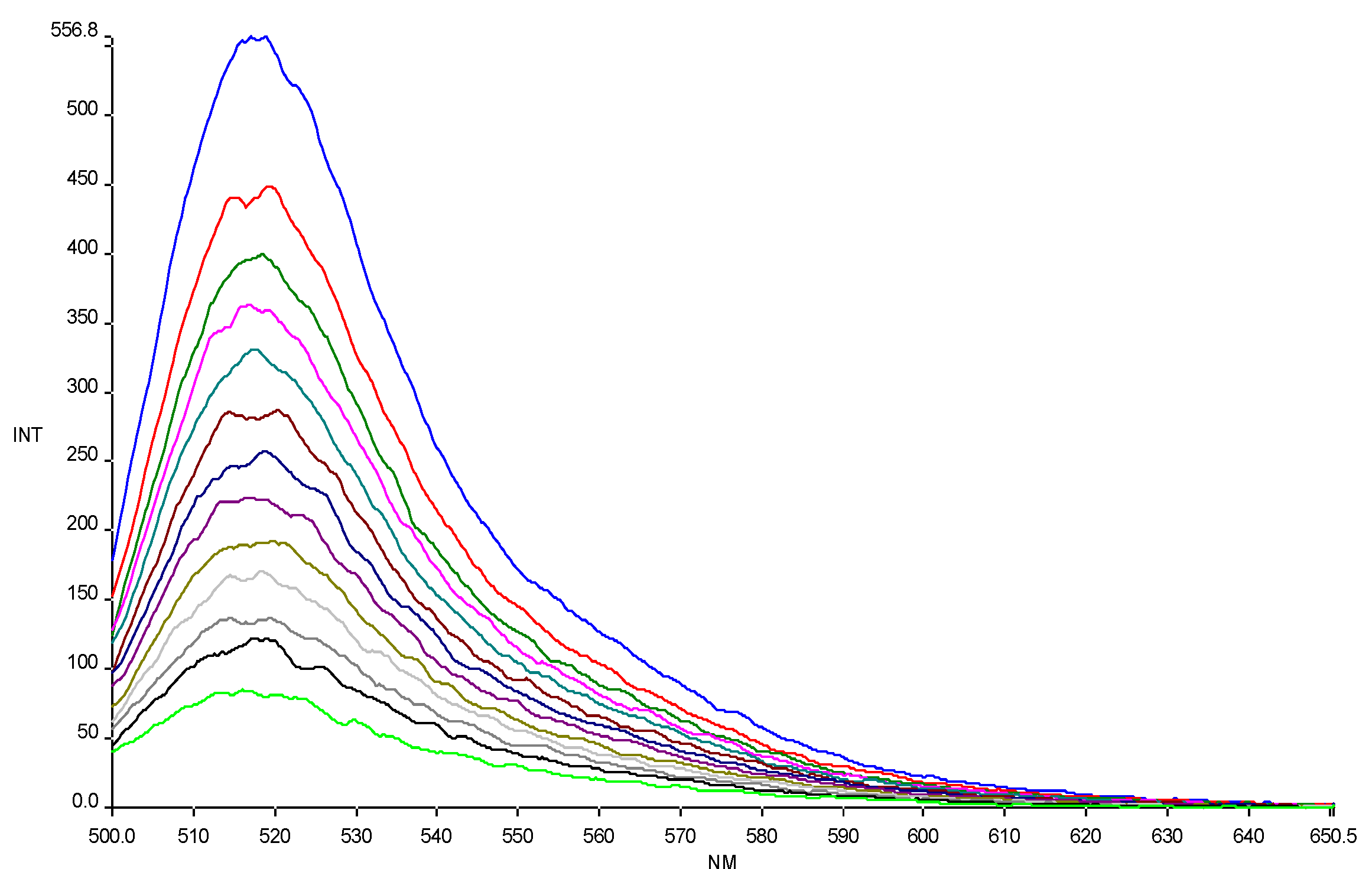
The affinity of CPPs, alone and conjugated to HS1pY, towards phospholipid vesicles was assessed by titration of carboxyfluoresceinyl-labeled peptide solutions with liposomes of different composition (an example of titration is reported in
Figure 1). As shown in
Figure 1, carboxyfluorescein emission is sensitive to environment change, showing a quenching when labeled peptides are in a more hydrophobic environment after addition of phospholipids [
16]. The fluorescence quenching was used to generate the binding isotherm of the labeled peptides and then to calculate the related partition coefficient (K
p).
Figure 1.
Quenching of carboxyfluorescein fluorescence emission in peptide 6a by DMPG SUVs (15.0 mM) titration. Excitation wavelength 485 nm. Peptide 0.3 µM in 5 mM Tris-HCl buffer, pH 6.8. Phospholipid/peptide molar ratio ranging from 0.00 to 1.57.
Figure 1.
Quenching of carboxyfluorescein fluorescence emission in peptide 6a by DMPG SUVs (15.0 mM) titration. Excitation wavelength 485 nm. Peptide 0.3 µM in 5 mM Tris-HCl buffer, pH 6.8. Phospholipid/peptide molar ratio ranging from 0.00 to 1.57.
