Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies

Abstract
:1. Introduction
2. Histone Deacetylases and Cancer

{kind=link}
{kind=link}
| HDAC | Localization | Deregulation in cancer | Tumor |
|---|---|---|---|
| Class I | |||
| HDAC1 | Nucleus | Overexpression/underexpression | Esophageal, colon, prostate, CTCL |
| HDAC2 | Nucleus | Overexpression/mutation | Prostate, colon, gastric, endometrial, CTCL |
| HDAC3 | Nucleus | Overexpression | Prostate, colon |
| HDAC8 | Nucleus | Overexpression | Colon |
| Class IIa | |||
| HDAC4 | Nucleus/Cytoplasm | Overexpression/underexpression/ mutation | Prostate, colon, breast |
| HDAC5 | Nucleus/Cytoplasm | Underexpression | Colon, AML |
| HDAC7 | Nucleus/Cytoplasm | Overexpression | Colon |
| HDAC9 | Nucleus/Cytoplasm | Overexpression/underexpression | Medulloblastomas, astrocytomas |
| Class IIb | |||
| HDAC6 | Predominantly Cytoplasm | Overexpression | Breast, AML, CTCL |
| HDAC10 | Predominantly Cytoplasm | Overexpression | Heptocellular Carcinoma |
| Class IV | |||
| HDAC11 | Nucleus/Cytoplasm | Overexpression | Breast |
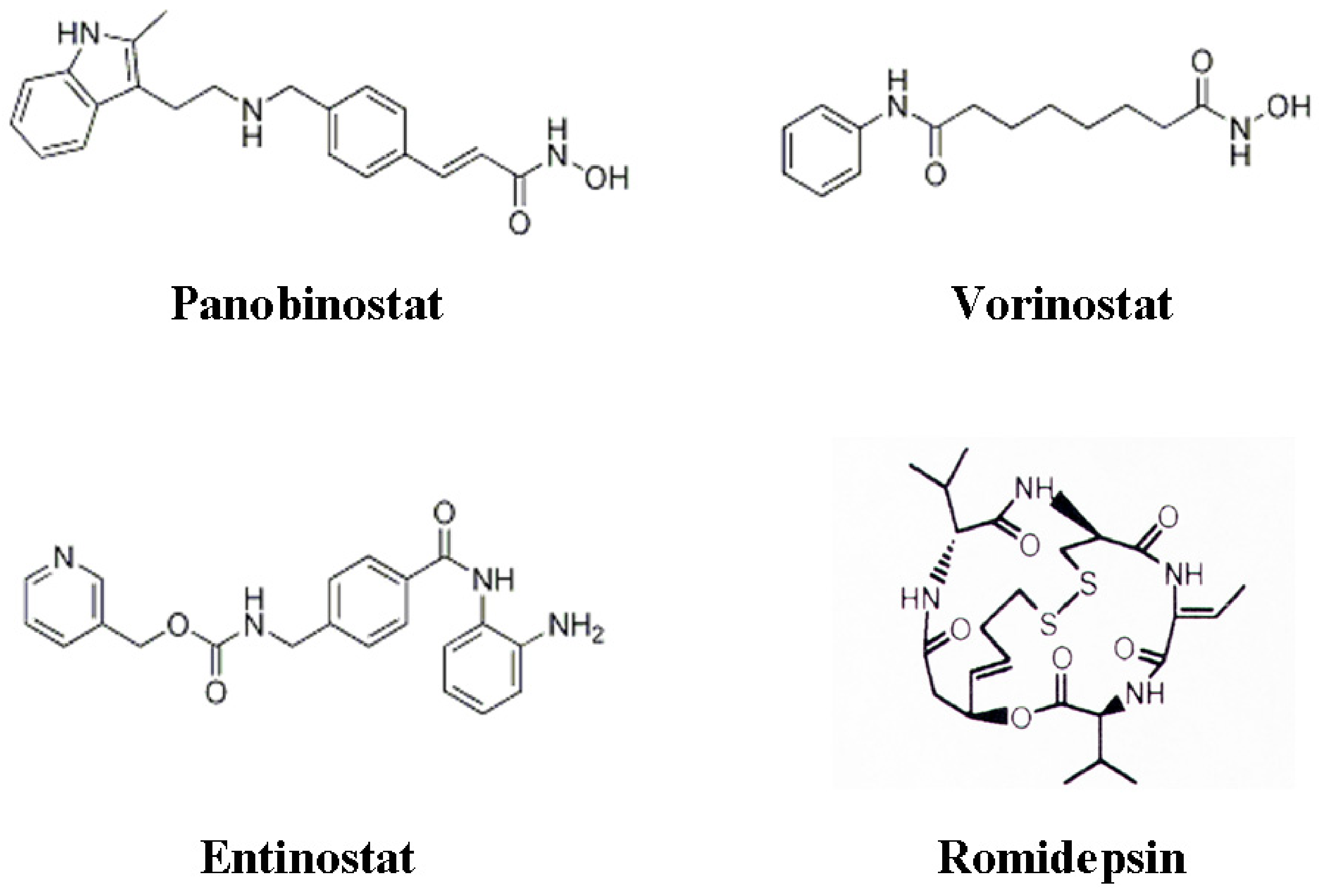
3. Histone Deacetylase Inhibitors
4. HDACIs and Angiogenesis
4.1. Combination Strategies with HDACI to Target Angiogenesis
| Gene | Target Cell | Activity on angiogenesis | Effect on gene transcription by HDAC inhibition [reference] |
|---|---|---|---|
| p53 | Cancer | Inhibits | Up-regulation [49] |
| pVHL | Cancer | Inhibits | Up-regulation [49,58] |
| HIF-1α | Cancer | Induces | Down-regulation [49,59,60] |
| VEGF | Cancer | Induces | Down-regulation [49,59,60,61] |
| Activin A | Cancer | Inhibits | Up-regulation [57] |
| bFGF | Cancer | Induces | Down-regulation [60,61] |
| Thrombospondin 1 | Cancer | Inhibits | Up-regulation [62,63] |
| MMP-2 | Cancer | Induces | Up-regulation [57] |
| MMP-9 | Cancer | Induces | Up-regulation [57] |
| RECK | Cancer | Inhibits | Up-regulation [57] |
| Neurofibromin2 | Cancer | Inhibits | Up-regulation [58,64] |
| Ang1 | Cancer | Induces | Down-regulation [38] |
| Connective tissue growth factor | Cancer | Inhibits | Up-regulated [63] |
| Fibroblast growth factor 19 | Cancer | Induces | Down-regulated [63] |
| VEGF receptor 1 | Endothelial | Induces | Down-regulation [57] |
| VEGF receptor 2 | Endothelial | Induces | Down-regulation [57] |
| Neuropilin-1 | Endothelial | Induces | Down-regulation [57] |
| Semaphoring III | Endothelial | Inhibits | Up-regulation [65] |
| Tie2 | Endothelial | Induces | Down-regulation [59] |
| Ang2 | Endothelial | Induces | Down-regulation [59] |
| eNOS | Endothelial | Induces | Down-regulation [66,67,68] |
| VEGFD | Endothelial | Induces | Down-regulation [57] |
| Clusterin | Endothelial | Inhibits | Up-regulation [69] |
| Fibrillin1 | Endothelial | Inhibits | Up-regulation [69] |
| Quiescin Q6 | Endothelial | Inhibits | Up-regulation [69] |
| PDGF-B | Endothelial | Inhibits | Up-regulation [56] |
| PDGFR-β | Endothelial | Inhibits | Up-regulation [56] |
| Survivin | Endothelial | Induces | Down-regulation [59] |
5. HDACIs and Autophagy
| Biological effect/gene | Pathway | Effect on gene transcription by HDAC inhibition [reference] |
|---|---|---|
| Autophagy | ||
| Beclin-1 | Aggresome | Up-regulated [86] |
| ATG-7 | Aggresome | Up-regulated [86] |
| ROS production/activity | ||
| TBP2 | ROS | Up-regulated [104] |
| Thioredoxin | ROS | Up-regulated [104] |
| Apoptosis | ||
| TRAIL | Extrinsic apoptosis | Up-regulated [105,106,107,108] |
| DR5 | Extrinsic apoptosis | Up-regulated [105,106,107,108] |
| DR4 | Extrinsic apoptosis | Up-regulated [109] |
| Fas | Extrinsic apoptosis | Up-regulated [106,110] |
| FasL | Extrinsic apoptosis | Up-regulated [106,110] |
| TNFα | Extrinsic apoptosis | Up-regulated [111] |
| c-FLIP | Extrinsic apoptosis | Down-regulated [101,103] |
| Bcl2 | Intrinsic apoptosis | Down-regulated [112] |
| BclXL | Intrinsic apoptosis | Down-regulated [31,113] |
| Bclw | Intrinsic apoptosis | Down-regulated [114] |
| Mcl-1 | Intrinsic apoptosis | Down-regulated [31,113] |
| XIAP | Intrinsic apoptosis | Down-regulated [115,116] |
| Caspase-3 | Intrinsic apoptosis | Up-regulated [31] |