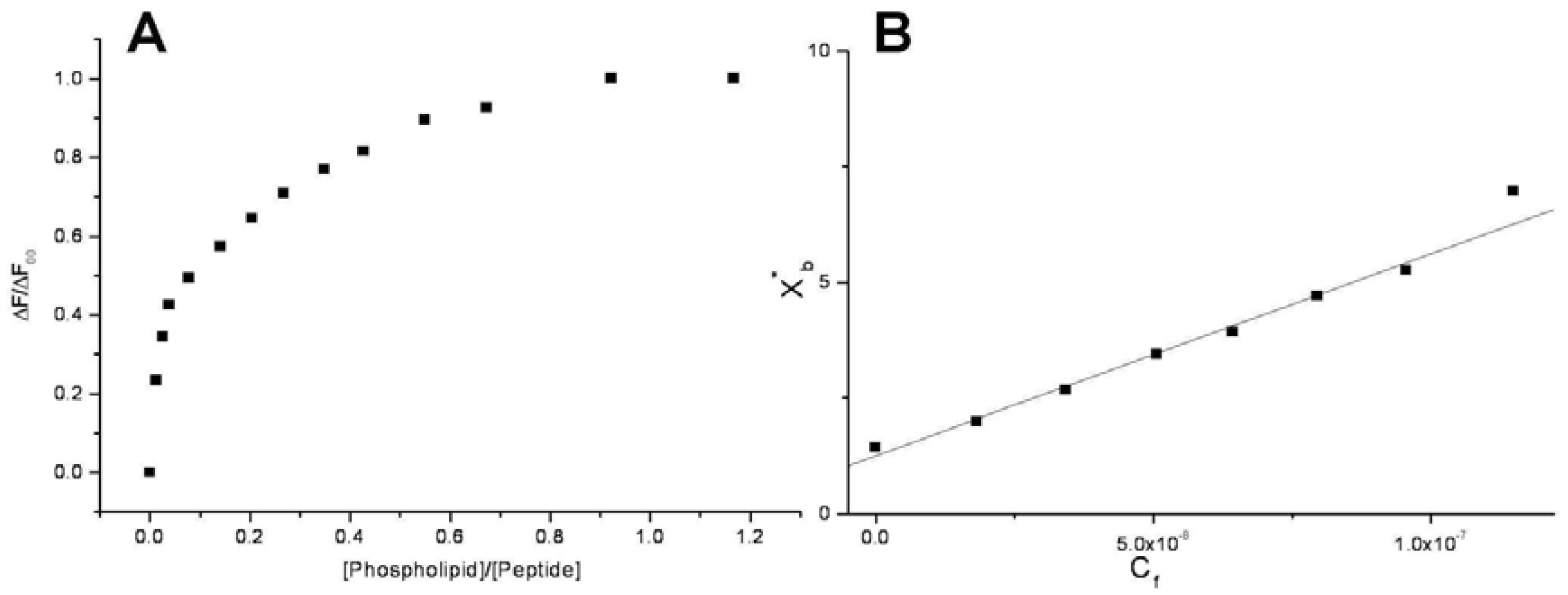
Conventional binding curves were obtained by plotting the ΔF/ΔF
∞ as a function of lipid-to-peptide molar ratio (
Figure 2A). Assuming that the peptides were initially portioned only over the outer leaflet of SUVs, conventional binding isotherm (
Figure 2B) was constructed plotting the correct molar ratio of bound peptide per 60% of the total lipid (X
b*) vs. the equilibrium concentration of free peptide in the solution (C
f). The surface partition coefficients K
p are estimated by extrapolating the initial slopes of the curves to zero C
f values and the resulted values are summarized in
Table 2 and in
Figure 3.
Figure 2.
Saturation (A) and binding isotherm (B) curves of peptide 6a (0.3 µM) titrated by DMPG SUVs (15.0 mM) in 5 mM Tris-HCl buffer, pH 6.8. Calculated Kp is 1.5x109, with an R value of 0.995 for the linear fit. Reported values represent means of three separate experiments.
Figure 2.
Saturation (A) and binding isotherm (B) curves of peptide 6a (0.3 µM) titrated by DMPG SUVs (15.0 mM) in 5 mM Tris-HCl buffer, pH 6.8. Calculated Kp is 1.5x109, with an R value of 0.995 for the linear fit. Reported values represent means of three separate experiments.
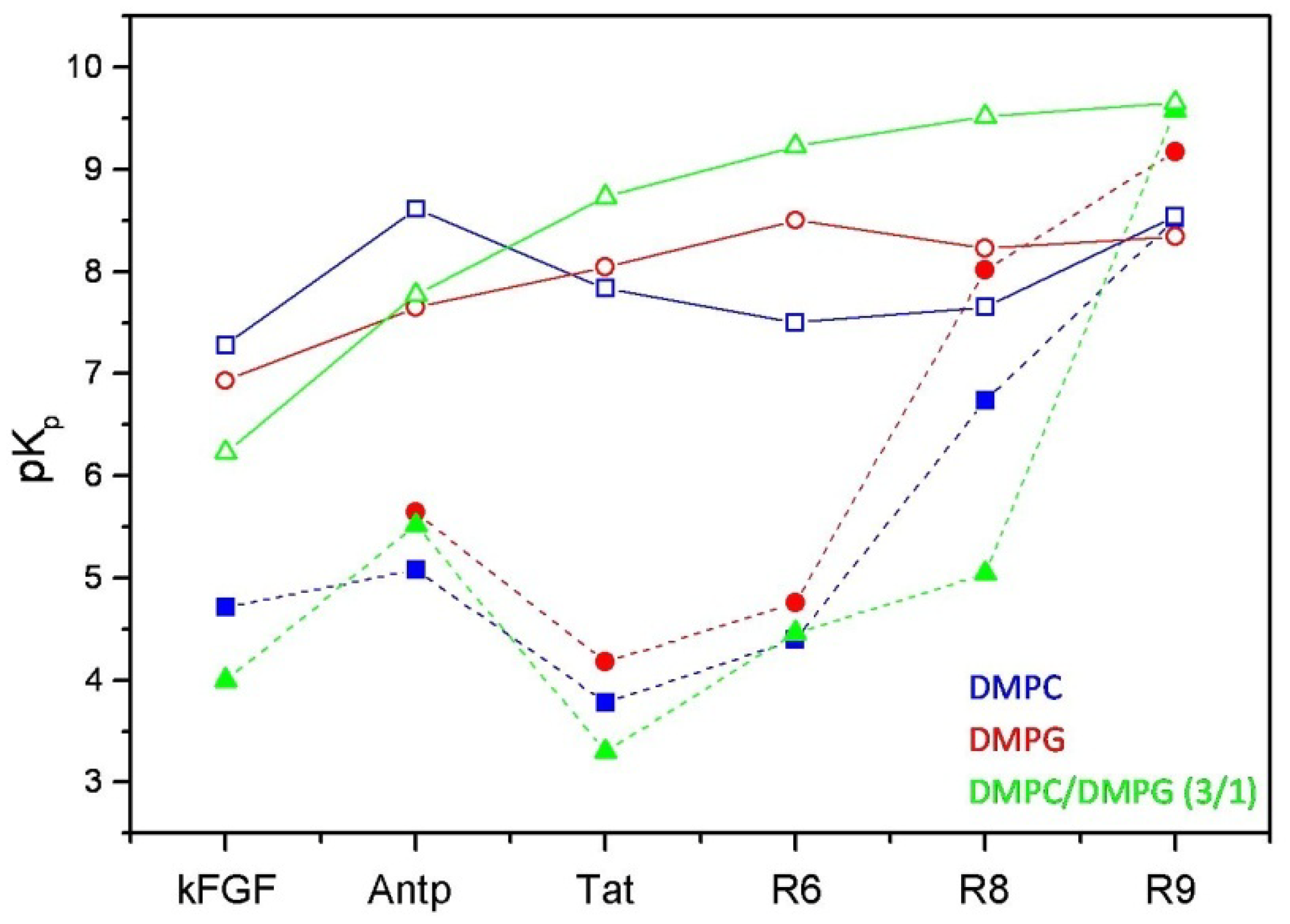
Figure 3.
Comparison between the logarithmic values of the surface partition coefficients (pKp) of CPPs and corresponding conjugated peptides. CPPs are represented by open symbols, meanwhile CPP-HS1pY peptides are represented by solid symbols.
Figure 3.
Comparison between the logarithmic values of the surface partition coefficients (pKp) of CPPs and corresponding conjugated peptides. CPPs are represented by open symbols, meanwhile CPP-HS1pY peptides are represented by solid symbols.
The calculated values (
Table 2) confirm the strong dependence of the peptide-lipid interactions on the nature and composition of both vesicles and peptides. As shown in
Figure 3, reporting the logarithmic values of K