| Apaf-1 | Intrinsic apoptosis | Up-regulated 31] |
| Bak | Intrinsic apoptosis | Up-regulated [31,104,113] |
| Bid | Intrinsic apoptosis | Up-regulated/cleaved [116,117,118] |
| Bim | Intrinsic apoptosis | Up-regulated/phosphorylated [104,113,118,119,120] |
| Bmf | Intrinsic apoptosis | Up-regulated [104,121] |
| Bax | Intrinsic apoptosis | Up-regulated/phosphorylated [119] |
| Noxa | Intrinsic apoptosis | Up-regulated [120] |
| Puma | Intrinsic apoptosis | Up-regulated [122] |
| AVEN | Intrinsic apoptosis | Down-regulated [63] |
| Survivin | Intrinsic/Extrinsic apoptosis | Down-regulated [123] |
6. HDACIs and Apoptosis
6.1. Combination Strategies with HDACI to Target Apoptosis

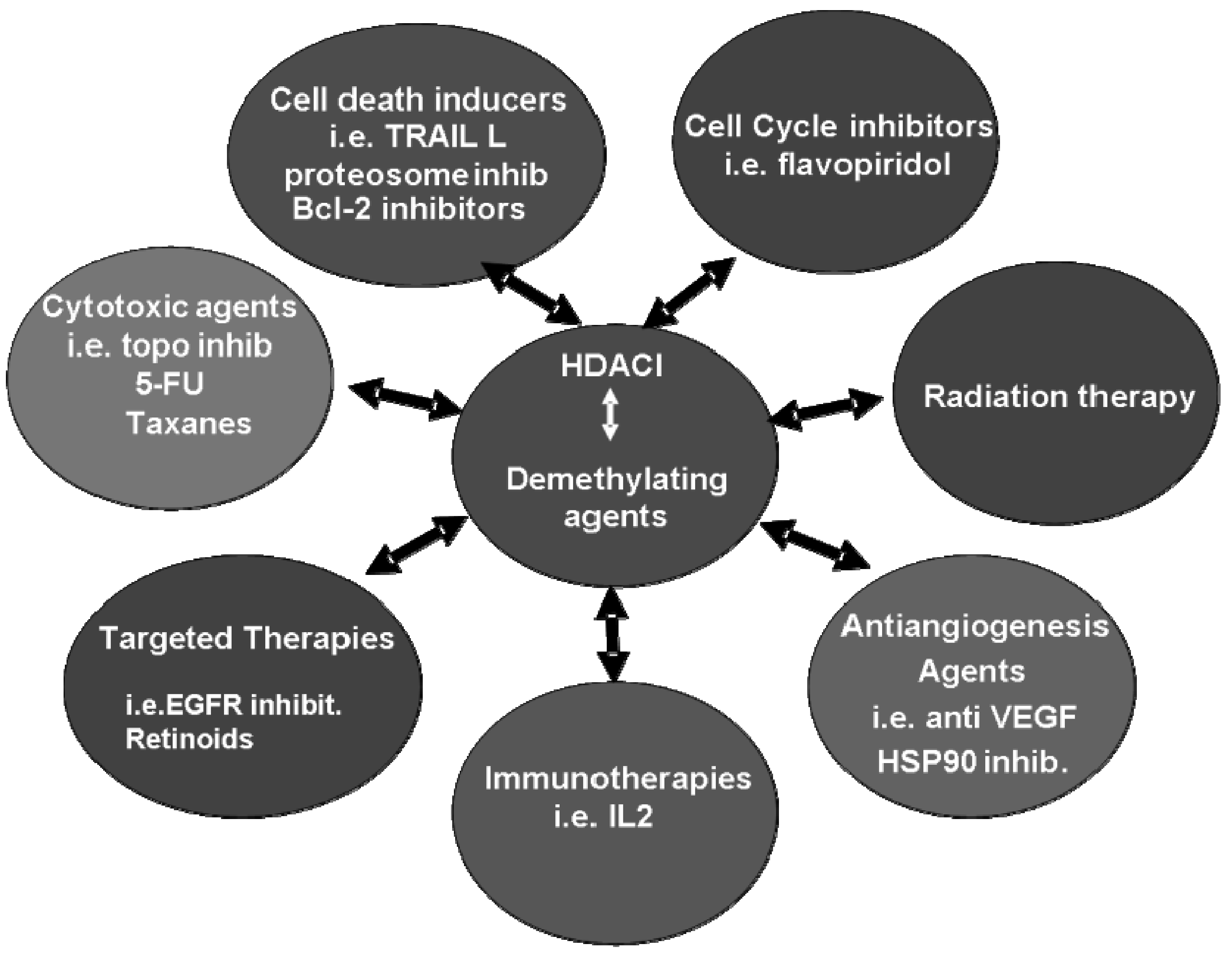
7. Clinical Combination Strategies including HDACI
| HDACI | Combination | Phase | Disease | Patient number | Response |
|---|---|---|---|---|---|
| Vorinostat | Carboplatin/Paclitaxel | I | Advanced solid tumors | 25/28 patients available for evaluation | NSCLC patients were best responders; PR (53%), SD (21%) |
| FOLFOX | I | Refractory colorectal cancer | 21 patients enrolled | Study resulting in a determined vorinostat MTD of 300 mg 2× daily in combination with FOLFOX | |
| Doxorubicin | I | Solid tumors | 24/32 patients available for evaluation | PR (8%; prostate and breast cancer patients); SD (8%; melanoma patients) | |
| Docetaxel | I | CRPC and NSCLC | NA | Study terminated due to excessive DLTs | |
| Gemcitibine/cisplatinum | I | Metastatic NSCLC | 19/28 patients available for evaluation | PR (47%) | |
| Erlotinib | I | Refractory NSCLC | 9 patients available for evaluation | SD (67%) | |
| Bortezomib | I | Refractory solid tumors | 29 patients available for evaluation | Study resulted in a determined vorinostat MTD of 300 mg BID with bortzomid dosed at 1.3 mg/m2. Evidence of clinical activity was observed | |
| Bevacizumab | II | Stage IV clear cell renal carcinoma | 32/34 patients available for evaluation | 18% objective responses (1× CR; 5× PR), 67% (SD). Median progression free survival: 5.3 months Overall survival: 16.2 months | |
| Sorafenib | I | Advanced solid tumors | 12/17 patients available for evaluation | 1 unconfirmed PR; 9 SD (minor responses). MTD/RP2D in combination recommended is 300 mg vorinostat QD d 1–14 with 400 mg sorafenib BID d 1–21 (21 day cycles). | |
| Flavopiridol | I | Advanced solid tumors | 31/34 patients evaluable for evaluation | Concluded that intermittent pulsing of high dose vorinostat in combination with flavopiridol is achievable without increased toxcities. RP2D is 800 mg vorinostat (3 days; d 1–3) with 30 mg/m2 flavopiridol (30min followed by 30 mg/m2 every over 4h every 14d). | |
| Romidepsin | Gemcitibine | I | Advanced solid tumors | 33 patients available for evaluation | SD (36%) |
| Bortezamib | II | Refractory/relapsed multiple myeloma | 5 patients currently enrolled | Concluded that this combination is active and further patient recruitment is currently underway | |
| Entinostat | Erlotinib | I | Advanced NSCLC | 9 patients available for evaluation | PR (11%) and SD (11%) |
| 5-azacitidine | II | Relapsed advanced NSCLC | 25 patients currently enrolled | CR (4%) and SD (8%); remaining patients had PD | |
| Aromatase inhibitor therapy | II | ER+ breast cancer | 27 patients enrolled | 1 confirmed PR; 1 SD > 6 months. Concluded this combination demonstrated clinical benefit. | |
| Panobinostat | Trastuzumab | I | HER2 positive metastatic breast cancer | 18 patients enrolled | Preliminary data indicates this combination to be well tolerated and displays clinical activity |
| Lenalidomide/ dexamethasome | I | Relapsed/refractory multiple myeloma | 22 patients enrolled | Combination well tolerated with indications of clinical efficacy | |
| Docetaxel | Ib | Chemotherapy naïve CRPC | 21 patients enrolled | Minimal DLTs have been observed with some patients achieving a biochemical response indicated by reduced PSA levels | |
| Epirubicin | I | Solid tumors | 10 patients | Patient cohort treated with 50 mg panobinostat reported to date and concluded that sequence combination of panobinostat and epirubicin is well tolerated. |

8. Concluding Remarks
References
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar]
- Campas-Moya, C. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today (Barc) 2009, 45, 787–795. [Google Scholar] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar]
- Ellis, L.; Hammers, H.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009, 280, 145–153. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene. 2007, 26, 5420–5432. [Google Scholar]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; Tsuneyoshi, M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; Kristiansen, G. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer. 2008, 98, 604–610. [Google Scholar]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar]
- Halkidou, K.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Nuclear accumulation of histone deacetylase 4 (HDAC4) coincides with the loss of androgen sensitivity in hormone refractory cancer of the prostate. Eur. Urol. 2004, 45, 382–389, author reply 389. [Google Scholar]
- Park, J.M.; Lee, G.Y.; Choi, J.E.; Kang, H.G.; Jang, J.S.; Cha, S.I.; Lee, E.B.; Kim, S.G.; Kim, C.H.; Lee, W.K.; Kam, S.; Kim, D.S.; Jung, T.H.; Park, J.Y. No association between polymorphisms in the histone deacetylase genes and the risk of lung cancer. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1841–1843. [Google Scholar]
- Cebrian, A.; Pharoah, P.D.; Ahmed, S.; Ropero, S.; Fraga, M.F.; Smith, P.L.; Conroy, D.; Luben, R.; Perkins, B.; Easton, D.F.; Dunning, A.M.; Esteller, M.; Ponder, B.A. Genetic variants in epigenetic genes and breast cancer risk. Carcinogenesis 2006, 27, 1661–1669. [Google Scholar]
- Ozdag, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; Collins, V.P.; Bowtell, D.; Kouzarides, T.; Brenton, J.D.; Caldas, C. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics 2006, 7, 90. [Google Scholar]
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F.; Herranz, M.; Palacios, J.; Arango, D.; Orntoft, T.F.; Aaltonen, L.A.; Schwartz, S., Jr.; Esteller, M. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; Szabo, S.; Buckhaults, P.; Farrell, C.; Meeh, P.; Markowitz, S.D.; Willis, J.; Dawson, D.; Willson, J.K.; Gazdar, A.F.; Hartigan, J.; Wu, L.; Liu, C.; Parmigiani, G.; Park, B.H.; Bachman, K.E.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar]
- Marquard, L.; Gjerdrum, L.M.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Ralfkiaer, E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 2008, 53, 267–277. [Google Scholar]
- Zelent, A.; Guidez, F.; Melnick, A.; Waxman, S.; Licht, J.D. Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 2001, 20, 7186–7203. [Google Scholar]
- Pandolfi, P.P. Transcription therapy for cancer. Oncogene 2001, 20, 3116–3127. [Google Scholar]
- Lin, R.J.; Sternsdorf, T.; Tini, M.; Evans, R.M. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001, 20, 7204–7215. [Google Scholar]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 2000, 184, 1–16. [Google Scholar]
- Bhalla, K.N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin. Oncol. 2005, 23, 3971–3993. [Google Scholar]
- Bereshchenko, O.R.; Gu, W.; Dalla-Favera, R. Acetylation inactivates the transcriptional repressor BCL6. Nat. Genet. 2002, 32, 606–613. [Google Scholar]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar]
- Schrump, D.S. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin. Cancer Res. 2009, 15, 3947–3957. [Google Scholar]
- Miremadi, A.; Oestergaard, M.Z.; Pharoah, P.D.; Caldas, C. Cancer genetics of epigenetic genes. Hum. Mol. Genet. 2007, 16, (Special No. 1). R28–R49. [Google Scholar]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Hideshima, T.; Akiyama, M.; Chauhan, D.; Munshi, N.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Richon, V.M.; Marks, P.A.; Anderson, K.C. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl. Acad. Sci. USA 2004, 101, 540–545. [Google Scholar]
- Moore, P.S.; Barbi, S.; Donadelli, M.; Costanzo, C.; Bassi, C.; Palmieri, M.; Scarpa, A. Gene expression profiling after treatment with the histone deacetylase inhibitor trichostatin A reveals altered expression of both pro- and anti-apoptotic genes in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta 2004, 1693, 167–176. [Google Scholar]