p of respective peptides, a first distinctive feature of un-conjugated CPPs is their different selectivity towards neutral (zwitterionic) or negatively charged SUVs. Taking into account of this behavior, CPPs may be separated into two groups, interconnecting different CPP families. kFGF and Nle
54-Antp show an high affinity towards DMPC SUVs, meanwhile Tat and oligoarginines show an high affinity towards mixed DMPC/DMPG (3:1 molar ratio) SUVs, and this behavior is strongly related to the number (R6 < R8 < R10) of arginine residues.
Table 2.
Surface partition coefficients (Kp) of the investigated peptides in SUVs of different composition. The standard errors are less than 5% (n.d.: not determined).
Table 2.
Surface partition coefficients (Kp) of the investigated peptides in SUVs of different composition. The standard errors are less than 5% (n.d.: not determined).
| Peptide | pI | DMPC | DMPG | DMPC/DMPG |
|---|
| 1 | 5.57 | 1.9 × 107 | 8.6 × 106 | 1.7 × 106 |
| 1a | 2.11 | 5.2 × 104 | n.d. | 1.0 × 104 |
| 2 | 12.31 | 4.1 × 108 | 4.5 × 107 | 6.0 × 107 |
| 2a | 7.06 | 1.2 × 105 | 4.4 × 105 | 3.3 × 105 |
| 3 | 12.70 | 6.9 × 107 | 1.1 × 108 | 5.4 × 108 |
| 3a | 8.83 | 6.0 × 103 | 1.5 × 104 | 2.0 × 103 |
| 4 | 12.70 | 3.2 × 107 | 3.2 × 108 | 1.7 × 109 |
| 4a | 5.63 | 2.5 × 104 | 5.7 × 104 | 2.9 × 104 |
| 5 | 12.85 | 6.6 × 107 | 1.7 × 108 | 3.3 × 109 |
| 5a | 9.55 | 5.5 × 106 | 1.1 × 108 | 1.1 × 106 |
| 6 | 12.95 | 3.5 × 108 | 2.2 × 108 | 4.5 × 109 |
| 6a | 11.10 | 3.3 × 108 | 1.5 × 109 | 3.8 × 109 |
The relationship between K
p constants and peptide composition can be expressed taking into account of the peptide p
I values (
Table 2). Apparently, peptides 2-6, characterized by very similar p
I values, show a linear deviation of K
p values in presence of negatively charged DMPG or DMPC/DMPG SUVs. Interestingly, the K
p values of Nle
54-Antp (K
p DMPC > K
p mixed > K
p DMPG), as well as the comparison of the K
p values of Tat and R6, having the same p
I (12.70), show the influence of hydrophobic residues in addition to the positively charged ones in the binding process.