- Glaser, K.B.; Staver, M.J.; Waring, J.F.; Stender, J.; Ulrich, R.G.; Davidsen, S.K. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol. Cancer Ther. 2003, 2, 151–163. [Google Scholar]
- Gray, S.G.; Qian, C.N.; Furge, K.; Guo, X.; Teh, B.T. Microarray profiling of the effects of histone deacetylase inhibitors on gene expression in cancer cell lines. Int. J. Oncol. 2004, 24, 773–795. [Google Scholar]
- Crabb, S.J.; Howell, M.; Rogers, H.; Ishfaq, M.; Yurek-George, A.; Carey, K.; Pickering, B.M.; East, P.; Mitter, R.; Maeda, S.; Johnson, P.W.; Townsend, P.; Shin-ya, K.; Yoshida, M.; Ganesan, A.; Packham, G. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem. Pharmacol. 2008, 76, 463–475. [Google Scholar]
- Schrump, D.S.; Fischette, M.R.; Nguyen, D.M.; Zhao, M.; Li, X.; Kunst, T.F.; Hancox, A.; Hong, J.A.; Chen, G.A.; Kruchin, E.; Wright, J.J.; Rosing, D.R.; Sparreboom, A.; Figg, W.D.; Steinberg, S.M. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin. Cancer Res. 2008, 14, 188–198. [Google Scholar]
- Ellis, L.; Pan, Y.; Smyth, G.K.; George, D.J.; McCormack, C.; Williams-Truax, R.; Mita, M.; Beck, J.; Burris, H.; Ryan, G.; Atadja, P.; Butterfoss, D.; Dugan, M.; Culver, K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin. Cancer Res. 2008, 14, 4500–4510. [Google Scholar]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar]
- Vervoorts, J.; Luscher-Firzlaff, J.; Luscher, B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006, 281, 34725–34729. [Google Scholar]
- Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Simms-Waldrip, T.; Fu, C.; Sakamoto, K.M. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008, 68, 2557–2560. [Google Scholar]
- Simms-Waldrip, T.; Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Fu, C.; Sakamoto, K.M. The aggresome pathway as a target for therapy in hematologic malignancies. Mol. Genet. Metab. 2008, 94, 283–286. [Google Scholar]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.E.; Lee, S.W.; Moon, E.J.; Kim, H.S.; Lee, S.K.; Chung, H.Y.; Kim, C.W.; Kim, K.W. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar]
- Fath, D.M.; Kong, X.; Liang, D.; Lin, Z.; Chou, A.; Jiang, Y.; Fang, J.; Caro, J.; Sang, N. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. J. Biol. Chem. 2006, 281, 13612–13619. [Google Scholar]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell Biol. 2006, 26, 2019–2028. [Google Scholar]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 2004, 279, 41966–41974. [Google Scholar]
- Mottet, D.; Bellahcene, A.; Pirotte, S.; Waltregny, D.; Deroanne, C.; Lamour, V.; Lidereau, R.; Castronovo, V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 2007, 101, 1237–1246. [Google Scholar]
- Liu, T.; Kuljaca, S.; Tee, A.; Marshall, G.M. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat. Rev. 2006, 32, 157–165. [Google Scholar]
- Kwon, H.J.; Kim, M.S.; Kim, M.J.; Nakajima, H.; Kim, K.W. Histone deacetylase inhibitor FK228 inhibits tumor angiogenesis. Int. J. Cancer 2002, 97, 290–296. [Google Scholar]
- Qian, D.Z.; Wang, X.; Kachhap, S.K.; Kato, Y.; Wei, Y.; Zhang, L.; Atadja, P.; Pili, R. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004, 64, 6626–6634. [Google Scholar]
- Sasakawa, Y.; Naoe, Y.; Noto, T.; Inoue, T.; Sasakawa, T.; Matsuo, M.; Manda, T.; Mutoh, S. Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem. Pharmacol. 2003, 66, 897–906. [Google Scholar]
- Zgouras, D.; Becker, U.; Loitsch, S.; Stein, J. Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem. Biophys. Res. Commun. 2004, 316, 693–697. [Google Scholar]
- Kang, J.H.; Kim, M.J.; Chang, S.Y.; Sim, S.S.; Kim, M.S.; Jo, Y.H. CCAAT box is required for the induction of human thrombospondin-1 gene by trichostatin A. J. Cell. Biochem. 2008, 104, 1192–1203. [Google Scholar]
- LaBonte, M.J.; Wilson, P.M.; Fazzone, W.; Groshen, S.; Lenz, H.J.; Ladner, R.D. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med. Genomics 2009, 2, 67. [Google Scholar]
- Mie Lee, Y.; Kim, S.H.; Kim, H.S.; Jin Son, M.; Nakajima, H.; Jeong Kwon, H.; Kim, K.W. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochem. Biophys. Res. Commun. 2003, 300, 241–246. [Google Scholar]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; Castronovo, V. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar]
- Rossig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Forstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 2002, 91, 837–844. [Google Scholar]
- Michaelis, M.; Suhan, T.; Cinatl, J.; Driever, P.H.; Cinatl, J., Jr. Valproic acid and interferon-alpha synergistically inhibit neuroblastoma cell growth in vitro and in vivo. Int. J. Oncol. 2004, 25, 1795–1799. [Google Scholar]
- Michaelis, M.; Michaelis, U.R.; Fleming, I.; Suhan, T.; Cinatl, J.; Blaheta, R.A.; Hoffmann, K.; Kotchetkov, R.; Busse, R.; Nau, H.; Cinatl, J., Jr. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol. Pharmacol. 2004, 65, 520–527. [Google Scholar]
- Hellebrekers, D.M.; Melotte, V.; Vire, E.; Langenkamp, E.; Molema, G.; Fuks, F.; Herman, J.G.; Van Criekinge, W.; Griffioen, A.W.; van Engeland, M. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007, 67, 4138–4148. [Google Scholar]
- Yu, C.; Friday, B.B.; Lai, J.P.; McCollum, A.; Atadja, P.; Roberts, L.R.; Adjei, A.A. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin. Cancer Res. 2007, 13, 1140–1148. [Google Scholar]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Verheul, H.M.; Salumbides, B.; Van Erp, K.; Hammers, H.; Qian, D.Z.; Sanni, T.; Atadja, P.; Pili, R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin. Cancer Res. 2008, 14, 3589–3597. [Google Scholar]
- Murakami, J.; Asaumi, J.; Maki, Y.; Tsujigiwa, H.; Kuroda, M.; Nagai, N.; Yanagi, Y.; Inoue, T.; Kawasaki, S.; Tanaka, N.; Matsubara, N.; Kishi, K. Effects of demethylating agent 5-aza-2(')-deoxycytidine and histone deacetylase inhibitor FR901228 on maspin gene expression in oral cancer cell lines. Oral Oncol. 2004, 40, 597–603. [Google Scholar]
- Hellebrekers, D.M.; Castermans, K.; Vire, E.; Dings, R.P.; Hoebers, N.T.; Mayo, K.H.; Oude Egbrink, M.G.; Molema, G.; Fuks, F.; van Engeland, M.; Griffioen, A.W. Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 2006, 66, 10770–10777. [Google Scholar]
- Suuronen, T.; Nuutinen, T.; Ryhanen, T.; Kaarniranta, K.; Salminen, A. Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2007, 357, 397–401. [Google Scholar]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A., 2nd; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; Giles, F.J.; Nawrocki, S.T. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2009. [Google Scholar]
- Crighton, D.; Wilkinson, S.; O'Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar]
- Crighton, D.; O'Prey, J.; Bell, H.S.; Ryan, K.M. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ. 2007, 14, 1071–1079. [Google Scholar]
- Rosenbluth, J.M.; Mays, D.J.; Pino, M.F.; Tang, L.J.; Pietenpol, J.A. A gene signature-based approach identifies mTOR as a regulator of p73. Mol. Cell Biol. 2008, 28, 5951–5964. [Google Scholar]
- Rosenbluth, J.M.; Pietenpol, J.A. mTOR regulates autophagy-associated genes downstream of p73. Autophagy 2009, 5, 114–116. [Google Scholar]
- Oh, M.; Choi, I.K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–322. [Google Scholar]
- Walker, T.; Mitchell, C.; Park, M.A.; Yacoub, A.; Graf, M.; Rahmani, M.; Houghton, P.J.; Voelkel-Johnson, C.; Grant, S.; Dent, P. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol. Pharmacol. 2009, 76, 342–355. [Google Scholar]
- Cao, Q.; Yu, C.; Xue, R.; Hsueh, W.; Pan, P.; Chen, Z.; Wang, S.; McNutt, M.; Gu, J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int. J. Biochem. Cell Biol. 2008, 40, 272–283. [Google Scholar]
- Hrzenjak, A.; Kremser, M.L.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; Johnstone, R.W. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar]
- Meech, S.J.; Edelson, R.; Walsh, P.; Norris, D.A.; Duke, R.C. Reversible resistance to apoptosis in cutaneous T cell lymphoma. Ann. N. Y. Acad. Sci. 2001, 941, 46–58. [Google Scholar]
- Cory, S.; Adams, J.M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. Apoptosis: mechanisms and relevance in cancer. Ann. Hematol. 2005, 84, 627–639. [Google Scholar]