These results fit with the data reported by Liu and Deber [
17], and may be summarized as follows: hydrophobic peptides, i.e. kFGF, bind and insert into lipid vesicles predominantly via hydrophobic interaction between the lipid fatty acid acyl chains and the hydrophobic peptide segment, showing high affinity towards DMPC vesicles. On the other hand, the insertion into phospholipid vesicles of peptides with low hydrophobic character may be viewed as a two step process, in which the primary electrostatic attraction (between the anionic lipid heads and the cationic side-chains of basic residues) is essential for the binding process, driving the peptides to the membrane surface. Subsequently, the hydrophobic interaction with the lipid acyl chains stabilizes the peptide-membrane interaction and affects peptide insertion.
The analysis of the binding isotherms by means of which the K
p were determined, provides useful information on the organization of the peptide within the membrane. Isotherm shapes are almost straight lines, suggesting a simple partition process. They differ from those observed for several pore-forming polypeptides, for which initially the curves are flat, but then their slopes rise sharply (about 100 fold) once a threshold concentration is achieved [
18].
The conjugation with HS1pY strongly altered both conformational and binding properties of the tested CPPs. The remarkable K
p differences between vector and HS1pY conjugated peptides are mainly related to the electrostatic repulsion among the negative charges of HS1pY side-chains and the surface charges of phospholipid vesicles, confirming the important role played by electrostatic forces in the interaction of peptides with phospholipid membranes. In particular kFGF-HS1pY did not interact with DMPG vesicles, and the K
p determined in the presence of mixed SUVs is about one half of that of the parent peptide
1. This behavior is shared by the other CPPs (see
Table 2 and
Figure 3), in particular is noteworthy the low affinity of the Tat peptide
2a. On the other hand, the positive effect on lipid affinity of the high number of arginine residues is also highlighted by the K
p of the R10 derivative (
6a): only for this peptide the K
p values of free and conjugated form are comparable.
2.2. Membrane Permeability Induced by Peptides
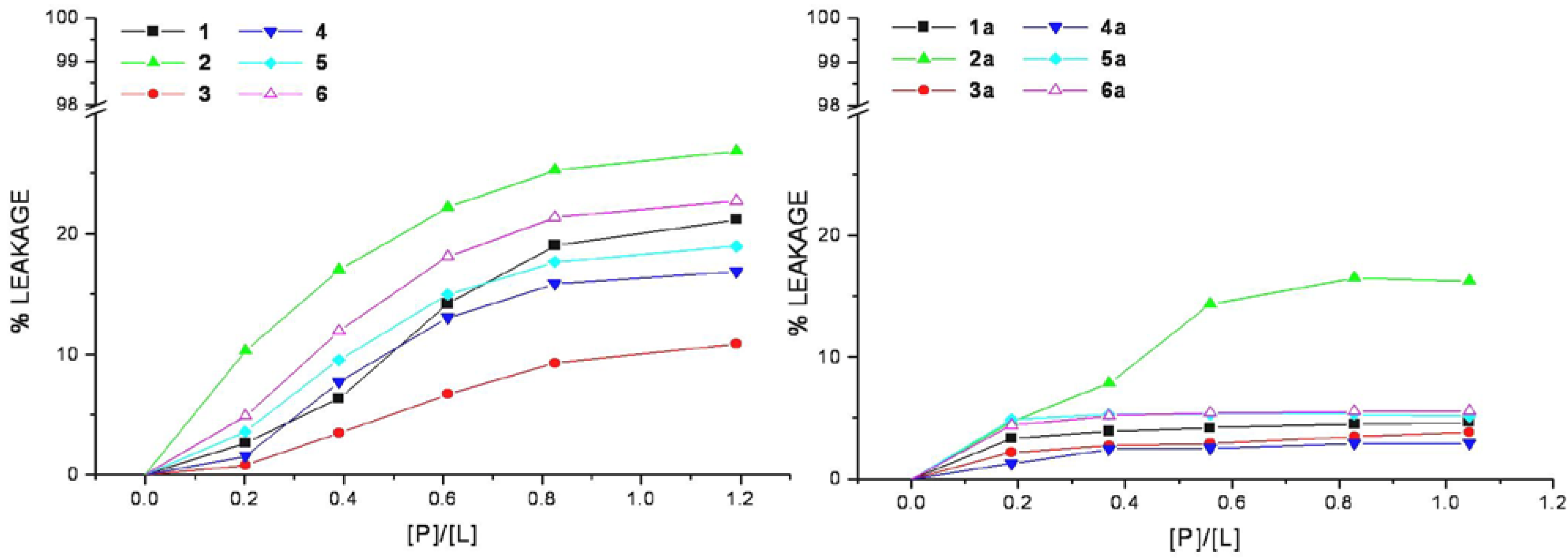
The efficiency of cell-penetrating peptides and corresponding HS1pY conjugates to perturb the lipid packing and causing leakage of vesicular contents has been examined. Increasing amount of selected peptide was added to mixed DMPC/DMPG vesicles containing the fluorescent dye calcein. An increase in fluorescence intensity due to the relief from the self-quenching of the calcein molecule concentrated (70 mM) within SUVs indicates the dye release.
The relation between the apparent percent of leakage and the peptide-to-lipid molar ratio was reported in
Figure 4 and indicated that none of the CPPs induces a significant leakage of dye from vesicles. In the same way even at very high peptide/lipid molar ratio (>1), the corresponding conjugates cannot significantly permeabilize vesicles as indicated by a low percentage of leakage (<20%).
Figure 4.
Membrane permeabilization induced by peptides. The level of membrane permeabilization was estimated by the percent of leakage of calcein from mixed DMPC/DMPG SUVs, in Tris-HCl buffer 20 mM, NaCl 150 mM, and EDTA 1 mM, pH 7.4, at 25 °C.
Figure 4.