- Rossi, D.; Gaidano, G. Messengers of cell death: apoptotic signaling in health and disease. Haematologica 2003, 88, 212–218. [Google Scholar]
- Thornberry, N.A.; Lazebnik, Y. Caspases: enemies within. Science. 1998, 281, 1312–1316. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin. Ther. Targets 2005, 9, 809–824. [Google Scholar]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar]
- Dokmanovic, M.; Marks, P.A. Prospects: histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 293–304. [Google Scholar]
- Lindemann, R.K.; Gabrielli, B.; Johnstone, R.W. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle 2004, 3, 779–788. [Google Scholar]
- Fantin, V.R.; Loboda, A.; Paweletz, C.P.; Hendrickson, R.C.; Pierce, J.W.; Roth, J.A.; Li, L.; Gooden, F.; Korenchuk, S.; Hou, X.S.; Harrington, E.A.; Randolph, S.; Reilly, J.F.; Ware, C.M.; Kadin, M.E.; Frankel, S.R.; Richon, V.M. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008, 68, 3785–3794. [Google Scholar]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer. 2008, 8, 782–798. [Google Scholar]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.; Sharkey, J.; Anthony, D.A.; Banks, K.M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; Bolden, J.E.; Takeda, K.; Yagita, H.; Secrist, J.P.; Smyth, M.J.; Johnstone, R.W. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl. Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar]
- Earel, J.K., Jr.; VanOosten, R.L.; Griffith, T.S. Histone deacetylase inhibitors modulate the sensitivity of tumor necrosis factor-related apoptosis-inducing ligand-resistant bladder tumor cells. Cancer Res. 2006, 66, 499–507. [Google Scholar]
- Kauh, J.; Fan, S.; Xia, M.; Yue, P.; Yang, L.; Khuri, F.R.; Sun, S.Y. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One 2010, 5, e10376. [Google Scholar]
- Xu, W.; Ngo, L.; Perez, G.; Dokmanovic, M.; Marks, P.A. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc. Natl. Acad. Sci. USA 2006, 103, 15540–15545. [Google Scholar]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; Weisz, A.; de Lera, A.R.; Gronemeyer, H.; Altucci, L. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar]
- Singh, T.R.; Shankar, S.; Srivastava, R.K. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene 2005, 24, 4609–4623. [Google Scholar]
- MacFarlane, M.; Inoue, S.; Kohlhaas, S.L.; Majid, A.; Harper, N.; Kennedy, D.B.; Dyer, M.J.; Cohen, G.M. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005, 12, 773–782. [Google Scholar]
- Imai, T.; Adachi, S.; Nishijo, K.; Ohgushi, M.; Okada, M.; Yasumi, T.; Watanabe, K.; Nishikomori, R.; Nakayama, T.; Yonehara, S.; Toguchida, J.; Nakahata, T. FR901228 induces tumor regression associated with induction of Fas ligand and activation of Fas signaling in human osteosarcoma cells. Oncogene 2003, 22, 9231–9242. [Google Scholar]
- Sutheesophon, K.; Nishimura, N.; Kobayashi, Y.; Furukawa, Y.; Kawano, M.; Itoh, K.; Kano, Y.; Ishii, H.; Furukawa, Y. Involvement of the tumor necrosis factor (TNF)/TNF receptor system in leukemic cell apoptosis induced by histone deacetylase inhibitor depsipeptide (FK228). J. Cell Physiol. 2005, 203, 387–397. [Google Scholar]
- de Ruijter, A.J.; Meinsma, R.J.; Bosma, P.; Kemp, S.; Caron, H.N.; van Kuilenburg, A.B. Gene expression profiling in response to the histone deacetylase inhibitor BL1521 in neuroblastoma. Exp. Cell Res. 2005, 309, 451–467. [Google Scholar]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol. Cancer Ther. 2004, 3, 425–435. [Google Scholar]
- Sanda, T.; Okamoto, T.; Uchida, Y.; Nakagawa, H.; Iida, S.; Kayukawa, S.; Suzuki, T.; Oshizawa, T.; Miyata, N.; Ueda, R. Proteome analyses of the growth inhibitory effects of NCH-51, a novel histone deacetylase inhibitor, on lymphoid malignant cells. Leukemia 2007, 21, 2344–2353. [Google Scholar]
- Rosato, R.R.; Maggio, S.C.; Almenara, J.A.; Payne, S.G.; Atadja, P.; Spiegel, S.; Dent, P.; Grant, S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol. Pharmacol. 2006, 69, 216–225. [Google Scholar]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar]
- Peart, M.J.; Tainton, K.M.; Ruefli, A.A.; Dear, A.E.; Sedelies, K.A.; O'Reilly, L.A.; Waterhouse, N.J.; Trapani, J.A.; Johnstone, R.W. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003, 63, 4460–4471. [Google Scholar]
- Lindemann, R.K.; Newbold, A.; Whitecross, K.F.; Cluse, L.A.; Frew, A.J.; Ellis, L.; Williams, S.; Wiegmans, A.P.; Dear, A.E.; Scott, C.L.; Pellegrini, M.; Wei, A.; Richon, V.M.; Marks, P.A.; Lowe, S.W.; Smyth, M.J.; Johnstone, R.W. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc. Natl. Acad. Sci. USA 2007, 104, 8071–8076. [Google Scholar]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar]
- Inoue, S.; Riley, J.; Gant, T.W.; Dyer, M.J.; Cohen, G.M. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia 2007, 21, 1773–1782. [Google Scholar]
- Zhang, Y.; Adachi, M.; Kawamura, R.; Imai, K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006, 13, 129–140. [Google Scholar]
- Li, P.; Wang, D.; Yao, H.; Doret, P.; Hao, G.; Shen, Q.; Qiu, H.; Zhang, X.; Wang, Y.; Chen, G. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene 29, 3153–3162. [PubMed]
- Mahalingam, D.; Medina, E.C.; Esquivel, J.A., 2nd; Espitia, C.M.; Smith, S.; Oberheu, K.; Swords, R.; Kelly, K.R.; Mita, M.M.; Mita, A.C.; Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin. Cancer Res. 16, 141–153. [PubMed]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar]
- Whitecross, K.F.; Alsop, A.E.; Cluse, L.A.; Wiegmans, A.; Banks, K.M.; Coomans, C.; Peart, M.J.; Newbold, A.; Lindemann, R.K.; Johnstone, R.W. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood 2009, 113, 1982–1991. [Google Scholar]
- Park, C.M.; Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K.C.; Nimmer, P.; Shoemaker, A.R.; Song, X.; Tahir, S.K.; Tse, C.; Wang, X.; Wendt, M.D.; Yang, X.; Zhang, H.; Fesik, S.W.; Rosenberg, S.H.; Elmore, S.W. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 2008, 51, 6902–6915. [Google Scholar]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; Roberts, L.; Tahir, S.K.; Xiao, Y.; Yang, X.; Zhang, H.; Fesik, S.; Rosenberg, S.H.; Elmore, S.W. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 3, 5. [PubMed]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar]
- Ramalingam, S.S.; Parise, R.A.; Ramanathan, R.K.; Lagattuta, T.F.; Musguire, L.A.; Stoller, R.G.; Potter, D.M.; Argiris, A.E.; Zwiebel, J.A.; Egorin, M.J.; Belani, C.P. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin. Cancer Res. 2007, 13, 3605–3610. [Google Scholar]
- Fakih, M.G.; Pendyala, L.; Fetterly, G.; Toth, K.; Zwiebel, J.A.; Espinoza-Delgado, I.; Litwin, A.; Rustum, Y.M.; Ross, M.E.; Holleran, J.L.; Egorin, M.J. A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin. Cancer Res. 2009, 15, 3189–3195. [Google Scholar]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; Daud, A. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar]
- Schneider, B.J.; Bradley, D.; Smith, D.C.; Egorin, M.; Kalemkerian, G.; Dunn, R.; Daignault, S.; Hussain, M. Phase I study of vorinostat plus docetaxel in patients with solid tumor maligancies. J. Clin. Oncol. 2009, 27, Abstr. 2528. [Google Scholar]
- Tredaniel, J.; Descourt, R.; Moro-Sibilot, D.; Misset, J.; Gachard, E.; Garcia-Vargas, J.; Roben, E.; Zalcman, G. Vorinostat in combination with gemcitibine and cisplatinum in patients with advanced non-small cell lung cancer (NSCLC): A phase I dose-escalation study. J. Clin. Oncol. 2009, 27, Abstr. 8049. [Google Scholar]
- Reguart, N.; Cardona, A.F.; Isla, D.; Cardenal, F.; Palmero, R.; Carrasco-Chaumel, E.; Rolfo, C.; Massuti, B. Phase I trial of vorinostat in combination with erlotinib in advanced non-small cell lung cancer (NSCLC) patients with EGFR mutations after erlotinib progression. J. Clin. Oncol. 2009, 27, Abstr. e19057. [Google Scholar]
- Ninan, J.A.; Bailey, H.; Kolesar, J.; Marnocha, R.; Eickoff, J.; Wright, J.; Espinoza-Delgado, I.; Aberti, D.; Wilding, G.; Schelman, W. A phase I study of vorinostat in combination with bortezomib in refractory solid tumors. J. Clin. Oncol. 2009, 27, Abstr. 2531. [Google Scholar]
- Doss, H.H.; Jones, S.F.; Infante, J.R.; Spigel, D.R.; Willcutt, N.; Lamar, R.; Barton, J.; Keegan, M.; Burris, H.A. A phase I trial of romidepsin in combination with gemcitabine in patients with pancreatic and other advanced solid tumors. J. Clin. Oncol. 2008, 26, Abstr. 2567. [Google Scholar]