Membrane permeabilization induced by peptides. The level of membrane permeabilization was estimated by the percent of leakage of calcein from mixed DMPC/DMPG SUVs, in Tris-HCl buffer 20 mM, NaCl 150 mM, and EDTA 1 mM, pH 7.4, at 25 °C.
Peptide-induced leakage of calcein entrapped in vesicles can be used to monitor membrane perturbation related to peptide toxicity. As shown in
Figure 4, all the selected CPPs are nontoxic even at extremely elevated peptide-to-lipid ratios causing no or little calcein leakage. This property remains unaltered in the corresponding conjugated peptides, rather they show a low lytic capability in comparison to parent peptides, this may be related to the decrease of lipid affinity.
These data furnish useful information on the peptide-lipid interactions in the context of the interaction models that currently exist. In agreement with the low-lytic properties of peptides in vesicle systems and with the data obtained from the binding isotherms, it is possible to assert that the tested peptides do not form pores in the manner of certain antimicrobial peptides. Consequently, our data do not support the “carpet” and the “barrel-stave” mechanism models both inducing pore-formation [
18,
19].
2.3. Monitoring the Secondary Structure of Peptides by Circular Dichroism.
The conformational state of cell-penetrating peptides and their HS1pY conjugates in membrane mimicking environments and bound to phospholipid vesicles has been studied by CD spectroscopy.
In aqueous buffer solution the CD spectra of free CPPs were characterized by the absence of a dominating secondary structure at room temperature. On the other hand, at low temperature Tat and oligoarginine (peptides
3 – 6) exhibited a CD pattern that closely resembles that of an extended left-handed polyproline II (PP
II) helix conformation [
20,
21].
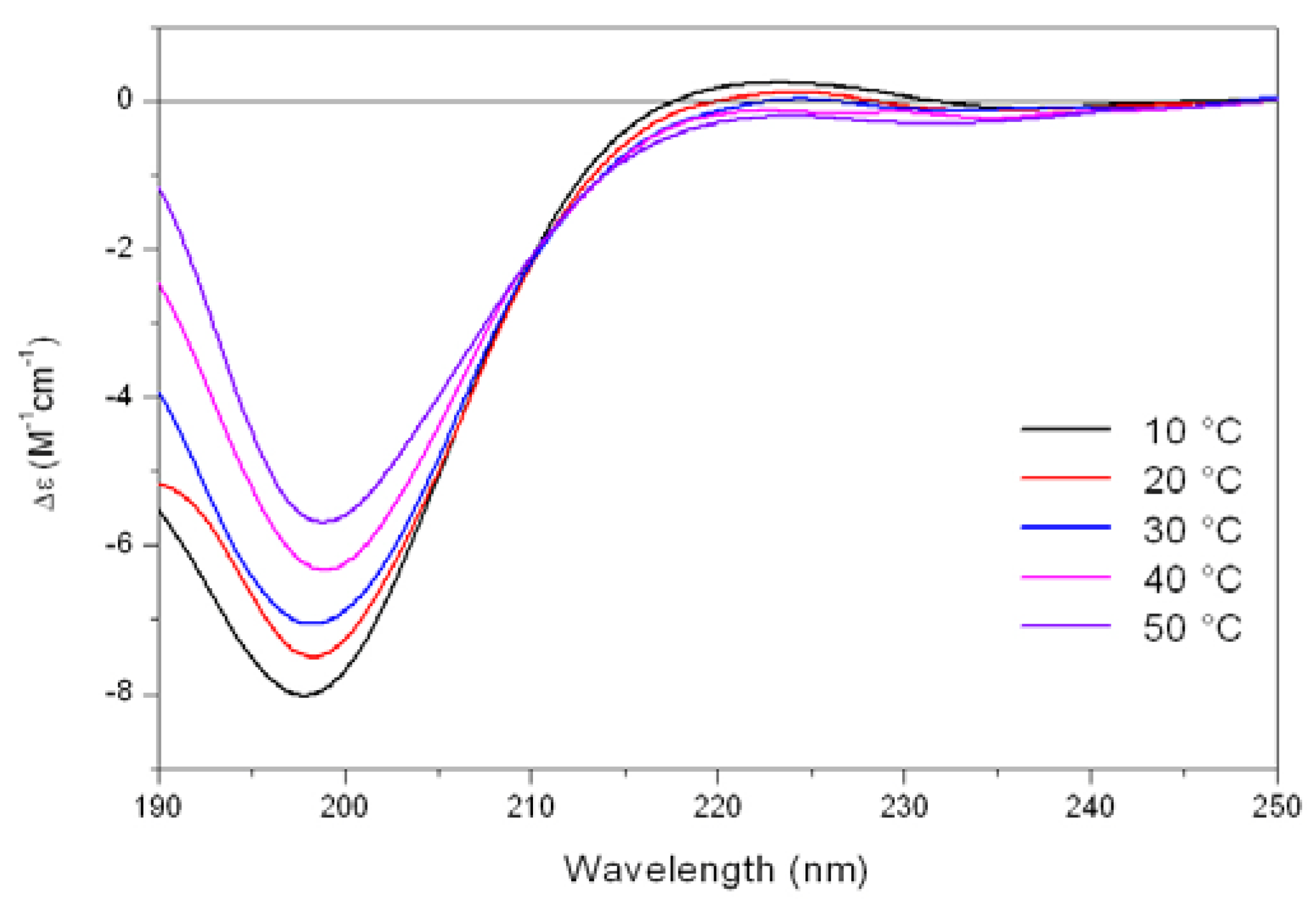
As shown in
Figure 5, the CD spectrum of Tat at low temperature (10 °C) is characterized by a strong negative band at 196 nm and a weak positive band at about 223 nm. On raising the temperature, the 223 nm positive band disappears, meanwhile the absolute value of negative band at 196 nm decreases. In addition, an isodichroic point at 210-215 nm was observed permitting to describe this system as a combination of two different states, where the low-temperature form was best described by a PP
II like conformation.
Figure 5.
Far-UV CD spectra of Tat peptide (13.5 mM) in Tris-HCl buffer (5mM, pH 6.8) as a function of increasing temperature.
Figure 5.
Far-UV CD spectra of Tat peptide (13.5 mM) in Tris-HCl buffer (5mM, pH 6.8) as a function of increasing temperature.
The propensity of charged peptides to adopt an extended left-handed 3
1-helix was initially proposed by Krimm and Mark [
22] who demonstrated as electrostatic interactions would favor a helical rather than an unordered structure in ionized polypeptides. In these cases, when the steric energy is taken into consideration, the favored conformation is a left-handed helix having 2.5 - 3.0 residues per turn, where residues at positions
i and
i+3 lie on the same edge of the helix. These results were confirmed by CD studies on ionized poly(Glu)
n or poly(Lys)
n by Holzwarth and Doty [
23] which showed that the spectra of these compounds differ significantly from those characteristic of unordered polypeptides, while resembling that of the PP
II helix. In this way, the uncharged residues of Tat are located on the same edge of the helix, whereas the Arg and Lys residues are mostly located on the other two sides of the PP
II helix, and the adopted spatial disposition is suitable for the interaction with the cellular membrane representing the first step of the internalization process.
Ho
et al. [
24], using LINUS protein structure and GRASP molecular surface predictive programs, suggested that this peptide might have similarities with the amphipatic α-helix structure present in many plasma membrane fusing peptides, like toxins, and designed a potent cell penetrating peptide (33-fold increase in translocation) derived from Tat, containing only three arginine residues at the 4, 7 and 10 positions. In contrast with this finding, other groups reported lack of α-helicity for Tat as detected by CD measurements [
11,
25], confirming our results.
In SDS micelles and in 9:1 v/v TFE/buffer, the CD spectra of kFGF and Nle54-Antp peptides (peptides 1 and 2) were characterized by two negative bands at 222 and 208 nm, respectively, and a positive maximum at 193 nm, characteristic of the presence of α-helical structures. Also the Tat peptide (peptide 3) adopted an α-helix conformation in 90% TFE solution, meanwhile in micellar SDS solution its behavior closely resembles that showed in aqueous solution: a left-handed PPII like conformation at low temperature and an unordered conformation at room temperature (20). On the contrary, the CD spectra of oligoarginine peptides in both 90% TFE and in micellar SDS solution were characterized by a negative band at about 200 nm at all tested temperatures, characteristic of the irregular conformation of the peptide chain.
In the presence of phospholipid SUVs, CPPs exhibited a conformational behavior completely different from that showed in membrane mimicking environment (SDS micelles or TFE). In particular, the Nle
54-Antp peptide (peptide
2) showed a largely random conformation in presence of zwitterionic DMPC vesicles, whereas in presence of anionic DMPG SUVs the peptide adopted mainly a β-sheet structure. In mixed DMPC/DMPG vesicles the CD spectrum strongly resembled that of an α-helix where the negative band is centered at 222 nm (data not shown). On the contrary, the kFGF peptide (peptide
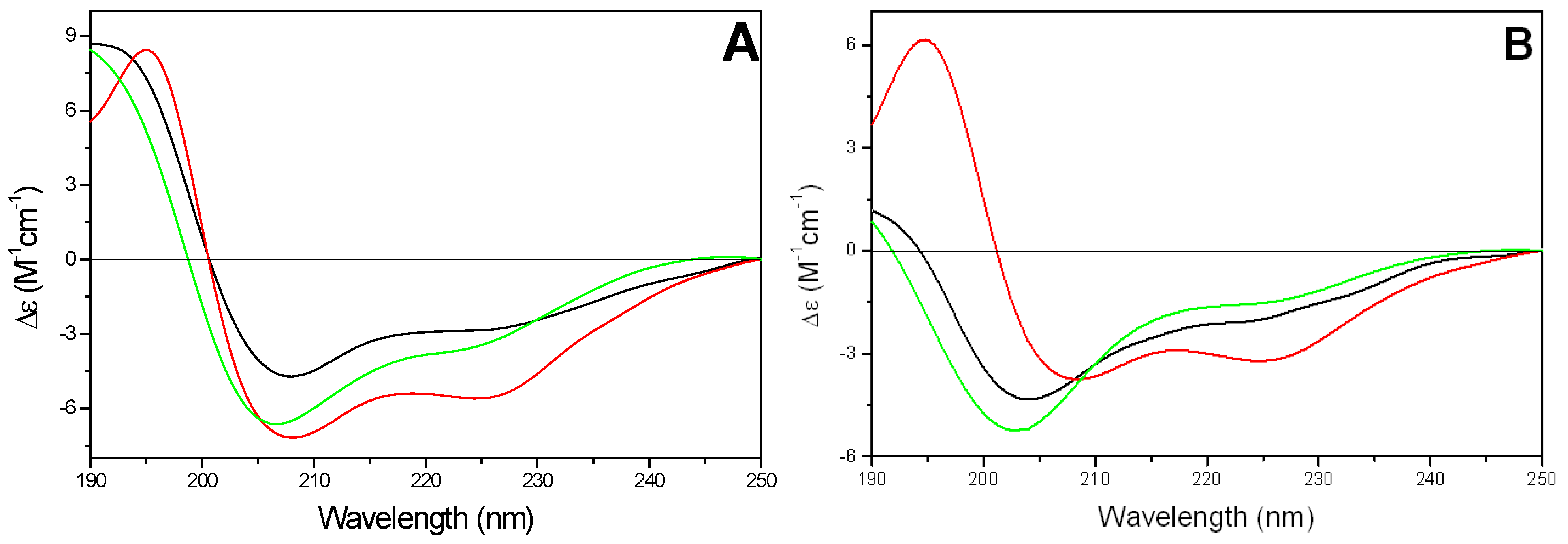
1) adopted mainly an α-helix conformation in all tested phospholipids (
Figure 6A). The amount of α-helix conformation was not significantly affected by the change of temperature below and above lipid gel to liquid crystal phase transition (about 23 °C, data not shown). The estimated helicities were 35-40% in the presence of either DMPG or DMPC/DMPG vesicles and 55-60% in the presence of DMPC SUVs, respectively.