- Berenson, J.R.; Yellin, O.; Mapes, R.; Eades, B.; Abaya, C.D.; Strayer, A.; Nix, D.; Swift, R.A. A phase II study of a 1-hour infusion of romidepsin combined with bortezomib for multiple myeloma (MM) patients with relapsed or refractory disease. J. Clin. Oncol. 2009, 27, Abstr. e19508. [Google Scholar]
- Konduri, K.; Spira, A.I.; Jotte, R.M.; Boyd, T.; Gaffar, Y.A.; Reynolds, C.; Witta, S.E. Results from a phase I safety lead-in study investigating the combination of erlotinib and the histone deacetylase inhibitor entinostat in patients with advanced NSCLC. J. Clin. Oncol. 2009, 27, Abstr. e14545. [Google Scholar]
- Juergens, R.A.; Vendetti, F.; Coleman, B.; Sebree, R.S.; Rudek, M.A.; Belinsky, S.; Brock, M.; Herman, J.; Baylin, S.; Rudin, C.M. Interim analysis of a phase II trial of 5-azacitidine (5AC) and entinostat (SNDX-275) in relapsed advanced lung cancer (NSCLC). J. Clin. Oncol. 2009, 27, Abstr. 8055. [Google Scholar]
- Conte, P.; Campone, M.; Pronzato, P.; Amadori, D.; Frank, R.; Shuetz, F.; Rea, D.; Wardley, A.; Britten, C.; Elias, A. Phase I trial of panobinostat (LBH589) in combination with trastuzumab in pretreated HER2-positive metastatic breast cancer (mBC): Preliminary safety and tolerability results. J. Clin. Oncol. 2009, 27, Abstr. 1081. [Google Scholar]
- Spencer, A.; Taylor, K.; Lonial, S.; Mateos, M.V.; Jalaluddin, M.; Hazell, K.; Bourquelot, P.M.; San Miguel, J.F. Panobinostat plus lenalidomide and dexamethasone phase I trial in multiple myeloma (MM). J. Clin. Oncol. 2009, 27, Abstr. 8542. [Google Scholar]
- Rathkopf, D.E.; Chi, K.N.; Vaishampayan, U.; Hotte, S.; Vogelzang, N.; Alumkal, J.; Agrawal, M.; Hydam, T.M.; Fandi, A.; Scher, H.I. Phase Ib dose finding trial of intravenous panobinostat with docetaxel in patients with castration-resistant prostate cancer (CRPC). J. Clin. Oncol. 2009, 27, Abstr. 5064. [Google Scholar]
- Pili, R.; Lodge, M.; Verheul, H.; Mashtare, T.; Wahl, R.L.; Martin, J.E.; Espinoza-Delgado, I.; Liu, G.; Carducci, M.A. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in pre-treated patients with renal cell carcinoma: safety, efficacy and pharmacodynamic results. In ASCO 2010 Genitourinary Cancers Symposium, San Francisco, CA, USA, 5–7 March 2010; p. Abstr. 350.
- Dasari, A.; Gore, L.; Messersmith, W.A.; Diab, S.; Jimeno, A.; Weekes, C.D.; Lewis, K.D.; Drabkin, H.A.; Flaig, T.W.; Camidge, D.R. A phase I safety and tolerability study of vorinostat (V) in combination with sorafenib (S) in patients with advanced solid tumors, with exploration of two tumor-type specific expanded cohorts at the recommended phase II (renal and non-small cell lung carcinoma). J. Clin. Oncol. 2010, 28, Abstr. 2562. [Google Scholar]
- Dickson, M.A.; Rathkopf, D.E.; Grant, S.; Roberts, J.D.; Reid, J.M.; Ames, M.M.; McGovern, R.M.; Gonen, M.; Dials, H.J.; Schwartz, G.K. Phase I trial of pulse-dose vorinostat with flavopiridol in solid tumors. J. Clin. Oncol. 2010, 28, Abstr. e13511. [Google Scholar]
- Munster, P.N.; Petrou, P.; Ryan, C.J.; Jahan, T.M.; DuBois, S.G.; Rugo, H.S.; Chan, J.K.; Thurn, K.T.; Reinert, A.; Daud, A. A phase I trial of the histone deacetylase inhibitor panobinostat (LBH589) and epirubicin in patients with with solid tumor malignancies. J. Clin. Oncol. 2010, 28, Abstr. e13140. [Google Scholar]
- Wardley, A.M.; Stein, R.; McCaffrey, J.; Crown, J.; Malik, Z.; Rea, D.; Barrett-Lee, P.J.; Lee, G.T. Phase II data for entinostat, a class I selective inhibitor, in patients whose breast cancer is progressing on aromatase inhibitor therapy. J. Clin. Oncol. 2010, 28, Abstr. 1052. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ellis, L.; Pili, R. Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies. Pharmaceuticals 2010, 3, 2441-2469. https://doi.org/10.3390/ph3082441
Ellis L, Pili R. Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies. Pharmaceuticals. 2010; 3(8):2441-2469. https://doi.org/10.3390/ph3082441
Chicago/Turabian StyleEllis, Leigh, and Roberto Pili. 2010. "Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies" Pharmaceuticals 3, no. 8: 2441-2469. https://doi.org/10.3390/ph3082441