In the presence of zwitterionic or mixed vesicles the CD spectra of the Tat peptide were similar to those observed both in buffer and in micellar SDS solutions at room temperature. On the contrary, the CD pattern in presence of negatively charged DMPG vesicles closely resembled that obtained in 90% TFE solution, even if the negative band at 222 nm disappeared in a shoulder (data not shown).
The CD spectra of oligoarginines in presence of DMPC and mixed DMPC/DMPG SUVs were characterized by an evident positive band near 220 nm and a negative band at about 200 nm, which intensity decreased at the increasing of the percentage of negatively charged phospholipid.
Figure 6.
Far-UV CD spectra of kFGF (
1) (11.9 µM) (A) and of kFGF-HS1pY (
1a) (10.9 µM) (B) in presence of SUVs of different composition (
![Pharmaceuticals 03 01045 i001]()
).
Figure 6.
Far-UV CD spectra of kFGF (
1) (11.9 µM) (A) and of kFGF-HS1pY (
1a) (10.9 µM) (B) in presence of SUVs of different composition (
![Pharmaceuticals 03 01045 i001]()
).
The conjugation with HS1pY peptide strongly affected the CD spectra of all CPPs, even if a rationalization of its effect is not possible. The characteristic α-helical conformation of the kFGF peptide (peptide
1) decreased or disappeared. In particular, in the presence of negatively charged SUVs, the CD spectrum of peptide
1a was characterized by a negative band at 202-203 nm with a shoulder at about 225 nm. The intensity of the negative band was strongly correlated to the charge density of SUV: it was maximum in the presence of DMPG vesicles and decreased with increasing the amount of DMPC phospholipids in the SUVs (
Figure 6B).
A very similar behavior was observed for the Antp containing peptide 2a. The elongation with HS1pY decreased the helical contribute both in micellar SDS and in 90% TFE solutions. In presence of zwitterionic DMPC vesicles the conjugated peptide adopts a largely irregular conformation, whereas the CD spectra in presence of DMPG or mixed vesicles closely resemble those described for the corresponding kFGF derivative.
The conjugation of HS1pY with Tat (peptide 3a) stabilized the PPII like conformation in buffer and in DMPC vesicles. On the contrary, the propensity to adopt an ordered structure in presence of negatively charged SUVs (DMPG or mixed vesicles) as well as in membrane mimicking environments (SDS and TFE) was strongly reduced, and the CD spectra are characterized by the presence of a negative band at about 200 nm.
The introduction of the HS1pY peptide to the C-terminal end of oligoarginine peptides (peptides 4a – 6a) induced very similar changes in the CD spectra recorded under different conditions. In buffer solution the CD spectra were characterized by the presence of two positive band at about 200 and 225 nm, meanwhile in micellar SDS solution the spectra showed a strong negative band at about 200 nm, with a shoulder at 225-230 nm. A very similar behavior was observable in the CD spectra in the presence of phospholipid vesicles. In particular, in the presence of DMPC or DMPG phospholipids the spectra were characterized by a positive band at 230 nm, suggesting the presence of a PPII helix conformation. Surprisingly, the CD spectra of peptides 4a, 5a and 6a in TFE solution were characterized by a very low intensity of the dichroic signal and by the presence of negative band at 205 nm and positive band at about 190 nm.
2.4. Delivery of HS1pY peptide into mammalian cells
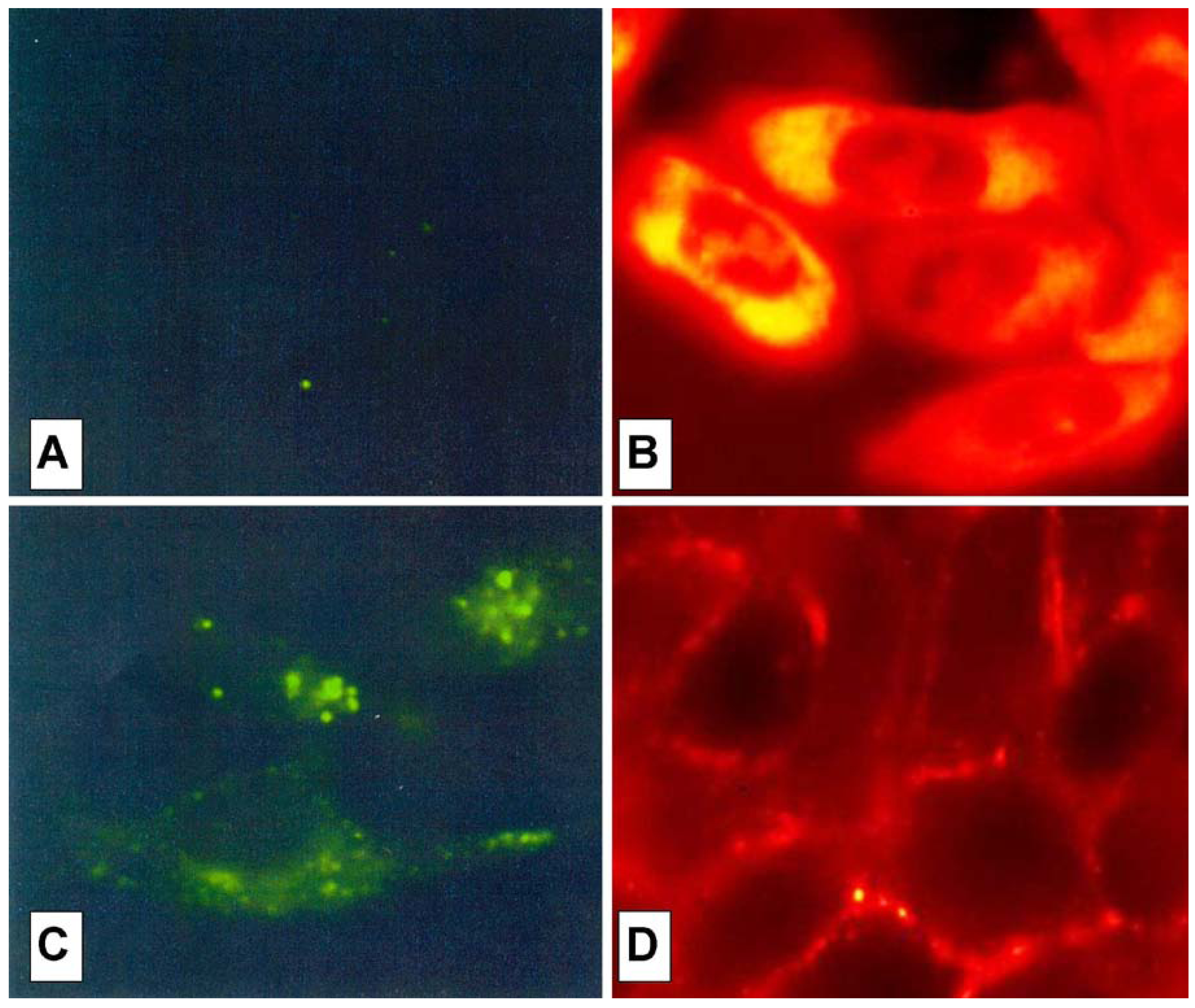
The cell peptide uptake was examined by confocal microscopy (
Figure 7), using the carboxyfluoresceinyl labeled peptides. Briefly, the peptides were added (12.5 µM) to the incubation medium of CHO (Chinese hamster ovary) cells, for 30 min, 1 h and 2 h at 37 °C. Cells were washed and peptide internalization detected by confocal microscopy. In these conditions, previous detected membrane-induced carboxyfluorescein quenching (see fluorescence studies) did not affect in efficient way the cell peptide uptake detection.
Surprisingly, the conjugation of the HS1pY peptide destroyed the cell-penetrating properties of kFGF and R6 constructs (peptides 1a and 4a), meanwhile all the other constructs (peptides 2a, 3a, 5a and 6a) were internalized by cells. Maximal internalization was reached after 30 min; prolonged incubation times did not increase fluorescence visible inside cells (data not shown). Fluorescence was apparent throughout the cytoplasm and the nuclei of the cells and appeared to localize mainly to the nucleus. Works are in progress to identify the organelle(s) that is accessed with conjugated peptides.
Figure 7 also shows that, while the parent phospho-decapeptide (peptide
7) could not penetrate cells at 4 °C (panel A), few fluorescence spots were visible inside control cells incubated at 37 °C, probably as a consequence of an endocytotic process (panel C).
The comparison of these data with the data of our biophysical investigation suggested that the single interaction with the cell membrane and its binding affinity, evaluated by fluorescence studies on vesicle models, per se do not confer specific translocation features, and more important, do not govern cell-penetrating capability.
Recent works [
26,
27] showed that the major route for Tat and Nle
54-Antp peptide-mediated cellular uptake of cargo is endocytosis, rather than a translocation mechanism. In addition, reevaluation of the mechanism of internalization of cationic CPPs demonstrated that uptake was temperature-dependent indicative of a step requiring energy [
27] as well as reported for signal peptide vectors (kFGF) [
5,
6]. Cationic peptides that initially accumulated on the cell surface in small patches (a process that was temperature-independent) were then internalized in intracellular vesicular structures. One can hypothesize that the high positive charges of conjugated peptides promote the binding to the cell surface. In addition, CPPs uptake by endocytosis rather than translocation mechanism may explain our difficulty to demonstrate the ability of these cationic peptides to pass across a non-cellular phospholipid bilayer (data not shown).
Figure 7.
Confocal analysis of the HS1pY uptake. CHO cells were incubated at 4 °C (A) or 37 °C (B, C and D) for 30 min with 12.5 µM concentration of either the phosphodecapeptide (A and C) or the conjugated Tat (peptide 3a) (B) and kFGF (peptide 1a) (D). Preparation of the cells and details of the microscopy are described in the Experimental Section.
Figure 7.
Confocal analysis of the HS1pY uptake. CHO cells were incubated at 4 °C (A) or 37 °C (B, C and D) for 30 min with 12.5 µM concentration of either the phosphodecapeptide (A and C) or the conjugated Tat (peptide 3a) (B) and kFGF (peptide 1a) (D). Preparation of the cells and details of the microscopy are described in the Experimental Section.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





 ).
).

